Agencia Estatal Boletín Oficial del Estado






 Este texto consolidado es de carácter informativo y no tiene valor jurídico.
Este texto consolidado es de carácter informativo y no tiene valor jurídico.[Bloque 1: #pr]
Excelentísimos señores:
El Decreto 544/1972, de 9 de marzo, que modifica determinados artículos del Reglamento de Centrales Lecheras y otras Industrias Lácteas, encomienda a los Ministerios de Agricultura y de Comercio, en la nueva redacción dada al artículo 91, la fijación de normas para las determinaciones analíticas y pruebas de la leche respecto a su composición a nivel ganadero o de relación ganadero-industria, así como las relativas a la tipificación comercial del producto industrializado.
Para la redacción de las citadas normas se ha considerado conveniente adaptarse en lo posible a las aprobadas por Organismos internacionales especializados en la materia, con el fin de aprovechar la experiencia habida en su aplicación y de facilitar la confrontación de los resultados en las relaciones comerciales supranacionales a las que España está abocada.
En consecuencia, en virtud de lo dispuesto en el mencionado artículo 91 del Reglamento de Centrales Lecheras y otras Industrias Lácteas, y a propuesta de los Ministros de Agricultura y de Comercio, esta Presidencia del Gobierno ha resuelto:
[Bloque 2: #pr-2]
Se aprueban las normas de toma de muestras y análisis de la leche que figuran respectivamente en los anejos 1 y 2 de esta disposición. Tales normas se considerarán como métodos oficiales de análisis a efectos de lo previsto en el artículo 91 del Reglamento de Centrales Lecheras y otras Industrias Lácteas.
Los métodos citados en el apartado anterior se refieren a las determinaciones cuantitativas de los distintos componentes de la leche y de azúcar de la leche condensada. En los casos en que se trate de investigar la adición o sustitución de los elementos naturales de la leche por otros extraños a dicho producto, operaciones prohibidas por el Reglamento de Centrales Lecheras y otras Industrias Lácteas, así como cualquier otro tipo de aditivo que no esté expresamente autorizado, se utilizarán las técnicas más idóneas en cada caso, según criterio conjunto de los Departamentos interesados.
[Bloque 3: #se]
La presente disposición entrará en vigor a los treinta días de su publicación en el «Boletín Oficial del Estado».
Lo que comunico a VV. EE. para su conocimiento y efectos.
Dios guarde a VV. EE.
[Bloque 4: #fi]
Madrid, 7 de julio de 1972.
CARRERO
Excmos. Sres. Ministros de Agricultura y de Comercio.
[Bloque 5: #a1]
Uno. Ámbito de aplicación.
Se describen a continuación los procedimientos que deben seguirse para la toma de muestras de leche, con objeto de obtener de una unidad (por ejemplo, un recipiente que contenga leche a granel, un pequeño envase para su venta directa al público, etc.) una parte que sea lo más representativa posible de la misma.
Los métodos descritos constituyen un extracto de la norma B-1 del Código de Principios referentes a la Leche y a los Productos Lácteos y normas derivadas del Programa Conjunto FAO/OMS sobre Normas Alimentarias.
Dos. Instrucciones generales de carácter técnico.
1. Materiales.
Todos los materiales utilizados para la toma de muestras deberán estar secos y limpios y reunir las características establecidas para cada tipo de producto.
2. Recipientes.
2.1. Los recipientes para las muestras de productos líquidos deberán ser de un material apropiado, impermeable al agua y a las grasas (vidrio, metal inoxidable, plástico especialmente acondicionado), que permitan su esterilización si fuera necesario, y de forma y capacidad adecuadas para los tipos de leche de los que se han de tomar las muestras, en las condiciones que más adelante se definen para cada caso en particular. Tales recipientes deberán estar secos y limpios y se podrán cerrar herméticamente, bien por medio de un tapón de caucho o de plástico, bien mediante una cápsula de metal o material plástico que cierre a rosca, provista interiormente de un revestimiento asimismo plástico, impermeable a los líquidos, insoluble, no absorbente, inatacable por las grasas y que no altere la composición de los productos lácteos. Si se utilizan tapones de caucho, éstos se recubrirán de un material de las características antes descritas (que puede ser igualmente un plástico apropiado), antes de ser introducidos en la boca del recipiente que contiene la muestra. Podrán utilizarse también bolsas de plástico adecuadas.
2.2. Los recipientes para las muestras de productos sólidos o semisólidos serán cilíndricos, de boca ancha y deberán reunir asimismo todas las condiciones expuestas en el apartado anterior. De igual manera podrán utilizarse también bolsas de plástico adecuadas.
2.3. En el caso de tratarse de pequeños envases para la venta al público, servirá como muestra el contenido de dichos recipientes intactos y cerrados. A tal efecto se consideran pequeños envases aquellos cuya capacidad sea inferior a dos litros.
3. Técnica.
El método exacto para la toma de muestras, así como el peso o el volumen que habrán de tomarse como tal muestra varían según los distintos tipos de leche y la finalidad para la que se requieren. En consecuencia, la técnica correspondiente se define más adelante para cada caso en particular.
4. Conservación de las muestras.
4.1. A las muestras de productos lácteos líquidos, destinadas a análisis químicos, podrán añadírseles una sustancia conservadora adecuada. Dichos conservadores no afectarán al análisis subsiguiente o, en todo caso, deberá poder determinarse el valor exacto con el que inciden en los resultados para realizar las correcciones necesarias. La naturaleza y cantidad utilizada de las sustancias conservadoras se indicarán en la etiqueta y en los informes. Preferentemente se utilizará dicromato potásico, en la proporción de un gramo por litro, o formaldehído en la proporción de un gramo por cada dos litros y medio.
