Agencia Estatal Boletín Oficial del Estado






Suscrito el 25 de julio de 2017, Convenio de colaboración entre el Ministerio de Sanidad, Servicios Sociales e Igualdad (Dirección General de Salud Pública, Calidad e Innovación), y Gilead Sciences, S.L.U., en el contexto del estudio sobre evaluación de la factibilidad de la implementación de la profilaxis preexposición (PrEP), como estrategia de prevención de la infección por el VIH en población de alto riesgo en el Sistema Nacional de Salud, y en cumplimiento de lo dispuesto en el artículo 48.8 de la Ley 40/2015, de 1 de octubre, de Régimen Jurídico del Sector Público, procede la publicación en el «Boletín Oficial del Estado» de dicho convenio, que figura como anexo de esta Resolución.
Madrid, 27 de julio de 2017.–El Secretario General de Sanidad y Consumo, José Javier Castrodeza Sanz.
En Madrid a 25 de julio de 2017
REUNIDOS
De una parte, doña Elena Andradas Aragonés, Directora General de Salud Pública, Calidad e Innovación del Ministerio de Sanidad, Servicios Sociales e Igualdad, nombrada por Real Decreto 1049/2015, de 13 de noviembre, actuando en ejercicio de las competencias que le han sido delegadas de conformidad con lo dispuesto en el apartado Sexto.2, letra c), de la Orden SSI/131/2013, de 17 de enero.
De otra parte, doña Carmen María Río Presa, con DNI número 09365707-S, en calidad de Consejera Delegada de Gilead Sciences, S.L.U., sociedad constituida de acuerdo con la legislación española, con domicilio social en Madrid, calle Vía de los Poblados, 3, Parque Empresarial Cristalia, Edificio 7/8, planta 6.ª, Código Postal 28033, con CIF B80754799, en virtud de escritura elevada a público el 18 de mayo de 2012 ante el Notario del Ilustre Colegio de Madrid, D. Juan Carlos Carnicero Íñiguez, con el número 533 de su protocolo.
EXPONEN
Primero.–El Ministerio de Sanidad, Servicios Sociales e Igualdad, a través del Plan Nacional sobre el Sida de la Subdirección General de Promoción de la Salud y Epidemiología en la Dirección General de Salud Pública, Calidad e Innovación, establece a través de su Plan Estratégico de Prevención y Control del VIH y otras ITS 2013-2016 en su objetivo 4, disminuir la incidencia de infecciones por el VIH y otras ITS, que tanto la prevención combinada del VIH como la de otras ITS, son estrategias de salud pública, y que consecuentemente, tienen que incluir tanto políticas estructurales que favorezcan el desarrollo de los servicios de salud, como la mejora de las condiciones de vida de la población y contemplar igualmente acciones dirigidas a eliminar el estigma y la discriminación asociadas al VIH.
El Real Decreto 485/2017, de 12 de marzo, desarrolla la estructura orgánica básica del Ministerio de Sanidad, Servicios Sociales e Igualdad en su artículo 10, punto 3, establece que a la Dirección General de Salud Pública, Calidad e Innovación «le corresponde la elaboración de los sistemas de información, el impulso de planes de salud y programas de calidad en el Sistema Nacional de Salud, incluido el Plan Nacional sobre Sida, así como el análisis y evaluación del funcionamiento del sistema sanitario español y su comparación con otros sistemas sanitarios»
La prevención primaria debe dirigirse tanto a la población general como a grupos prioritarios por ser más vulnerables, a través de intervenciones específicas, combinando medidas preventivas dirigidas a reducir la transmisión del VIH y de otras ITS.
La profilaxis preexposición (PrEP) es una intervención biomédica dirigida a prevenir la transmisión del VIH en personas seronegativas para el VIH con alto riesgo de contraer la infección. Consiste en el uso diario, de un medicamento antirretroviral antes de la exposición al virus, y se acompaña de un paquete de medidas preventivas para mejorar la adherencia e incidir en el cambio de conductas. Existen estudios que han demostrado la eficacia de la PrEP como prevención de la transmisión del VIH junto con otras medidas preventivas.
La Agencia Europea del medicamento (EMA), emitió un informe positivo para la indicación de Truvada® (Tenofovir disoproxil 245 mg + Emtricitabina 200 mg), como tratamiento preventivo de la infección por el VIH, a dosis diaria de 1 comprimido acompañado de otras medidas de prevención, siendo aprobada la indicación por la Comisión Europea por procedimiento centralizado.
Segundo.–Que Gilead Sciences INC, es una compañía biotecnológica estadounidense dedicada a investigar, descubrir, desarrollar y comercializar fármacos para tratar enfermedades. Desde su fundación en 1987 y durante muchos años la compañía se ha dedicado principalmente a desarrollar medicamentos antivirales para tratar pacientes infectados con el VIH, la hepatitis y otras infecciones virales.
Que es la propietaria de la patente de Truvada® y mediante su filial, Gilead Sciences, S.L.U., comercializa y suministra esta presentación farmacológica en España.
Tercero.–Que es el deseo de ambas partes realizar actividades que permitan generar conocimiento respecto a estrategias de prevención de infección frente al VIH que pueden ser complementarias a las ya implantadas en el Sistema Nacional de Salud español.
Por todo ello, el Ministerio de Sanidad, Servicios Sociales e Igualdad (en adelante MSSSI) y Gilead Sciences, S.L.U. (en adelante GILEAD), establecen el siguiente Convenio, que se regirá por las siguientes
CLÁUSULAS
Puesta en marcha de un estudio para valorar la factibilidad de la implementación de la PrEP como estrategia de prevención de la infección por el VIH en población de alto riesgo en el Sistema Nacional de Salud», de ahora en adelante el «Estudio».
El MSSSI, a través del Plan Nacional sobre el Sida de la Subdirección General de Promoción de la Salud y Vigilancia en Salud Pública de la Dirección General de Salud Pública, Calidad e Innovación será el Promotor y Coordinador del Estudio y junto con los centros participantes, desarrollara las actividades necesarias para la consecución del objeto del mismo.
1. Correspondientes al MSSSI.
1.1 Diseñar el protocolo de estudio (Se adjunta en anexo A).
1.2 Diseñar una plataforma online para la recogida de datos incluidos en el Protocolo de estudio.
1.3 Gestionar la plataforma con tres entornos, centros participantes, Comunidades Autónomas y Ministerio.
1.4 Recoger, depurar y analizar los datos que los centros participantes en el Estudio introduzcan en la plataforma.
1.5 Convocar las reuniones que considere necesarias con las entidades participantes en el Estudio y solicitar la información adicional que determine necesaria para la adecuada ejecución del Estudio.
1.6 Elaborar el informe final de resultados y la publicación. En dicha publicación de los resultados, figurará por escrito la colaboración de GILEAD.
2. Correspondientes a GILEAD.
2.1 Donación del fármaco Truvada® 200 mg/245 mg comprimidos recubiertos con película (30 comprimidos C.N.: 650921) para la realización del Estudio. La donación consistirá en medicación para un máximo de 400 personas, a dosis de un comprimido al día durante 52 semanas de seguimiento, que supondría un máximo de 364 comprimidos por cada persona que concluya el Estudio.
2.2 Realizará los trámites necesarios con las CCAA participantes en el Estudio para la donación del medicamento Truvada®, la identificación de las farmacias hospitalarias donde realizará el suministro de la medicación y las condiciones de entrega de la misma.
2.3 GILEAD suministrará la medicación a los servicios de farmacia de los hospitales identificados en el Protocolo del Estudio (anexo A). GILEAD queda exonerada de cualquier responsabilidad legal derivada del suministro de la medicación una vez realizada la entrega de la misma a los servicios de farmacia de los hospitales incluidos en el Protocolo de Estudio.
3.1 El promotor será responsable de la gestión de los datos de seguridad del Estudio así como de comunicar a las autoridades correspondientes, investigadores y comités éticos tanto los informes individuales como los informes periódicos de seguridad, de conformidad con toda la Legislación y disposiciones vigentes.
3.2 El promotor comunicará al Departamento de Farmacovigilancia (Drug Safety & Public Health, DSPH) de GILEAD, en la dirección indicada más adelante, cualquier posible problema de seguridad, enmienda del Protocolo o cambio en el Formulario de consentimiento informado que se derive de un problema de seguridad asociado a cualquier producto de GILEAD, en el periodo de quince (15) días naturales desde que tenga conocimiento de dicho acontecimiento.