4.2. No se añadirán sustancias conservadoras a las muestras de los productos lácteos sólidos, semisólidos o desecados. Dichas muestras se enfriarán rápidamente y se conservarán en cámara frigorífica a una temperatura entre 0º y 5 ºC hasta su análisis correspondiente, excepto cuando se trate de leches en polvo, que podrán conservarse a la temperatura ambiente.
5. Transporte.
Una vez tomadas, las muestras se llevarán al laboratorio lo antes posible. Durante el transporte se adoptarán las debidas precauciones para evitar que estén expuestas directamente al sol y, en el caso de productos perecederos, para no ser sometidos a temperaturas inferiores a 0 ºC ni superiores a 10 ºC.
Tres. Toma de muestras de las leches natural, certificada, higienizada y esterilizada.
1. Materiales.
1.1. Agitadores o émbolos para la mezcla de los líquidos a granel. Tales instrumentos deberán tener una superficie suficiente para provocar una buena agitación del producto y ser lo bastante ligeros en peso para que el operador pueda moverlos rápidamente en el líquido. Generalmente lo más apropiado es un agitador metálico de paletas anchas, provisto en su base de un gran disco perforado, y de suficiente longitud para llegar al fondo del recipiente. Para mezclar el contenido de recipientes de gran tamaño se aconseja la agitación por medios mecánicos.
1.2. Cuchara de tamaño adecuado para recoger la muestra.
2. Recipientes.
Véase lo indicado anteriormente en las instrucciones generales de carácter técnico (Dos, 2.1 y Dos, 2.3).
3. Técnicas.
3.1. En todos los casos se procederá a mezclar perfectamente el líquido. Dicha operación puede realizarse trasvasándolo repetidamente de un recipiente a otro, o agitándolo manual o mecánicamente.
3.2. En el caso de grandes recipientes deberá mantenerse la agitación del contenido de los mismos hasta que el líquido se haya mezclado de manera homogénea.
3.3. Inmediatamente después de efectuada la mezcla se tomará la muestra con una cuchara.
3.4. Si en la práctica resulta difícil conseguir una homogeneidad perfecta, se tomarán muestras de diferentes lugares del recipiente, que en conjunto representen un volumen total de por lo menos 250 mililitros.
Cuatro. Toma de muestras de las leches concentrada, evaporada y condensada.
1. Materiales.
Agitador para el caso de recipientes (cántaras, bidones, etc.) que contengan el producto a granel. Generalmente, lo más apropiado es un agitador metálico de paletas anchas, provisto en su base de un gran disco perforado y de suficiente longitud para que llegue al fondo del recipiente.
2. Recipientes.
2.1. Los recipientes para las muestras deberán ser de boca ancha y sus tapas cerrar herméticamente (Dos, 2.1. y Dos, 2.2).
2.2. En el caso de tratarse de pequeños envases para la venta al público, la muestra consistirá en uno de tales envases, intacto y sin abrir (Dos, 2.3), y siempre que sea posible llevará, las marcas de identificación del fabricante.
3. Técnica.
3.1. El agitador se utilizará para desprender el producto que se haya adherido a las paredes y al fondo del recipiente y para mezclar cuidadosamente el contenido de este último. Se pasarán de dos a tres litros del producto bien mezclado a un recipiente más pequeño. En éste se repetirá la operación de agitación y se tomará una muestra de 250 mililitros por lo menos.
3.2. En el caso de pequeños envases para la venta al público, el que sirva de muestra no deberá abrirse hasta inmediatamente antes de proceder al análisis.
Cinco. Toma de muestras de la leche en polvo.
1. Material.
La toma de muestras se efectuará con una sonda limpia y seca de acero inoxidable, aluminio o aleación de aluminio.
2. Recipientes.
Las muestras se trasvasarán a recipientes limpios y secos, provistos de cierre hermético, que serán opacos cuando el tipo de análisis así lo exija.. Las dimensiones da tales recipientes serán lo suficientemente amplias para permitir la mezcla del contenido por medio de sacudidas.
3. Técnica.
La sonda se introducirá en el polvo a una velocidad constante de penetración. Cuando el tubo de la sonda llegue al fondo del envase, se retirará y su contenido se vaciará, inmediatamente en el recipiente destinado a la muestra. No deberá tocarse el polvo con los dedos. Se realizará uno o varios sondeos hasta obtener un peso de muestra total de 300 a 500 gramos.
[Bloque 6: #a2]
1 (a). Grasa
Leches natural, certificada, higienizada y esterilizada
1 (a).1. Principio.
Este método es aplicable a las leches natural, certificada, higienizada y esterilizada, enteras o parcialmente desnatadas, definidas en el Reglamento de Centrales Lecheras y otras Industrias Lácteas.
Se entiende por contenido en materia grasa de las leches natural, certificada, higienizada y esterilizada, el porcentaje en masa de las sustancias determinadas por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL - 1A: 1969 de la Federación Internacional de Lechería.
El contenido en materia grasa se determina gravimétricamente, por extracción de la citada materia grasa de una solución alcohólico-amoniacal del tipo de leche de que se trate, mediante éter dietílico y éter de petróleo, evaporación de los disolventes y pesado del residuo, según el principio del método de Röse-Gottlieb.
1 (a).2. Material y aparatos.
1 (a).2.1. Balanza analítica.
1 (a).2.2. Probetas o matraces de extracción adecuados, provistos de tapones de vidrio esmerilado, de tapones de corcho u otros dispositivos de cierre inatacables por los disolventes utilizados. Los tapones de corcho serán de buena calidad y se tratarán sometiéndolos sucesivamente a extracciones con éter dietílico y con éter de petróleo. Después se introducirán durante veinte minutos, por lo menos, en agua a una temperatura de 60 ºC o superior, y se dejarán enfriar también en agua, con objeto de que estén saturados cuando se utilicen.
1 (a).2.3. Matraces de paredes delgadas y bases planas, de una capacidad de 150 a 250 mililitros.
1 (a).2.4. Estufa de desecación, bien ventilada y controlada termostáticamente (ajustada para que funcione a una temperatura de 102 ± 2ºC), o una estufa de desecación por vacío (temperatura 70º - 75ºC, presión menor de 50 milímetros de Hg.).
1 (a).2.5. Materiales para facilitar la ebullición, exentos de materia grasa, no porosos ni deleznables al ser utilizados, como, por ejemplo, perlas de vidrio o trozos de carburo de silicio [el empleo de estos materiales os facultativo, véase el apartado 1 (a).4.3].
1 (a).3. Reactivos.
Todos los reactivos deberán ser de calidad pura para análisis y no dejar en la evaporación mayor cantidad de residuos que la autorizada para el ensayo en blanco [1 (a).4.2]. En caso necesario, los reactivos podrán destilarse de nuevo en presencia de un gramo aproximadamente de mantequilla deshidratada por 100 mililitros, de disolvente. El agua que se utilice deberá ser destilada o, por lo menos, de igual pureza que el agua destilada.
1 (a).3.1. Solución de amoníaco, de un 25 por 100 aproximadamente, en masa por volumen, de NH3 (densidad aproximada a 20 ºC, 0,91 gramos por mililitro), o una solución más concentrada conociéndose dicha concentración.
1 (a).3.2. Etanol deI 96 ± 2 por 100 en volumen o, en su defecto, etanol desnaturalizado con metanol, etil-metil-cetona, benceno o éter de petróleo.
1 (a).3.3. Eter dietílico, exento de peróxidos.
Para el ensayo de los peróxidos, añadir a 10 mililitros de éter dietílico contenidos en una pequeña probeta con tapón de vidrio, previamente enjuagada con un poco de éter, un mililitro de solución al 10 por 100 de yoduro potásico, recién preparada. Agitar y dejar reposar durante un minuto. No debe aparecer coloración amarilla en ninguna de las dos capas.
El éter dietílico puede mantenerse exento de peróxidos, añadiendo una lámina de cinc húmeda, que deberá sumergirse completamente en una solución ácida diluida de sulfato de cobre durante un minuto y después lavarse con agua. Por litro de éter dietílico, utilizar una superficie de 80 centímetros cuadrados aproximadamente de lámina de cinc cortada en bandas lo suficientemente largas para que lleguen, por lo menos hasta el centro del recipiente.
1 (a).3.4. Éter de petróleo, de puntos de ebullición entre 30º y 60ºC.
1 (a).3.5. Disolvente mixto, preparado poco tiempo antes de su utilización, mezclando volúmenes iguales de éter dietílico y éter de petróleo. (Se podrá sustituir la mezcla de disolventes en aquellos casos en que su utilización esté prevista por éter dietílico o por éter de petróleo.)
1 (a).4. Procedimiento.
1 (a).4.1. Preparación de la muestra.
Poner la muestra a una temperatura de 20ºC. Mezclarla cuidadosamente hasta obtener una distribución homogénea de la materia grasa. No agitar muy enérgicamente para evitar la formación de espuma en la leche o el batido de la materia grasa.
Si resulta dificultoso dispersar la capa de nata, calentar lentamente hasta 35º - 40ºC, mezclando cuidadosamente y teniendo la precaución de reincorporar a la muestra la nata que pudiera haberse adherido a las paredes del recipiente. Enfriar rápidamente la muestra hasta la temperatura ambiente. Si se desea se puede utilizar un homogeneizador apropiado para facilitar la dispersión de la grasa.
Si se separa materia grasa líquida o se observa la presencia de partículas blancas de forma irregular adheridas a las paredes del recipiente que contiene la muestra, el análisis no dará resultados correctos.
1 (a).4.2. Ensayo en blanco.
Al mismo tiempo que se determina el contenido en materia grasa de la muestra, efectuar un ensayo en blanco con 10 mililitros de agua destilada en lugar de la muestra, empleando el mismo tipo de aparato de extracción, los mismos reactivos en las mismas cantidades y siguiendo el mismo procedimiento que se describe a continuación. Si el resultado del ensayo en blanco excede de 0,5 miligramos, habrá que comprobar los reactivos, y aquel o aquéllos que resulten impuros deberán sustituirse o purificarse.
1 (a).4.3. Determinación.
Secar el matraz [si se desea con algún material 1 (a).2.5, que facilite una ebullición moderada durante la subsiguiente eliminación de los disolventes] en la estufa durante un intervalo de media a una hora. Dejar que se enfríe el matraz hasta la temperatura ambiente de la balanza y, una vez enfriado, pesarlo con una aproximación de 0,1 miligramo.