3.3 El promotor notificará todos los acontecimientos adversos graves (AAG), así como todos los informes de situaciones especiales y seroconversiones en relación con cualquier producto de GILEAD al Departamento de Farmacovigilancia de GILEAD en el periodo de quince (15) días naturales desde que tenga conocimiento de dicha información de seguridad y de conformidad con toda la Legislación vigente. Todos los informes dirigidos a GILEAD en virtud de esta sección deben enviarse a la atención de:
Departamento de Farmacovigilancia GILEAD.
Fax: 91 378 98 41.
Teléfono: 91 771 24 80.
E-mail: farmacovigilancia.spain@GILEAD.com.
3.4 Tras la solicitud de GILEAD, el promotor aportará cualquier información adicional que se requiera para realizar las evaluaciones médicas de cualquier información de seguridad proporcionada a GILEAD. El promotor proporcionará a GILEAD toda la ayuda razonable aportando cualquier otra información que GILEAD solicite. GILEAD enviará dichas solicitudes de información adicional a:
Dirección General de Salud Pública, Calidad e Innovación.
Ministerio de Sanidad, Servicios Sociales e Igualdad.
Paseo del Prado, 18-20, 28071 Madrid.
3.5 El promotor será responsable de preparar los informes periódicos de seguridad necesarios para el Estudio. A fin de evitar dudas, GILEAD no desea recibir copias de estos informes de periódicos de seguridad.
3.6 Una vez finalizado el estudio, el promotor enviará al Departamento de Farmacovigilancia de GILEAD un informe de reconciliación de datos en el que se incluirán todos los informes de seguridad previamente enviados. Como mínimo, la lista debe incluir el número de protocolo, el número de identificación del sujeto, el número de referencia del caso, el producto de GILEAD y la descripción del acontecimiento.
3.7 Con excepción de los informes periódicos de seguridad, el promotor proporcionará a GILEAD una copia de todos los informes relacionados presentados a las autoridades reguladoras, así como cualquier correspondencia con dichas autoridades relacionada con el Estudio.
Las definiciones de esta cláusula vienen recogidas en el anexo B.
Para el seguimiento y evaluación de este Convenio, se constituirá una Comisión Paritaria de Seguimiento compuesta por representantes de ambos partícipes, que tendrá las siguientes funciones:
– Revisar la ejecución objeto del presente Convenio de Colaboración.
– Interpretar, en caso de duda, el contenido del presente Convenio de Colaboración, y en consecuencia, proponer las decisiones oportunas acerca de las variaciones o cambios aconsejables para la mejor ejecución de las actuaciones.
– El seguimiento y dirección de la ejecución de las actuaciones contempladas en el Convenio de Colaboración.
La composición de la Comisión Paritaria de Seguimiento será la siguiente:
Dos personas en representación de la Dirección General de Salud Pública, Calidad e Innovación del Ministerio de Sanidad, Servicios Sociales e Igualdad.
Dos personas en representación de GILEAD.
Esta Comisión Paritaria de Seguimiento se reunirá al menos dos veces al año y, en cualquier caso, cuando una de las partes lo solicite. Las reuniones irán precedidas de la correspondiente agenda donde consten los puntos a tratar. Se levantará acta de cada una de las reuniones, recogiendo los acuerdos alcanzados. Estas actas se circularán a los miembros de la Comisión para que en el plazo de 5 días formulen los comentarios que estimen oportunos. El acta final deberá remitirse como máximo en el plazo de 15 días desde que tuvo lugar la reunión de la Comisión.
El presente Convenio no conlleva contraprestación económica por parte del MSSSI. En el caso de GILEAD, la aportación económica estará en relación con el coste de la donación de la medicación.
El presente Convenio producirá sus efectos al día siguiente de su firma, una vez inscrito en el Registro Electrónico estatal de órganos e instrumentos de Cooperación del sector público estatal y publicados en el Boletín Oficial del Estado, y concluirá a la finalización del Estudio, previsible para el 15 de diciembre de 2018, considerando el seguimiento de 52 semanas desde el inicio del mismo del último participante.
No obstante, las partes, de mutuo acuerdo, podrán acordar su prórroga, formalizando a tal efecto, con anterioridad a la fecha de vencimiento del mismo, la oportuna adenda con las condiciones de la prórroga, de acuerdo con el artículo 49 de la Ley 40/2015, de Régimen Jurídico del Sector Público.
Sin perjuicio de su extinción por el cumplimiento de las actuaciones que constituyen su objeto, y en base al artículo 51 de la Ley 40/2015, de 1 de octubre, el presente Convenio podrá resolverse por alguna de las siguientes causas:
1. El transcurso del plazo de vigencia del convenio sin haberse acordado la prórroga del mismo.
2. El acuerdo unánime de todos los firmantes.
3. El incumplimiento de las obligaciones y compromisos asumidos por parte de alguno de los firmantes.
En este caso, cualquiera de las partes podrá notificar a la parte incumplidora un requerimiento para que cumpla en un plazo de 30 días con las obligaciones o compromisos que se consideran incumplidos. Este requerimiento será comunicado al responsable del mecanismo de seguimiento, vigilancia y control de la ejecución del Convenio y a las demás partes firmantes.
Si trascurrido el plazo indicado en el requerimiento persistiera el incumplimiento, la parte que lo dirigió notificará a las partes firmantes la concurrencia de la causa de resolución y se entenderá resuelto el convenio.
4. Por decisión judicial declaratoria de la nulidad del convenio.
En caso de resolución del Convenio, las partes quedan obligadas al cumplimiento de sus respectivos compromisos hasta la fecha en que ésta se establezca, no afectando a la finalización de las actuaciones que en tal momento se hubieran comenzado a ejecutar.
Todo ello sin perjuicio de la aplicación de lo previsto en el artículo 52 de la Ley 40/2015, en lo que resulte procedente.
El presente Convenio, así como las actividades que se realicen en el desarrollo del mismo, se regirán por las cláusulas firmadas entre ambas partes en el presente documento.
El presente Convenio tiene naturaleza administrativa, quedando excluido del Texto Refundido de la Ley de Contratos del Sector Público, al amparo del artículo 4.1.d), excepto para la resolución de las dudas que pudieran presentarse, supuesto en el que resultarán de aplicación los principios contenidos en el citado texto legal.
El Convenio se ajusta igualmente a lo establecido en la Ley 40/2015, de 1 de octubre, de Régimen Jurídico del Sector Público y en particular en cuanto a su contenido mínimo (artículo 49).
Cualquier discrepancia que surja en relación a la interpretación, desarrollo, modificación y efectos que pudieran derivarse de la aplicación del presente Convenio, deberán solventarse con carácter previo, por la Comisión cuya composición se recoge en la cláusula tercera del presente Convenio, encargada de regular y coordinar las actividades del mismo. Aquellas discrepancias que no se resuelvan en el marco de la Comisión Paritaria, se someterán a la jurisdicción contencioso-administrativa, de conformidad con lo dispuesto en la Ley 29/1998, de 13 de julio, reguladora de la citada Jurisdicción.
En cumplimiento de la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal y del Reglamento 1720/2007 que desarrolla dicha Ley, las partes intervinientes se comprometen a utilizar los datos de carácter personal a los que pudiera tener acceso con objeto de este Convenio conforme a dicha normativa y demás disposiciones vigentes, adoptando las medidas de seguridad legalmente exigidas, a fin de garantizar la confidencialidad de los datos personales que pudiesen ser tratados.
Los derechos de explotación, en todas las modalidades, de la documentación que se produzca como consecuencia de las actuaciones contempladas en este Convenio, y sin perjuicio de lo dispuesto en la Sección segunda del Capítulo III del Título II Libro I de la Ley de Propiedad Intelectual, aprobada por Real Decreto Legislativo 1/1996, de 12 de abril, serán propiedad del MSSSI.
El MSSSI y GILEAD declaran que han realizado y realizarán sus actividades en relación con el presente Convenio cumpliendo en todo momento con la legislación española contra la corrupción (esto es, Ley Orgánica 10/1995, de 23 de noviembre, del Código Penal y sus modificaciones). A estos efectos, el MSSI y Gilead declaran que disponen de los procedimientos necesarios para evitar el soborno y corrupción por parte de sus representantes.
En cumplimiento de la Ley 40/2015, de 1 de octubre, de Régimen Jurídico del Sector Público, artículo 49, sobre «Contenido de los convenios», la modificación del contenido del convenio requerirá acuerdo unánime de los firmantes.
En prueba de conformidad, ambas partes firman el presente documento, por duplicado, en el lugar y fecha ut supra.–La Directora General de Salud Pública, Calidad e Innovación, Elena Andradas Aragonés.–La Consejera Delegada de Gilead Sciences, Carmen María Río Presa.
Introducción
La PrEP es una intervención biomédica dirigida a prevenir la transmisión del VIH en personas seronegativas para el VIH con alto riesgo de contraer la infección. Consiste en el uso diario, de un medicamento antirretroviral antes de la exposición al virus, y se acompaña de un paquete de medidas preventivas para mejorar la adherencia e incidir en el cambio de conductas.
La Agencia Europea del medicamento (EMA), emitió un informe positivo para la indicación de Truvada® (Tenofovir disoproxil 245 mg + Emtricitabina 200 mg comprimidos recubiertos con película) como tratamiento preventivo de la infección por el VIH, a dosis diaria de 1 comprimido, siendo aprobada la indicación por la Comisión Europea por procedimiento centralizado.
Diferentes estudios han demostrado su eficacia, dependiendo de multitud de variables, siendo en alguno de ellos cercanas al 86%1,2. Estos datos se han conseguido debido a la alta adherencia y al uso combinado de otras medidas de prevención.
Tal y como se recoge en el Plan Estratégico de Prevención y Control del VIH y otras ITS 2013-2016, se ha constatado un repunte de algunos comportamientos de riesgo y un menor uso del preservativo, como parece indicar el incremento de las ITS. Los HSH son un grupo de población especialmente afectado por el VIH en todo el mundo, y la epidemia está reemergiendo entre ellos en los países con más recursos. Coordinar intervenciones estructurales, biomédicas y conductuales que incorporen estrategias eficaces, capaces de alcanzar a una mayor proporción de HSH, podría reducir de forma importante la incidencia del VIH entre ellos3,4.
Tanto la prevención combinada del VIH como la de otras ITS, son estrategias de salud pública, y consecuentemente, tienen que incluir tanto políticas estructurales que favorezcan el desarrollo de los servicios de salud, como la mejora de las condiciones de vida de la población. En la prevención del VIH y otras ITS es fundamental considerar medidas de reducción de las desigualdades sociales y territoriales en salud. Las intervenciones propuestas deben ser accesibles, incluyendo el aspecto económico, especialmente entre las poblaciones renuentes a la prevención y las que se encuentran en situaciones de vulnerabilidad.
Se plantea por tanto, un estudio de implementación que permita responder a las preguntas operativas relacionadas con la introducción de esta medida, así como la identificación de posibles efectos colaterales de la misma, a tener en cuenta en la planificación de servicios.
Referencias
1. Molina, J-M, Capitant ,C, Spire, B, Pialoux ,G, Cotte, L, Charreau, I, et al. On-Demand Preexposure Prophylaxis in Men at High Risk for HIV-1 Infection. N Engl J Med. 2015 Dec 3;373(23):2237-46.
2. McCormack, S, Dunn, DT, Desai, M, Dolling DI, Gafos, M, Gilson, R, et al. Pre-exposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial. Lancet Lond Engl. 2016 Jan 2;387(10013):53-60.
3. Sullivan, PS et al. Successes and challenges of HIV prevention in men who have sex with men. Lancet. 2012 July 28; 380(9839): 388-399.35.
4. Fernández de Mosteyrín, S, et al. Prácticas y percepción del riesgo en hombres con infección por el virus de la inmunodeficiencia humana que tienen sexo con otros hombres. Enferm Infecc Microbiol Clin. 2013. http://dx.doi.org/10.1016/j.eimc.2013.04.01736
Objetivos
El objetivo principal y general del estudio es evaluar la factibilidad de la implementación de la PrEP en la estrategia general de prevención de la infección por el VIH en población de alto riesgo en el Sistema Nacional de Salud.
Los objetivos concretos son los siguientes:
– Evaluar la factibilidad de llevar a cabo en España un programa de prevención del VIH en el que se integre la PrEP con el cumplimiento de las condiciones de uso autorizadas.
– Evaluar de la posible interferencia de la PrEP con el resto de acciones que se llevan a cabo en el ámbito comunitario en España para la prevención de la infección VIH en población de alto riesgo.
– Evaluar la factibilidad de llevar a cabo la PrEP utilizando los circuitos asistenciales existentes: reclutamiento de posibles candidatos, evaluación del circuito, seguimiento de los sujetos, derivación a atención especializada.
– Evaluar el impacto de la PrEP en el dispositivo asistencial actual en sujetos con alto riesgo de infección VIH.
– Evaluar el impacto económico de la implementación de la PrEP
Participantes y métodos
Se trata de un estudio post-autorización, de diseño observacional, prospectivo promovido por la Administración Sanitaria y clasificado por la Agencia Española del Medicamento y Productos sanitarios (AEMPS) como como EPA-AS código DIR-TRU-2017-01.
Participantes
Se incluirán un máximo de 400 hombres que tienen sexo con hombres (HSH) y personas transexuales, que acepten participar de forma voluntaria y por escrito en el estudio, una vez explicadas las características del mismo y que cumplan los siguientes:
Criterios de inclusión:
1. Edad mayor o igual a 18 años y menor de 65 años.
2. Confirmación de prueba negativa reciente de VIH (menos de 1 semana) por prueba antigénica/ ELISA de 4.ª generación.
3. HSH y personas transexuales, con riesgo sustancial de infección por el VIH, por lo que deben de cumplir al menos, dos de los siguientes criterios:
a. Más de 10 parejas diferentes en el último año.
b. Práctica de sexo anal sin protección en el último año.
c. Consumo de drogas recreativas en el último año.
d. Uso de Profilaxis post-exposición en el último año.
e. Al menos una ITS bacteriana en el último año.
4. Consentimiento informado para participar en el estudio firmado por el sujeto participante.
Criterios de exclusión:
1. Personas con diagnóstico conocido de VIH
2. Personas que pudieran estar en periodo ventana de seroconversión, infección activa o reciente sugerente de la misma.
3. Filtrado glomerular calculado (CockcroftGault) menos de 60ml/min o fosfato sérico menor de 1,5.
4. Personas con antecedentes de osteoporosis o fracturas por fragilidad ósea.
5. Personas coinfectados con VHB o VHC o con sospecha de hepatopatía (evaluación analítica).
6. Personas con alta sospecha de no adherencia (faltas reiteradas a las citas programadas o recogida de resultados o aquellos que no puedan completar el seguimiento).
Variables de resultado (ver anexo 1)
1. Idoneidad del circuito asistencial.
a. Captación: Sujetos candidatos, proporción de sujetos que cumplen criterios de inclusión/exclusión, proporción de sujetos incluidos.
b. Seguimiento: Proporción de abandonos o retirada prematura de la PrEP y causas.
– Derivación a atención especializada y razones.
– VIH positivo.
– Reacciones adversas al fármaco (incluyendo deterioro función renal y otras).
– Visitas no realizadas y abandono por el paciente: razones.
2. Cumplimiento de las condiciones de la nueva indicación de uso de Truvada.
– Uso de preservativo: proporción de uso al inicio del estudio vs al final del seguimiento.
– Adherencia satisfactoria al tratamiento, mayor de un porcentaje pre-especificado de sujetos.
– Detección de otras ITS.
3. Factibilidad de la integración de la PrEP y sostenibilidad de la estrategia (anexo 2).
– Carga de trabajo adicional en el dispositivo asistencial: valoración del número de horas dedicadas a la PrEP por sujeto participante en relación con las horas dedicadas a la atención de esos sujetos en el caso de atención sin incluir la PrEP.
– Encuesta de satisfacción del paciente con el programa (Anexo 3).
– Encuesta de satisfacción de los profesionales con el programa (Anexo 5.)