Invertir tres o cuatro veces el recipiente que contiene la muestra preparada y pesar inmediatamente en el aparato de extracción, directamente o por diferencia de 10 a 11 gramos de la muestra bien mezclada, con una aproximación de un miligramo. Añadir 1,5 mililitros de la solución de amoníaco al 25 por 100, o un volumen equivalente de una solución más concentrada, y mezclar convenientemente. Añadir 10 mililitros de etanol y mezclar suavemente, pero de modo homogéneo, manteniendo abierto el aparato de extracción. Añadir 25 mililitros de éter dietílico, cerrar el aparato y agitarlo vigorosamente, invirtiéndolo varias veces, durante un minuto. Si es necesario, enfriar el aparato con agua corriente. Quitar el tapón cuidadosamente y añadir 25 mililitros de éter de petróleo, utilizando los primeros mililitros para enjuagar el tapón y el interior del cuello del aparato, dejando que los líquidos de los enjuagues penetren en el último. Cerrarlo, volviendo a colocar el tapón, y agitarlo e invertirlo repetidamente durante treinta segundos. Si no está previsto centrifugar en la operación descrita no agitar muy enérgicamente.
Dejar el aparato en reposo hasta que la capa líquida superior esté completamente límpida y claramente separada de la fase acuosa. Podrá efectuarse igualmente la separación mediante el uso de una centrífuga adecuada. Si se utiliza una centrífuga cuyo motor no sea trifásico, pueden producirse chispas y será preciso tomar las debidas precauciones para evitar una explosión o un incendio debido a la presencia de vapores de éter (por ejemplo, en caso de rotura de un tubo).
Quitar el tapón y enjuagarlo, así como también el interior de] cuello del aparato, con algunos mililitros de la mezcla de disolventes, y dejar que los líquidos de los enjuagues penetren en el aparato. Trasvasar con cuidado al matraz, lo más completamente posible, la capa superior por decantación o con la ayuda de un sifón. Si el trasvase no se efectúa mediante un sifón, tal vez sea necesario añadir un poco de agua para elevar la separación entre las dos capas, con objeto, de facilitar la decantación.
Enjuagar el exterior e interior del cuello del aparato, o el extremo y la parte inferior del sifón, con algunos mililitros de la mezcla de disolventes. Dejar deslizar los líquidos del enjuague del exterior del aparato dentro del matraz y los del interior del cuello y del sifón, dentro del aparato de extracción,
Proceder a una segunda extracción repitiendo las operaciones descritas, desde la adición del éter dietílico, pero utilizando sólo 15 mililitros de éter dietílico y 15 mililitros de éter de petróleo. Efectuar una tercera extracción procediendo como se indica anteriormente, pero omitiendo el enjuague final. Eliminar con cuidado por evaporación o destilación la mayor cantidad posible de disolvente (incluido el etanol). Si el matraz es de pequeña capacidad, será necesario eliminar un poco de disolvente después de cada extracción de la manera antes indicada.
Cuando ya no subsista olor a disolvente, calentar el matraz, tumbado, durante una hora en la estufa. Dejar que el matraz se enfríe hasta la temperatura ambiente de la balanza como se indicó y pesar con una aproximación de 0,1 miligramo. Repetir la operación calentando a intervalos de treinta y sesenta minutos hasta obtener una masa constante. Añadir de 15 a 25 mililitros de éter de petróleo para comprobar si la materia extraída es totalmente soluble. Calentar ligeramente y agitar el disolvente mediante un movimiento rotatorio hasta que toda la materia grasa se disuelva.
Si la materia extraída es totalmente soluble en el éter de petróleo, la masa de materia grasa será la diferencia entre las pesadas efectuadas. En caso contrario o de duda, extraer completamente la materia grasa contenida en el matraz, mediante lavados repetidos con éter de petróleo caliente, dejando que se deposite la materia no disuelta antes de cada decantación. Enjuagar tres veces el exterior del cuello del matraz. Calentar el matraz, tumbado, durante una hora en la estufa y dejar que se enfríe hasta la temperatura ambiente de la balanza, como lo indicado anteriormente, y pesar con una aproximación de 0,1 miligramo. La masa de la materia grasa será la diferencia entre la masa obtenida y la obtenida en esta pesada final.
1 (a).5. Cálculo.
La masa, expresada en gramos, de la materia grasa extraída es:
(M1 - M2) - (B1 - B2)
y el contenido en materia grasa de la muestra, expresado en porcentaje de la masa, es:
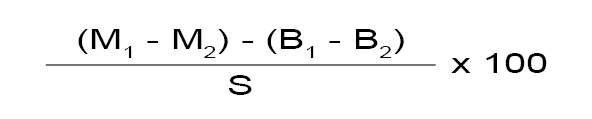
M1 = masa, en gramos, del matraz M, que contiene la materia grasa después de desecar hasta masa constante.
M2 = masa, en gramos, del matraz M sin materia grasa, o en el caso de presencia de materias insolubles, después de extraer completamente la materia grasa.
B1 = masa, en gramos, del matraz B del ensayo en blanco, después de desecar hasta masa constante.
B2 = masa, en gramos, del matraz B, o en el caso de presencia de materias insolubles, después de extraer completamente la materia grasa.
S = masa, en gramos, de la cantidad de muestra utilizada en la determinación.
La diferencia entre los resultados en dos determinaciones repetidas (resultados obtenidos simultáneamente o uno inmediatamente después de otro, por el mismo analista) no debe ser mayor de 0,03 g de materia grasa de 100 gramos de producto.
1 (a).6. Referencias.
1. Norma internacional FIL - IDF lA: 1969.
1 (b). Grasa
Leche desnatada
1 (b).1. Principio.
Este método es aplicable a las leches líquidas, no concentradas, y concretamente a la leche esterilizada desnatada, definida en el Reglamento de Centrales Lecheras y otras Industrias Lácteas.
Se entiende por contenido en materia grasa de la leche desnatada el porcentaje en masa de la cantidad total de lípidos y sustancias lipoides, determinada por el procedimiento expuesto a continuación, que corresponde al descrito en la norma F1L-22: 1963 de la Federación Internacional de Lechería.
El contenido en materia grasa se determina gravimétricamente por adición de amoníaco y alcohol a una cantidad conocida de leche desnatada, extracción por un disolvente de los lípidos y de las sustancias lipoides, evaporación del disolvente y pesado del residuo, obtenido por aplicación del método Röse-Gottlieb.
1 (b).2. Material y aparatos.
1 (b).2.1. Balanza analítica, de sensibilidad mínima 0,1 miligramos.
1 (b).2.2. Probetas o matraces de extracción apropiados provistos de tapones para cierre hermético (corcho o vidrio esmerilado).
1 (b).2.3. Erlenmeyer o matracas de fondo plano, de 150 a 250 mililitros de capacidad, con zona esmerilada para identificación.
1 (b).2.4. Materiales que faciliten la ebullición, exentos de materia grasa; por ejemplo, perlas de vidrio.
1 (b).2.5. Dispositivos apropiados para la evaporación de los disolventes.
1 (b).2.6. Estufa que permita obtener una temperatura constante de 102º - 104ºC o estufa de desecación por vacío.
1 (b).2.7. Pipetas graduadas de 10 mililitros.
1 (b).3. Reactivos.
1 (b).3.1. Etanol del 96 ± 1 por 100 en volumen o etanol desnaturalizado con metano] o con éter de petróleo.
1 (b).3.2. Éter dietílico, de punto de ebullición entre 34º y 35ºC, exento de peróxidos.
1 (b).3.3. Éter de petróleo, de puntos de ebullición entre 40º y 60ºC.
1 (b).3.4. Solución de amoníaco del 25 por 100 (densidad 0,91 a 20ºC) límpida e incolora.
Los reactivos no deben dejar residuos después de la evaporación.
1 (b).4. Procedimiento.
1 (b).4.1. Preparación de la muestra.
Mezclar la muestra cuidadosamente. En caso necesario, calentar la muestra a 35º - 40ºC y mezclar. Enfriar a 20ºC antes del análisis.
1 (3).4.2. Determinación.
Con una aproximación de 10 miligramos, pesar alrededor de 10 gramos, o introducir exactamente 10 milímetros (a 20º ± 2ºC), de leche desnatada en el aparato de extracción. Añadir un mililitro de la solución de amoniaco y mezclar cuidadosamente. Añadir 10 mililitros de etanol y mezclar el contenido. Añadir 25 mililitros de éter dietílico y, después de cerrar el aparato de extracción, mezclar el contenido agitando e invirtiendo varias veces durante un minuto. Añadir 25 mililitros de éter de petróleo, cerrar el aparato de extracción y mezclar el contenido agitando e invirtiendo repetidas veces.
Dejar reposar el aparato de extracción por lo menos dos horas o centrifugar (por lo menos cinco minutos a 500-600 revoluciones por minuto) hasta que la capa éter dietílico-éter de petróleo esté totalmente límpida y completamente separada de la fase acuosa. Trasvasar, lo más completamente posible, la capa éter dietílico-éter de petróleo por decantación, o con ayuda de un dispositivo de sifón (teniendo cuidado de no traspasar la fase acuosa), a un Erlenmeyer o matraz de fondo plano, que contenga un material destinado a facilitar la ebullición, previamente desecado y pesado.
Realizar una segunda extracción utilizando 15 mililitros de éter dietílico y 15 mililitros de éter de petróleo, siguiendo el procedimiento indicado. Trasvasar al mismo matraz la capa éter dietílico éter de petróleo como se indica anteriormente. Destilar con cuidado los disolventes contenidos en el matraz.
Después de la evaporación de los disolventes, secar la materia grasa durante una hora en estufa de vacío a 70º - 75ºC (presión inferior a 50 milímetros de mercurio), o en una estufa de presión normal a 102º - 104ºC, colocando el matraz en posición horizontal. Dejar enfriar el matraz y pesar cuando haya alcanzado la temperatura ambiente. Continuar el proceso de desecación hasta peso constante (desecación en vacío) o hasta un ligero aumento del peso (desecación a presión normal). En el último caso, tomar para el cálculo el último valor encontrado antes del aumento, del peso. Si se considera necesario, la materia grasa puede volverse a disolver en éter de petróleo para comprobar el resultado del análisis.
1 (b).4.3. Ensayo en blanco.
Para el control de los reactivos es necesario efectuar un análisis en blanco siguiendo exactamente la forma de operar indicada y utilizando 10 milímetros de agua en lugar de leche desnatada. El ensayo en blanco no deberá indicar más que una cantidad inapreciable.
Para determinar la influencia de las variaciones de temperatura y humedad del aire, pesar un matraz y tratarlo como el utilizado para el análisis, pero sin llenarlo con los disolventes.
1 (b).5. Cálculo.
En el cálculo de porcentaje de materia grasa se tendrán en cuenta, si es necesario, los valores encontrados en los ensayos en blanco. Si la cantidad de leche tomada para el análisis se ha tomado con ayuda de una pipeta, es necesario tener en cuenta la densidad de la leche.
La diferencia entre los resultados en dos determinaciones repetidas no debe ser mayor de 0,005 gramos de materia grasa por 100 gramos de producto.