Coordinación del estudio y centros participantes
Promotor y coordinador del estudio
|
Centro |
Investigador principal |
Cargo |
|
|---|---|---|---|
|
Promotor del estudio. |
Ministerio de Sanidad Servicios Sociales e Igualdad. |
Elena Andradas. |
Directora General de Salud Pública, Calidad e Innovación. |
|
Coordinadores del estudio. |
S.G de Promoción de la Salud y Epidemiología. |
Araceli Arce. |
Subdirectora G. de Promoción de la Salud y Epidemiología. |
|
Plan Nacional sobre el SIDA. |
Begoña Rodríguez. Ortiz de Salazar. Olivia Castillo. Rosa Polo. |
S.G. Adjunta de Promoción de la Salud y Epidemiología. Jefa de Área de Prevención. Jefa de Área Asistencial. |
Centros participantes en el estudio
|
CC.AA. |
Investigador principal |
Centro |
Investigador asociado |
|---|---|---|---|
|
Cataluña. |
Joan Colom, Subdirector General de drogas. Director en funciones. |
Unidad de ITS Drassanes/Hospital Universitari Vall d’Hebron. |
M.ª Jesús Barberá Gracia, Coordinadora de la Unidad de ITS Drassanes. |
|
Checkpoint/Hospital Germans Trias i Pujol. |
Bonaventura Clotet, Jefe Unidad de VIH. |
||
|
País Vasco. |
Antonio Arraiza Arantza Arrillaga, Coordinador del Plan de Sida e ITS. |
Centro de ITS de San Sebastián/Hospital Donosti. José Antonio Iribarren, Jefe de Servicio de Infecciosas. Hospital de Donosti. |
|
Material y métodos
Diseño: estudio post-autorización observacional prospectivo promovido por la Administración Sanitaria y clasificado por la AEM como como EPA-AS código DIR-TRU-2017-01.
Ámbito: CC.AA. de Cataluña y País Vasco.
Periodo de estudio: 52 semanas de seguimiento desde la inclusión en el estudio.
Identificación de las personas susceptibles de PrEP: serán los centros comunitarios y otros centros adscritos al SNS, los propios centros de ITS y los hospitales participantes, los que identificaran a las personas susceptibles de uso de la PrEP de entre aquellas que acuden a dichos centros de forma habitual o espontáneamente. Una vez identificadas, se informará a la persona de los objetivos del estudio y de la necesidad de cumplir los requisitos recogidos en el protocolo para poder participar en el mismo. En caso de aceptación del participante, se remitirán al centro de ITS correspondiente y designado para la selección de los participantes donde se objetivará si cumple los criterios para poder ser incluido en el protocolo de estudio.
Selección de los participantes: se hará en los centros de ITS y otros centros designados por la CCAA, con vinculación directa con un hospital con farmacia oficialmente reconocida los que recibirán a las personas identificadas como posibles participantes. En el centro de ITS/otros centros, se seleccionará al participante que cumpla los criterios de inclusión y que esté dispuesto a participar en el estudio y a recibir la profilaxis. Se le explicará de forma detenida que es la PrEP, cuales son las medidas preventivas asociadas y cuál es el objetivo del estudio.
A los sujetos que cumplan con todos los criterios de inclusión y ninguno de los de exclusión y que firmen el consentimiento para participar en el estudio, se procederá a incluirlos en el protocolo e iniciar la profilaxis con Truvada® 1 comprimido una vez al día. En el caso de que no firmen el consentimiento o que decidan retirarlo en cualquier momento, sus datos no serán a partir de entonces incorporados a la base de datos del estudio, pero ello no afectará a su atención sanitaria ni a la relación con su médico, que decidirá la estrategia terapéutica más adecuada para el sujeto de acuerdo con la práctica clínica habitual.
La adherencia deberá ser prioritaria desde el inicio de las entrevistas y se medirá mediante el cuestionario SMAQ (Simplified Medication AdherenceQquestionnaire) (anexo 4) y con contaje de pastillas.
Desde que se realiza la analítica basal hasta el día 1 de inicio, se deberá establecer un periodo de 21 días de diferencia con el objeto de que al repetir el VIH, se haya cumplido ya, el posible periodo ventana. Durante este tiempo se recomendará a la persona extremar las medidas de prevención y usar de forma consistente el preservativo. Esta misma recomendación se mantendrá a lo largo de todo el estudio.
Toda esta información deberá darse de forma verbal y por escrito para que no exista duda de la comprensión de la misma (Anexo 6). El participante deberá firmar un consentimiento de que acepta los términos del estudio quedando reflejado que podrá abandonar el mismo si así lo desea en cualquier momento.
Población y muestra: Se establece un periodo de inclusión de 3 meses, siendo un reclutamiento competitivo hasta un total de 400 personas, estableciendo un mínimo de 75 personas por CCAA.
Una vez que se ha constatado que la persona cumple los criterios de inclusión (previa realización de los estudios analíticos correspondientes según Tabla 1 y anexo 2) y que ha firmado el consentimiento informado (Anexo 6), se incluirán sus datos en la ficha del protocolo destinada para ello y se remitirá al participante a la farmacia de referencia para la dispensación del fármaco.
Análisis: El seguimiento de los participantes se realizará en los centros de ITS y/o en el Centro Comunitario designado por la CCAA que disponga de autorización de la misma y que esté ligada a un Hospital. Este seguimiento se realizará basalmente y a las 4, 12, 24, 36, 48 y 52 semanas. En la tabla 1 se recogen todas las determinaciones/actuaciones y cumplimentaciones a realizar en cada una de las semanas de seguimiento durante el protocolo.
Dispensación de la medicación: Tal y como viene recogido en la ficha técnica, el medicamento Truvada® es de dispensación hospitalaria. Sin embargo, existen excepciones siendo la Comunidad Autónoma la que puede tomar la decisión de dispensación de fármacos fuera del ámbito hospitalario. Por ello, si la Comunidad Autónoma lo autoriza, se podrá dispensar en otros centros sanitarios que dispongan de farmacia acreditada. Para ello, y si se autorizara, ambos centros se pondrán en contacto y establecerán los acuerdos necesarios para la dispensación de la medicación, acuerdos que deberán comunicar al promotor del estudio para su conformidad. Si no se produjera esta autorización, será la farmacia del hospital la que dispense la medicación.
Recogida de datos: El MSSSI se encargará de la creación un portal informático donde estarán recogidos todos los anexos para poder realizar la inclusión y seguimiento de los participantes de forma centralizada.
Criterios de retirada del estudio: Cualquier causa que viole el protocolo del estudio supondrá la exclusión del participante del mismo. Se incluyen los siguientes supuestos:
1. Positividad de la serología a VIH.
2. No asistencia a las citas programadas (dado que las citas se realizarán cada 3 meses, la falta a una de las citas supondrá la retirada del estudio).
3. No recogida de la medicación en la fecha indicada que suponga la ausencia de toma de la medicación durante más de 48h.
4. Falta de adherencia a la medicación (Cuestionario Smaq).
5. Presencia de algún efecto adverso.
6. Por decisión del participante.
7. Por decisión del profesional.
Evaluación económica
En la tabla 2, se anotará el coste de cada prueba por persona y revisión.
Se incluirán para la evaluación los costes directos, es decir, los controles analíticos periódicos (cada 3 meses), que se estiman en 550 €/ usuario/ año para las pruebas de control incluidas en el protocolo. Esto supondría un total de 220.000€ para el total de personas incluidas (400), asumiendo que todos se mantuvieran hasta la finalización del estudio.
Basados en los resultados de los ensayos clínicos realizados hasta ahora, la pérdida de personas por abandono o retirada por efectos adversos, oscila entre el 8 y el 19%. Esto supondría una pérdida de 32 a 76 personas con una disminución en el coste de 18.000 a 41.000 €.
La medicación Truvada®, será donada por la compañía GILEAD SCIENCE para la realización del estudio. Esta donación se realizará directamente a las farmacias acreditadas de los centros participantes en el estudio que hayan sido designadas por las CCAA.
Ni el promotor, ni los coordinadores ni los investigadores recibirán compensación económica por la realización de este estudio. Tampoco recibirán compensación económica las personas que decidan participar en el mismo.
Aspectos éticos y legales
Se garantizará el cumplimiento de la Ley Orgánica 15/1999, del 13 de diciembre, de Protección de Datos de Carácter Personal para proteger el derecho a la intimidad de los participantes. Se respetarán en todo momento las Buenas Prácticas Clínicas de la investigación y se garantizará el anonimato de las personas y hospitales o centros asistenciales participantes tanto en la construcción de las bases de datos como en cualquier tipo de difusión externa de los resultados. Se respetará la Declaración de Helsinki y sucesivas actualizaciones. Los datos recogidos para el estudio estarán identificados mediante un código y sólo el profesional que incluya al participante podrá relacionar dichos datos. Los resultados obtenidos solo se podrán publicar de forma anónima y nunca de forma individual. La propiedad de los datos es del MSSSI y no se podrá publicar ni parcial ni totalmente ninguna información procedente del Estudio sin autorización expresa del Ministerio.
Al tratarse de un estudio observacional, y de acuerdo con lo estipulado en la normativa vigente, no se asignará de antemano al sujeto del estudio a una estrategia terapéutica o profiláctica determinada, la decisión de prescribir Truvada® como PrEP no se tomará junto con la de incluir al sujeto en el estudio, ni se aplicarán procedimientos de diagnóstico o seguimiento a los sujetos del estudio que vayan más allá de la práctica clínica habitual.
Al tratarse de un estudio observacional el aseguramiento se realizará a través del SNS, confirmandose que todos los participantes disponen de tarjeta sanitaria.
Previo al inicio del estudio el protocolo se ha sometido a valoración del respectivo Comité Ético de Investigación Clínica (CEIC). Todos los participantes en el estudio deberán firmar un consentimiento informado por escrito.
Todos los participantes en la elaboración del Protocolo de estudio y los profesionales participantes en el mismo, deberán firmar una declaración de intereses según modelo autorizado por la Dirección General de Salud Pública del MSSSI.
|
Variables de resultados |
Indicador |
Fórmula |
Objetivo |
|---|---|---|---|
|
Idoneidad del circuito asistencial |
|||
|
Captación: Sujetos candidatos. |
Proporción de sujetos que cumplen criterios de inclusión. |
Número de personas que cumplen criterios de inclusión x 100/número de personas inicialmente seleccionados. |
|
|
Proporción de sujetos que cumplen criterios de exclusión. |
Número de personas que cumplen criterios de exclusión x 100/número de personas inicialmente seleccionados. |
||
|
Proporción de sujetos incluidos. |
Número de personas finalmente incluidos x 100/número de personas inicialmente seleccionados. |
||
|
Seguimiento. |
Proporción de retirada prematura de la PrEP. |
Proporción de personas con retirada prematura de la PrEP de los personas incluidos. |
|
|
Proporción de abandonos de la PrEP. |
Número de personas que abandonan la PrEP. Anexo 2. |
||
|
Casos de VIH positivo durante el estudio. |
Número de infecciones por VIH durante el estudio X 100/número de participantes en el estudio. Anexo 2. |
||
|
Reacciones adversas a la medicación. |
Sí/No. Anexo 2. |
||
|
Proporción de personas con efectos adversos. |
Número de personas con efectos adversos X 100/ número de personas participantes en el estudio. Anexo 2. |
||
|
Derivación al especializada. |
Sí/No. |
||
|
Proporción de participantes derivados a especializada. |
Número de personas derivadas al médico especialista X 100/ número de personas participantes en el estudio. |
||
|
Motivo de la derivación. |
|||
|
Porcentaje de cumplimentación de visitas y de abandonos. |
Porcentaje de personas completan las visitas. |
Número de personas con visitas completas X 100/número de participantes en el estudio. |
|
|
Porcentaje de participantes perdidos al final del estudio. |
Número de personas perdidas en el seguimiento durante el estudios X 100/número de participantes en el estudio. |
<20% |
|
|
Porcentaje de cumplimentación a las 4 semanas del estudio. |
Número de participantes en la semana 4 X 100/ Número de personas que inician el estudio. |
||
|
Porcentaje de cumplimentación a las 12 semanas del estudio. |
Número de participantes en la semana 12 X 100/ Número de personas que inician el estudio. |
||
|
Porcentaje de cumplimentación a las 24 semanas del estudio. |
Número de participantes en la semana 24 X 100/ Número de personas que inician el estudio. |
||
|
Porcentaje de cumplimentación a las 36 semanas del estudio. |
Número de participante en la semana 36 X 100/ Número de personas que inician el estudio. |
||
|
Porcentaje de cumplimentación a las 48 semanas del estudio. |
Número de participante en la semana 48 X 100/ Número de personas que inician el estudio. |
||
|
Porcentaje de cumplimentación a las 52 semanas del estudio. |
Número de participantes en la semana 52 X 100/ Número de personas que inician el estudio. |
||
|
Barreras y/o dificultades a la hora de realizar la captación, intervención y seguimiento de la PrEP. |
Anexo 3. |
||
|
Cumplimiento de las condiciones de uso de TRUVADA junto con otras medidas de prevención. |
|||
|
Detección y tratamiento de ITS. |
Casos de ITS durante el estudio. |
Número de infecciones de transmisión sexual durante el estudio. |
|
|
Número de personas con infecciones de transmisión sexual durante el estudio X 100/número de participantes en el estudio. |
|||
|
Porcentaje de tratamiento de ITS. |
Número de infecciones de transmisión sexual tratadas durante el estudio X 100/número de infecciones de transmisión sexual durante el estudio. |
>95% |
|
|
Proporción de personas que utilizan el preservativo como medida de prevención combinada. |
Uso de preservativo como medida combinada. Anexo 3. |
Sí/No. |
|
|
Número de personas que refieren el uso del preservativo como medida combinada X 100/Número de participantes del estudio. |
|||
|
Adherencia al tratamiento. |
Proporción de personas con adherencia a al tratamiento durante el estudio. |
Número de personas con adherencia a FARV X 100/Número de participantes del estudio. |
|
|
¿Ha habido adherencia? |
Anexo 4. |
||
|
¿Cuál ha sido el grado de adherencia? |
Anexo 4. |
||
|
Factibilidad de la integración de la PREP |
|||
|
Carga de trabajo adicional en el dispositivo asistencial. |
Tiempo que necesitó el profesional médico para la atención del participante en relación con tiempo dedicado a la atención de esas personas en el caso de atención, sin incluir la PrEP. |
Numero de minutos necesarios para la atención del participante incluido en la PrEP. Anexo 2. |
|
|
Tiempo que necesito el profesional de enfermería o auxiliar para la atención del participante en relación con tiempo dedicado a la atención de esas personas en el caso de atención sin incluir la PrEP. |
Numero de minutos necesarios para la atención del participante incluido en la PrEP. Anexo 2. |
||
|
Tiempo que necesitaron otros profesionales sanitarios para la atención del participante en relación con tiempo dedicado a la atención de esos personas en el caso de atención sin incluir la PrEP. |
Numero de minutos necesarios para la atención del participante incluido en la PrEP. Anexo 2. |
||
|
Percepción de facilidad de uso de los circuitos de actuación recogidos en la encuesta de satisfacción. |
Condiciones de intimidad y confidencialidad estructural. |
Sí/No. |
|
|
Existe relación del centro con el hospital de referencia y su farmacia. |
Sí/No. |
||
|
Satisfacción del participante con el programa. |
Anexo 3. |
||
|
Satisfacción de los profesionales con el programa. |
Anexo 5. |
||
|
Factibilidad. |
||||
|
Número total de personas atendidas del estudio por parte de profesional médico: |
||||
|
Total de tiempo dedicado a la atención de los participantes del estudio por parte del profesional médico (En minutos): |
||||
|
Número total de personas atendidas del estudio por parte de profesional de enfermería o auxiliar de enfermería: |
||||
|
Total de tiempo dedicado a la atención de los participantes del estudio por parte del profesional de enfermería o auxiliar de enfermería (En minutos): |
||||
|
Número total de personas atendidas del estudio por parte de otros profesionales sanitarios: |
||||
|
Total de tiempo dedicado a la atención de los participantes del estudio por parte del otros profesionales sanitarios (En minutos): |
||||
|
Casos nuevos de VIH. |
||||
|
Número de nuevos casos de VIH durante el periodo de estudio: |
||||
|
Seguimiento. |
||||
|
Número de participantes perdidos del estudio: |
||||
|
Número de personas en la semana 4 de seguimiento del estudio: |
||||
|
Número de personas en la semana 12 de seguimiento del estudio: |
||||
|
Número de personas en la semana 24 de seguimiento del estudio: |
||||
|
Número de personas en la semana 34 de seguimiento del estudio: |
||||
|
Número de personas en la semana 48 de seguimiento del estudio: |
||||
|
Número de personas en la semana 52 de seguimiento del estudio: |
||||
|
Efectos adversos*. |
||||
|
¿Ha habido algún efecto adverso?: |
SÍ |
NO |
||
|
Número de participante con efectos adversos: |
||||
* En caso de aparecer alguna alteración se remitirá al participante al hospital y se comunicará al departamento de farmacovigilancia AEM www.notificaram.es
Por favor responda a estas preguntas del grado de satisfacción en relación con la asistencia recibida considerando (1) como nada satisfecho, (2) poco satisfecho, (3) medianamente satisfecho y (4) muy satisfecho.