1 (b).6. Referencias.
1. Norma Internacional FIL-IDF 22: 1963.
1 (c). Grasa
Leches concentrada, evaporada y condensada
1 (c).1. Principio.
Este método es aplicable a las leches concentrada, evaporada y condensada, enteras o desnatadas, definidas en el Reglamento de Centrales Lecheras y otras Industrias Lácteas.
Se entiende por contenido en materia grasa de las leches concentrada, evaporada y condensada el porcentaje en masa de las sustancias determinadas por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL-13A: 1969 de la Federación Internacional de Lechería.
El contenido en materia grasa se determina gravimétricamente por extracción de la citada materia grasa de una solución alcohólico-amoniacal del tipo de leche de qua se trate, mediante éter dietílico y éter de petróleo, evaporación de los disolventes y pesado del residuo, según el principio del método de Róse-Gottlieb.
1 (c).2. Material y aparatos.
1 (c).2.1. Balanza analítica.
1 (c).2.2. Probetas o matraces de extracción adecuados provistos de tapones de vidrio esmerilado, de tapones de corcho u otros dispositivos de cierre inatacables por los disolventes utilizados. Los tapones de corcho serán de buena calidad y se tratarán sometiéndolos sucesivamente e extracciones con éter dietílico y con éter de petróleo. Después se introducirán, durante veinte minutos por lo menos, en agua a una temperatura de 60ºC o superior y se dejarán enfriar también en agua con objeto de que estén saturados cuando se utilicen.
1 (c).2.3. Matraces de paredes delgadas y bases planas de una capacidad de 150 a 250 mililitros.
1 (c).2.4. Estufa de desecación bien ventilada y controlada termostáticamente (ajustada para que funcione a una temperatura de 102 ± 2ºC o una estufa de desecación por vacío (temperatura 70º - 75ºC, presión menor de 50 milímetros de Hg.).
1 (c).2.5. Materiales para facilitar la ebullición, exentos de materia grasa, no porosos ni deleznable al ser utilizados, como, por ejemplo, perlas de vidrio o trozos de carburo de silicio (el empleo de estos materiales es facultativo).
1 (c).3. Reactivos.
Todos los reactivas deben ser de calidad pura para análisis y no dejar en la evaporación mayor cantidad de residuos que la autorizada para el ensayo en blanco. En caso necesario, los reactivos podrán destilarse de nuevo en presencia de un gramo aproximadamente de mantequilla deshidratada por 100 mililitros de disolvente. El agua que se utilice deberá ser destilada o por lo menos de igual pureza que el agua destilada.
1 (c).3.1. Solución de amoníaco de un 25 por 100 aproximadamente en masa por volumen de NH, (densidad aproximada a 20ºC, 0,91 gramos por mililitro) o una solución más concentrada, conociéndose dicha concentración.
1 (c).3.2. Etanol del 96 ± 2 por 100 en volumen, o etanol desnaturalizado con metanol, etil-metil-cetona, benceno o éter de petróleo.
1 (c).3.3. Éter dietílico exento de peróxidos.
Para el ensayo de los peróxidos, añadir a 10 mililitros de éter dietílico contenidos en una pequeña probeta con tapón de vidrio, previamente enjuagada con un poco de éter, un mililitro de solución al 10 por 100 en yoduro potásico recién preparada. Agitar y dejar reposar durante un minuto. No debe aparecer coloración amarilla en ninguna de las dos capas.
El éter dietílico puede mantenerse exento de peróxidos añadiendo una lámina de cinc húmeda, que deberá sumergirse completamente en una solución ácida diluida de sulfato de cobre durante un minuto y después lavarse con agua. Por litro de éter dietílico utilizar una superficie de 80 centímetros cuadrados aproximadamente de lámina de cinc cortada en bandas lo suficientemente largas para que lleguen, por lo menos, hasta el centro del recipiente.
1 (c).3.4. Éter de petróleo de puntos de ebullición entre 30º y 60ºC.
1 (c).3.5. Disolvente mixto preparado poco tiempo antes de su utilización mezclando volúmenes iguales de éter dietílico y éter de petróleo (se podrá sustituir la mezcla de disolventes, en aquellos casos en que su utilización esté prevista, por éter dietílico o por éter de petróleo).
1 (c).4. Procedimiento.
1 (c).4.1. Preparación de la muestra.
1 (c).4.1.1. Leche concentrada y leche evaporada.
Agitar e invertir el recipiente que contiene la muestra. Abrir y trasvasar lentamente la leche a un segundo recipiente (provisto de cierre hermético). Mezclar mediante trasvases sucesivos, teniendo cuidado de incorporar a la muestra toda la materia grasa u otro constituyente adheridos a las paredes o al fondo del primer recipiente. Finalmente, trasvasar la leche lo más completamente posible al segundo recipiente y cerrar este último.
En caso necesario, templar el envase original, cerrado, en baño María a 40º-60ºC. Sacarlo y agitar vigorosamente cada quince minutos. Al cabo de dos horas, retirar el envase y dejarlo enfriar hasta temperatura ambiente. Quitar totalmente la tapadera y mezclar cuidadosamente removiendo el contenido con una cuchara o una espátula (si se separa la materia grasa no se debe efectuar el análisis de la muestra).
En el caso de envases flexibles, abrirlos y trasvasar el contenido a un vaso. Rasgar los envases, despegar todas las materias adheridas a las paredes e introducirlas en el vaso.
1 (c).4.1.2. Leche condensada.