|
1 |
2 |
3 |
4 |
|
|---|---|---|---|---|
|
¿Considera que la información que se le ha dado en el centro comunitario le ha ayudado a tomar la decisión? |
||||
|
¿El tiempo de espera hasta ser atendido en el centro de ITS ha sido el adecuado? |
||||
|
¿Considera que el profesional que le ha atendido le ha explicado correctamente el estudio y ha resuelto sus dudas? |
||||
|
¿Considera que el profesional que le ha atendido le ha explicado correctamente los pasos que debe seguir una vez incluido en el protocolo de estudio para recoger la medicación? |
||||
|
El profesional que le ha atendido, ¿Se ha preocupado porque usted entienda el procedimiento? |
||||
|
En la farmacia. ¿Considera que ha tenido que esperar mucho tiempo? |
||||
|
El profesional de farmacia ¿Conocía el estudio en el que usted participa? |
||||
|
El profesional de la farmacia que le ha atendido, ¿Le ha explicado como tomar la medicación? |
||||
|
El profesional de farmacia ¿Le ha indicado cuando debe recoger nuevamente la medicación? |
||||
|
¿Le han explicado lo que debe de hacer en caso de presentar algún problema que pudiera estar relacionado con la medicación del estudio? |
||||
|
¿Cuál es su grado de satisfacción con la PrEP? |
Se realizará la semana, 4,12, 24, 36, 48 y 52 tal y como consta en la tabla 1.
|
1. Alguna vez ¿olvida tomar la medicación? |
□ Sí □ No |
|
2. ¿Toma siempre los fármacos a la hora indicada? |
□ Sí □ No |
|
3. Alguna vez ¿deja de tomar los fármacos si se siente mal? |
□ Sí □No |
|
4. ¿Olvidó tomar la medicación durante el fin de semana? |
□ Sí □ No |
|
5. En la última semana ¿cuántas veces no tomó alguna dosis?2 |
A: ninguna B: 1 - 2 C: 3 - 4 D: 5 - 7 |
|
6. Desde la última visita ¿cuántos días completos no tomó la medicación? |
Días: ... |
Se realizará la semana, 4, 12, 24, 36, 48 y 52, tal y como consta en la tabla 1.
1. Se considera no adherente:
1: sí.
2: no.
3: sí
4: sí,
5: C, D.
6: más de seis días.
El cuestionario es dicotómico, cualquier respuesta en el sentido de no adherente se considera no adherente.
2. La pregunta 5 se puede usar como semicuantitativa:
A: 95 - 100 % adhesión.
B: 85-94%
C: 65-84%
D: 30-64%
Por favor responda a estas preguntas en relación con las barreras a la hora de realizar la captación, intervención y seguimiento de la PrEP, considerando (1) como nada de acuerdo, (2) poco de acuerdo, (3) medianamente de acuerdo y (4) muy de acuerdo.
|
Captación |
1 |
2 |
3 |
4 |
|---|---|---|---|---|
|
El número de personas a los que se les ha ofrecido participar en el estudio ha respondido positivamente. |
||||
|
En caso de que la respuesta haya sido 1, explique por qué: |
||||
|
La proporción de sujetos que cumplen los criterios de inclusión ha sido la esperada |
||||
|
En caso de que la respuesta haya sido 1, explique por qué: |
||||
|
Intervención |
1 |
2 |
3 |
4 |
|
Las condiciones de intimidad y confidencialidad se han mantenido durante todo el proceso. |
||||
|
En caso de que la respuesta haya sido 1, explique por qué: |
||||
|
¿Dispone su centro de personal suficiente para atender a estas personas? |
||||
|
En caso de que la respuesta haya sido 1, explique por qué: |
||||
|
¿Considera adecuado el circuito de actuación diseñado para el estudio? |
||||
|
En caso de que la respuesta haya sido 1, explique por qué: |
||||
|
Seguimiento |
1 |
2 |
3 |
4 |
|
El número de abandonos de la medicación es inferior al 20% |
||||
|
En caso de que la respuesta haya sido 1, explique por qué: |
||||
|
La cumplimentación de las visitas supera el 85% |
||||
|
En caso de que la respuesta haya sido 1, explique por qué: |
||||
|
Considera que esta medida de prevención en su población asistida, ¿ha facilitado el abandono del preservativo? |
||||
|
En caso de que la respuesta haya sido 1, explique por qué: |
||||
|
¿La relación con el hospital ha sido ágil? |
||||
|
En caso de que la respuesta haya sido 1, explique por qué: |
||||
|
Observaciones: |
||||
Se realizará la semana, 4, 24, 48 y 52, tal y como consta en la tabla 1.
Tabla 1. Procedimientos del estudio. Seguimiento de los participantes
|
Intervenciones |
Basal |
* Día 1 |
Semana 4 |
Semana 12 |
Semana 24 |
Semana 36 |
Semana 48 |
Semana 52 |
|---|---|---|---|---|---|---|---|---|
|
Serología a VIH. |
X |
X |
X |
X |
X |
X |
X |
X |
|
Consentimiento informado (anexo 6). |
X |
|||||||
|
Historial médico. |
X |
|||||||
|
Examen físico completo. |
X |
X |
||||||
|
Examen genital, rectal y faríngeo. |
X |
X |
X |
X |
X |
X |
X |
|
|
Medicaciones y drogas concomitantes. |
X |
X |
X |
X |
X |
X |
X |
X |
|
Uso del preservativo como medida de prevención combinada. |
X |
X |
X |
X |
X |
X |
X |
|
|
Perfil hematológico y hepático ᴪ. |
X |
X |
X |
X |
X |
X |
||
|
Detección de ITS (sífilis, gonorrea, clamidia, VPH). |
X |
X |
X |
X |
X |
|||
|
Serología a VHB& y VHC. |
X |
X |
X |
|||||
|
Estimación del filtrado glomerularᴪ |
X |
X |
X |
X |
X |
X |
X |
|
|
Sistemático de orina con glucosuria, sedimento y cociente proteínas/creatinina ᴪ. |
X |
X |
X |
X |
X |
X |
||
|
Medición de la adherencia Cuestionario SMAQ (anexo 4). |
X |
X |
X |
X |
X |
X |
||
|
Recogida de información sobre la facilidad de cumplimiento del circuito de actuación (anexo 2). |
X |
X |
X |
X |
X |
X |
||
|
Consejo asistido. |
X |
X |
X |
X |
X |
X |
X |
X |
|
Satisfacción del paciente (anexo 3). |
X |
X |
X |
X |
X |
X |
||
|
Barreras a la hora de realizar la captación, intervención y seguimiento (anexo 5). |
X |
X |
X |
X |
||||
|
Cumplimentación de formulario de seguimiento (anexo 2). |
X |
X |
X |
X |
X |
X |
||
|
Cumplimentación de tabla de coste (tabla 2). |
X |
X |
X |
X |
X |
X |
X |
X |
* Desde que se realiza la analítica basal hasta el día 1 de inicio, se deberá establecer un periodo de 21 días de diferencia con el objeto de que al repetir el VIH, se haya cumplido ya, el posible periodo ventana. Durante este tiempo se recomendará a la persona extremar sus medidas de prevención y usar de forma consistente el preservativo.
ᴪ En caso de aparecer alguna alteración se remitirá al paciente al hospital y se comunicará al departamento de farmacovigilancia.
& En caso de personas con vacunación completa y correcta titulación para el virus B, no será necesario repetir la serología.
– El profesional a cargo deberá reevaluar la PrEP en cada visita y decidirá si el participante debe continuar o no con la misma. Para ello utilizará los criterios de exclusión y las variables de resultados.
– En caso de exclusión del estudio se especificará claramente el motivo del mismo.
– En caso de seroconversión a VIH, se remitirá al participante al centro hospitalario de referencia para inicio de TAR.