Abrir el recipiente que contiene la muestra y mezclar cuidadosamente la leche con una cuchara o una espátula. Imprimir a este instrumento un movimiento rotativo ascendente y descendente de manera que las capas superiores e inferiores se mezclen bien con el resto del contenido. Tener cuidado de reincorporar a la muestra toda la masa de leche que pudiera haberse adherido a las paredes y a los fondos del recipiente. Trasvasar la leche lo más completamente posible a un segundo recipiente (provisto de cierre hermético) y cerrar este último.
En caso necesario, templar el bote original, cerrado, en baño María a 30º - 40ºC. Abrir el bote, desprender toda la leche adherida a las paredes del mismo, trasvasar a una cápsula lo suficientemente grande para permitir un manejo cuidadoso y mezclar hasta que toda la masa sea homogénea.
En el caso de tubos flexibles, abrirlos y trasvasar el contenido a un vaso. Rasgar los tubos, despegar todas las materias adheridas a las paredes e introducirlas en el vaso.
1 (c).4.2. Ensayo en blanco.
Al mismo tiempo que se determina el contenido en materia grasa de la muestra, efectuar un ensayo en blanco con 10 mililitros de agua destilada en lugar de la muestra, empleando el mismo tipo de aparato de extracción, los mismos reactivos en las mismas cantidades y siguiendo el mismo procedimiento que se describe a continuación. Si el resultado del ensayo en blanco excede de 0,5 miligramos, habrá que comprobar los reactivos y aquel o aquellos que resulten impuros deberán sustituirse o purificarse.
1 (c).4.3. Determinación.
Proceder como en 1 (a).4.3, excepto que se pesa de 4 a 5 gramos de la maestra, añadiendo a continuación 7 mililitros de agua, agitando suavemente y calentando ligeramente (40º - 50ºC) hasta la dispersión total del producto.
1 (c).5. Cálculo.
Como en 1 (a).5.
1 (c).6. Referencias.
1. Norma Internacional FIL-IDF 13A: 1969.
1 (d). Grasa
Leche en polvo
1 (d).1. Principio.
Este método es aplicable a las leches en polvo, entera, parcialmente desnatada y desnatada, definidas en el Reglamento de Centrales Lecheras y otras Industrias Lácteas.
Se entiende por contenido en materia grasa de la leche en polvo el porcentaje en masa de las sustancias determinadas por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL-9A: 1969 de la Federación Internacional de Lechería.
El contenido en materia grasa se determina gravimétricamente por extracción de la citada materia grasa de una solución alcohólico-amoniacal de leche en polvo mediante éter dietílico y éter de petróleo, evaporación de los disolventes y pesado del residuo, según el principio del método de Röse-Gottlieb.
1 (d).2. Material y aparatos.
1 (d).2.1, 2, 3, 4, 5 y 8 como 1 (a).2.1, 2, 3, 4, 5 y 6.
1 (d).3. Reactivos.
1 (d).3.1, 2, 3, 4 y 5 como 1 (a).3.1, 2, 3, 4 y 5.
1 (d).4. Procedimiento.
1 (d).4.1. Preparación de la muestra.
Trasvasar la leche en polvo a un recipiente limpio y seco (provisto de cierre hermético) de una capacidad que corresponda aproximadamente a dos veces el volumen del polvo. Cerrar enseguida el recipiente y mezclar cuidadosamente la leche en polvo agitándolo e invirtiéndolo repetidamente. Durante la preparación de la muestra deberá evitarse, en la medida de lo posible, la exposición de la leche en polvo al aire atmosférico con objeto de reducir al mínimo la absorción de humedad.
1 (d) 4.2. Ensayo en blanco.
Como en 1 (a).4.2.
1 (d).4.3. Determinación.
Como en 1 (a).4.3, excepto que se pesa aproximadamente un gramo de leche entera en polvo o 1,5 gramos de leche parcialmente desnatada en polvo o de leche desnatada en polvo añadiendo 10 mililitros de agua y agitando hasta la dispersión total del polvo de leche. Después de añadir la solución de amoniaco, calentar en baño María a una, temperatura de 60º - 70ºC durante quince minutos, agitando de vez en cuando, y enfriar a continuación con agua corriente, si se desea.
1 (d).5. Cálculo.
Como en 1 (a) 5.
1 (d)a. Referencias.
1. Norma Internacional FIL-IDF 9A: 1969.
2. Proteínas
2.1. Principio.
Se entiende por contenido en proteínas de la leche el contenido en nitrógeno expresado en porcentaje en peso y multiplicado por el factor de conversión, que se determina por el método expuesto a continuación, el cual corresponde al descrito en la norma FIL-20: 1962 de la Federación Internacional de Lechería.
Este método es aplicable a las leches no alteradas, natural, certificada, higienizada, esterilizada y a las reconstituidas asimismo no alteradas posteriormente de las leches concentrada, evaporada, condensada y en polvo.
La determinación del nitrógeno total se realiza por aplicación del método Kjeldahl: una determinada cantidad pesada de leche se trata con ácido sulfúrico en presencia de óxido de mercurio como catalizador con objeto de transformar el nitrógeno de los compuestos orgánicos en nitrógeno amoniacal. El amoníaco se libera por adición de sosa cáustica, se destila y se recoge a una solución de ácido bórico. A continuación se valora el amoniaco.
2.2. Material y aparatos.
2.2.1. Balanza analítica de un miligramo de sensibilidad mínima.
2.2.2. Aparato de digestión que permita mantener el matraz Kjeldahl en una posición inclinada y provisto de un sistema de calentamiento que no afecte más que a la parte del matraz ocupada por el líquido.
2.2.3. Matraz Kjeldahl de 500 mililitros de capacidad.
2.2.4. Refrigerante Liebig de tubo interior rectilíneo.
2.2.5. Un tubo de salida con bulbo de seguridad esférico, conectado a la parte inferior del refrigerante por unión esmerilada.
2.2.6. Una alargadera conectada al matraz Kjeldahl y al refrigerante Liebig por medio de goma. Uniones esmeriladas.
2.2.7. Un matraz Erlenmeyer de 500 mililitros de capacidad.
2.2.8. Probetas graduadas de 25, 50, 100 y 150 mililitros.
2.2.9. Bureta de 50 mililitros graduada a 0,1 mililitros.
2.2.10. Materiales para facilitar la ebullición. En la digestión, pequeños trozos de porcelana dura o perlas de vidrio, y en la destilación, pequeños trozos de piedra pómez recién calcinados.
2.3. Reactivos.
2.3.1. Sulfato potásico.
2.3.2. Óxido de mercurio, rojo.
2.3.3. Ácido sulfúrico concentrado (densidad, 1,84 a 20ºC).
2.3.4. Solución de sosa cáustica: 500 gramos de hidróxido sódico (NaOH) y 12 gramos de sulfuro de sodio (SNa2 . 9H2O) disueltos en 1.000 mililitros de agua destilada.
2.3.5. Solución de ácido bórico: 40 gramos de ácido bórico disuelto en 1.000 mililitros de agua destilada.
2.3.6. Ácido clorhídrico 0,1 N.
2.3.7. Indicador: Dos gramos de rojo de metilo y un gramo de azul de metileno disueltos en 1.000 mililitros de alcohol etílico del 96 por 100.
2.3.8. Solución de tetraborato de sodio para la valoración del ácido clorhídrico.
Los reactivos y las soluciones utilizadas no deben contener sustancias nitrogenadas.
2.4. Procedimiento.
2.4.1. Preparación de la muestra.
Antes del análisis poner la muestra a 20º ± 2ºC y mezclarla cuidadosamente. Si no se obtiene una dispersión homogénea de la materia grasa, calentarla lentamente a 40ºC, mezclar suavemente y enfriarla de nuevo a 20º ± 2ºC.
2.4.2. Determinación.
Introducir sucesivamente en el matraz Kjeldahl algunas perlas de vidrio o pequeños trozos de porcelana, alrededor de 10 gramos de sulfato potásico, 0,5 gramos de óxido de mercurio y alrededor de cinco gramos de leche exactamente pesados, con aproximación de un miligramo.
Añadir 20 mililitros de ácido sulfúrico y mezclar el contenido del matraz. Calentar cuidadosamente el matraz Kjeldahl sobre el dispositivo para la reacción hasta que no se forme espuma y el contenido se vuelva liquido. Continuar la reacción por calentamiento más intenso, hasta que el contenido del matraz esté perfectamente límpido e incoloro. Durante el calentamiento, agitar de vez en cuando el contenido del matraz. Cuando el líquido esté perfectamente límpido proseguir la ebullición durante una hora y media, evitando todo sobrecalentamiento local.
Dejar enfriar el contenido del matraz a la temperatura ambiente, añadir alrededor de 150 mililitros de agua destilada y algunos de piedra pómez, mezclar cuidadosamente y dejarlo todavía enfriar algo más. Con la ayuda de una probeta graduada, verter 50 mililitros de solución de ácido bórico en el matraz Erlenmeyer, añadir cuatro gotas de indicador y mezclar. Situar el matraz Erlenmeyer bajo el refrigerante, de manera que el extremo del tubo de salida se introduzca en la solución de ácido bórico. Con la ayuda de una probeta graduada añadir al contenido del matraz Kjeldahl 80 mililitros de la solución de sosa cáustica que contiene sulfuro. Durante esta operación, mantener el matraz inclinado, de tal manera que la sosa se deslice a lo largo de la pared del recipiente y que los líquidos no se mezclen. Conectar inmediatamente el matraz Kjeldahl al refrigerante por medio de la alargadera. Mezclar el contenido del matraz por agitación. Calentar a ebullición evitando la espuma. Proseguir la destilación hasta el momento en que el contenido del matraz presente ebullición a saltos. Regular el calentamiento de manera que la destilación dure por lo menos veinte minutos. Enfriar bien el destilado para evitar que se caliente la solución del ácido bórico. Poco tiempo antes de terminar la destilación, bajar el matraz Erlenmeyer para que el tubo de salida no esté introducido en la solución de ácido bórico. Detener el calentamiento; elevar el tubo de salida y enjuagar sus paredes exteriores e interiores con un poco de agua destilada. Valorar el destilado con ácido clorhídrico 0,1 N.
2.4.3. Ensayo en blanco.
Efectuar un ensayo en blanco, aplicando el método operatorio descrito, pero utilizando cinco mililitros de agua destilada en lugar de leche.
2.5. Cálculo.
2.5.1. Nitrógeno total.
Se calcula el contenido en nitrógeno total mediante la fórmula:
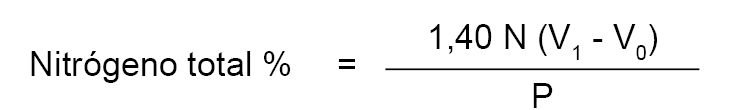
N = normalidad del ácido clorhídrico.
V1 = volumen en mililitros de ácido clorhídrico utilizado en la determinación