Tabla 2. Coste de las pruebas analíticas de laboratorio
|
Tipo de prueba |
Nombre en laboratorio |
Coste/ Ud. (€) |
Semana |
|---|---|---|---|
|
Ac+Ag VIH. |
|||
|
Perfil hematológico. |
|||
|
RPR. |
|||
|
Ac totales anti Treponema pallidum. |
|||
|
PCR de Chlamydia trachomatis y Neisseria gonorrhoeae. |
|||
|
Ag HBS. |
|||
|
CORE Ac- anti CORE. |
|||
|
AUSAB Ac anti Ag HBS. |
|||
|
Ag HBe. |
|||
|
Ac HBe. |
|||
|
Ac VHC. |
|||
|
Confirmativo VHC. |
|||
|
Ag CORE VHC. |
|||
|
Creatinina. |
|||
|
Filtrado Glomerular. |
|||
|
Anormal y sedimento en orina. |
|||
|
Glucosuria. |
|||
|
Cociente proteínas/ creatinina. |
|||
|
Subtotal. |
Título del estudio: Post-autorización, observacional prospectivo promovido por la Administración Sanitaria.
Promotor: Ministerio de Sanidad, Servicios Sociales e Igualdad.
Investigador principal: Nombre del Facultativo responsable en cada Centro Participante.
Centro: Subdirección General de Promoción de la Salud y Epidemiología. Dirección General de Salud Pública, Calidad e Innovación. Ministerio de Sanidad, Servicios Sociales e Igualdad. Paseo del Prado, 18-20, 28071 Madrid. Teléfono: 91 596 4196.
Introducción
Nos dirigimos a usted para informarle sobre un estudio de investigación en el que se le invita a participar. El estudio ha sido aprobado por un Comité de Ética de Investigación Clínico de acuerdo a la legislación vigente.
Nuestra intención es ofrecer una información correcta y suficiente para que comprenda y pueda decidir si desea o no participar en este estudio. Para ello lea esta hoja informativa con atención y nosotros le aclararemos las dudas que le puedan surgir.
Participación voluntaria
Le invitamos a participar en el estudio porque usted tiene alto riesgo de contraer la infección por VIH. Debe saber que su participación en este estudio es voluntaria y que puede decidir no participar.
Si decide participar y luego cambia su decisión, puede retirarse del estudio en cualquier momento comunicándoselo previamente a su médico.
En el caso de decidir no participar o dejar de hacerlo, sus datos no serán a partir de entonces incorporados a la base de datos del estudio, pero ello no afectará a su atención sanitaria ni a la relación con su médico, que decidirá la estrategia terapéutica más adecuada para usted de acuerdo con la práctica clínica habitual en su centro sanitario.
Si tiene alguna duda puede realizar todas las preguntas que considere oportunas.
Objetivo del estudio y procedimientos
La profilaxis pre-exposición (PrEP) es una intervención biomédica dirigida a prevenir la transmisión del VIH en personas seronegativas para el VIH con alto riesgo de contraer la infección. Consiste en el uso diario de un medicamento antirretroviral antes de la exposición al virus.
La Comisión Europea ha aprobado la indicación de Truvada® (Tenofovir disoproxil fumarato 300 mg + Emtricitabina 200 mg), a dosis diaria de 1 comprimido como tratamiento preventivo de la infección por el VIH, acompañado de otras medidas de prevención.
Para valorar la factibilidad de la PrEP en el Sistema Sanitario español, se ha puesto en marcha un estudio multicéntrico en el que se administrará Truvada® de forma diaria, a aquellas personas que cumplan los criterios de inclusión.
Para prevenir la transmisión de VIH y otras ITS se recomienda extremar las medidas de prevención y utilizar el preservativo. El profesional sanitario que le atienda, será el encargado de hacer las recomendaciones necesarias para extremar las medidas de prevención.
Una vez iniciada la PrEP y posteriormente, durante un año, se le realizará un seguimiento periódico en el centro donde se ha realizado el reclutamiento. Para ello se realizaran una historia clínica, un examen físico completo, analítica de control tanto antes de iniciar el estudio como posteriormente en las semanas 4, 12, 24, 36, 48 y 52 de iniciado el estudio. Así mismo, deberá rellenar diferentes encuestas de satisfacción que le serán entregadas por la persona que realizará su seguimiento.
Riesgos o molestias derivados de su participación
Tanto en los ensayos clínicos como en los estudios observacionales con PrEP puestos en marcha en los diferentes países se ha demostrado la seguridad de los mismos. Sin embargo, el uso de fármacos antirretrovirales no está exento de efectos adversos que, en el caso del Truvada®, pueden aparecer a nivel renal y a nivel óseo. Se ha descrito disminución del filtrado glomerular y disminución de la densidad mineral ósea que, en la mayoría de los casos, es reversible al suspender la medicación.
Los medicamentos antirretrovirales pueden ser incompatibles con otras medicinas y drogas, por lo que debe informar al médico de todos los medicamentos y drogas que esté tomando.
Beneficios potenciales
Es posible que su participación en el estudio suponga que usted no resulta infectado por el VIH pero no se puede garantizar que esto ocurra. En todo caso, su participación es muy importante ya que contribuirá a aumentar el conocimiento sobre la prevención de la infección.
Confidencialidad
Toda la información recogida para el estudio será estrictamente confidencial y se almacenará en un fichero de datos automatizados que está protegido de acuerdo con lo establecido en la Ley Orgánica 15/1999 de Protección de Datos de Carácter Personal y a su normativa de desarrollo. Los datos recogidos para el estudio estarán identificados mediante un código y sólo el investigador principal/colaboradores podrán relacionar dichos datos con usted. Los resultados obtenidos solo se podrán publicar de forma anónima y nunca de forma individual.
De acuerdo a lo que establece la legislación de protección de datos, usted puede ejercer los derechos de acceso, modificación, oposición y cancelación de datos, para lo cual deberá dirigirse a su médico del estudio.
Si usted decide retirar el consentimiento para participar en este estudio, ningún dato nuevo será añadido a la base de datos, pero sí se utilizarán los que ya se hayan recogido.
En caso de necesitar cualquier información o por cualquier otro motivo no dude en contactar con los profesionales que le han incluido en el estudio.
Consentimiento del participante
|
Título del estudio. |
Evaluación de la factibilidad de la implementación de la PrEP, como estrategia de prevención de la infección por el VIH en población de alto riesgo en el sistema nacional de salud. |
|
Centro. |
Yo,
Confirmo que:
– He leído la hoja de información que me han entregado.
– He podido hacer preguntas sobre el estudio.
– He recibido suficiente información sobre el estudio.
– Comprendo que mi participación es voluntaria.
– Comprendo que puedo retirarme del estudio cuando quiera, sin tener que dar explicaciones y sin que esto repercuta en mis cuidados médicos, y que puedo solicitar que la muestra no se utilice para la colección.
– Presto libremente mi conformidad para participar en el estudio.
Declaración del paciente:
El médico Dr./Dra. …………………………………………………. me ha explicado de forma satisfactoria qué es, cómo se realiza y para qué sirve la PrEP al VIH. También me ha explicado sus posibles riesgos. Soy consciente que la PrEP no siempre es eficaz, no existiendo garantías absolutas de que el resultado sea el más satisfactorio. He comprendido todo lo anterior perfectamente y doy mi consentimiento para que participar en el estudio.
Puedo retirar este consentimiento cuando lo desee.
Paciente D./D.ª ……………………………………………..., DNI……………………......…
Firma del paciente: Fecha:
Nombre del testigo:…………………………………............., DNI…………………………...
Firma del testigo: Fecha:
Firma del médico: …………………………….............……., N.º de colegiado …………...
Fecha:
Circuito de actuación
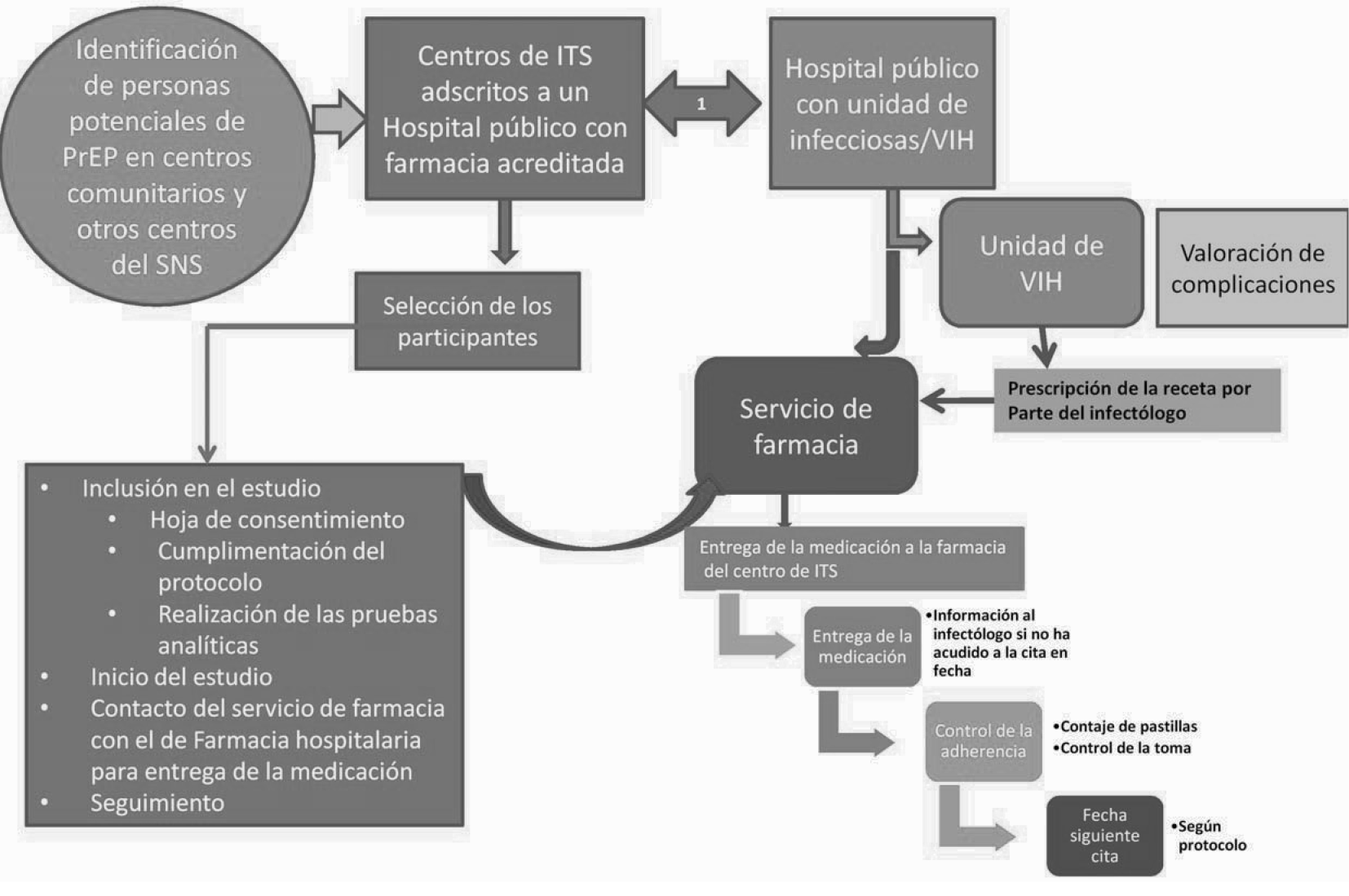
1. Se mantendrán reuniones periódicas para valorar la evolución de las personas incluidas. A las 12, 24, 36, 48 y 52 semanas se hará conjuntamente un análisis interno de la situación.
Abuso: Uso intencionado excesivo, esporádico o persistente, de un medicamento por parte de un paciente o un sujeto incluido en un ensayo clínico.
Acontecimiento Adverso («AE»): Cualquier acontecimiento médico que sufra un paciente o un sujeto incluido en un ensayo clínico que tome un medicamento, y que no tiene que tener una relación causal con el tratamiento. Un Acontecimiento Adverso (AE) por lo tanto puede ser cualquier signo desfavorable o involuntario (incluyendo, por ejemplo, un hallazgo de laboratorio anormal), síntoma o enfermedad temporal asociados con el uso de un medicamento tanto si se considera relacionado o no con el medicamento. Los AEs también pueden incluir complicaciones antes o después del tratamiento que ocurre como resultado de los procedimientos exigidos en el protocolo, falta de eficacia, sobredosis o informes de abuso/mal uso del medicamento. También se consideran AE los acontecimientos preexistentes que, como consecuencia de la participación en el estudio clínico, incrementa su gravedad o cambian su naturaleza.
Acontecimiento Adverso grave («SAE») / Reacción Adversa Grave («SAR»): cualquier acontecimiento o suceso médico indeseado que ocurre a cualquier dosis y:
a) Resulte en muerte; o
b) Amenaza la vida paciente;
NOTA: El término «amenazar la vida» en la definición de «grave» se refiere a un acontecimiento en que el paciente ha estado en peligro de muerte en el momento de suceder el acontecimiento; no se refiere a un acontecimiento que hipotéticamente podría haber causado la muerte si hubiese sido más grave; o
c) Requiere hospitalización o prolongación de la misma; o
d) Produce una incapacidad significativa o persistente; o
e) Produce una anomalía congénita o defecto de nacimiento; o
f) Es un acontecimiento o una reacción médicamente importante.
NOTA: acontecimiento médicamente importante: AEs que requiere de juicio médico y científico para determinar si es apropiado comunicarlo de manera expeditiva. Este tipo de acontecimientos no tienen por qué poner en peligro la vida del paciente ni causar la muerte u hospitalización, pero puede poner en peligro al paciente o puede requerir la intervención para prevenir otros problemas que resulten en SAEs. Se debe determinar si un acontecimiento es médicamente importante de acuerdo al juicio médico y científico. Algunos ejemplos de acontecimientos médicamente importantes son el tratamiento intensivo en urgencias o en casa debido a un broncoespasmo alérgico; discrasias sanguíneas o convulsiones que requieran hospitalización; o desarrollar dependencia al medicamento o abuso. Para evitar dudas, las infecciones resultantes de medicamentos contaminados se considerarán también un acontecimiento de importancia médica y sujeto comunicación expeditiva.
Error de medicación: Cualquier error involuntario en la prescripción, dispensación o administración de un medicamento, mientras que el medicamento está bajo el control de un profesional sanitario, un paciente o un consumidor.
Exposición al medicamento por razones del puesto de trabajo: Exposición a un medicamento como resultado de la ocupación profesional o no profesional.
Falta de eficacia: Situación donde hay un aparente fallo de un medicamento o tecnología médica sobre el pretendido efecto beneficioso esperado sobre los individuos en una población definida con un problema de salud determinado y bajo condiciones ideales de uso.
Inesperado: Un AE o AR donde la naturaleza o la gravedad de la reacción no es consistente con el término o descripción utilizado en el manual del investigador o en el etiquetado del medicamento.
Informes de embarazo: Mujer embarazada que entra en contacto con el medicamento directamente o vía semen de la pareja
Informe de situaciones especiales: cualquier a) Embarazo, b) Abuso, c) Error de Medicación, d)Mal uso e) Uso off-label, f) Sobredosis, g) Falta de eficacia, h) AE en lactantes tras exposición a la lactancia materna, i) AE asociado con reclamaciones de producto o que se derivan de exposición al medicamento por razones del puesto de trabajo. Para evitar dudas, esto se aplica a todos los informes, incluidos los informes en población pediátrica o anciana.
Mal uso: Cuando el medicamento se usa intencionadamente de forma inapropiada y en desacuerdo a las condiciones de autorización
Reacción Adversa («AR»): Acontecimiento médico (respuesta nociva e involuntaria) considerado causalmente relacionado con un medicamento bajo investigación o un medicamento autorizado a cualquier dosis administrada. Una reacción adversa puede ser causada por un error de medicación, uso fuera de lo que está previsto en el protocolo o de la información de prescripción (uso off-label), abuso o mal uso del medicamento, sobredosis o a la exposición laboral al medicamento.
Reclamaciones de producto: Quejas que surjan de posibles desviaciones en la fabricación, el embalaje o la distribución del medicamento.
Sobredosis: Administración de un medicamento en una cantidad, por administración o acumulada, superior a la máxima recomendada por protocolo o en la información de prescripción del medicamento. Las partes acuerdan que en el curso de la realización de un estudio clínico, los términos del Protocolo del estudio clínico (totalmente aprobado por todos los órganos aplicables) sustituyen al etiquetado local de los medicamentos.
Uso off-label: Cuando un medicamento se prescribe intencionalmente por un profesional sanitario para un propósito médico que no está de acuerdo con la información aprobada para el medicamento con respecto a la indicación, dosis, vía de administración o población de pacientes (por ejemplo los ancianos). Para evitar toda duda, el uso off-label no aplicará en los ensayos clínicos.
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid




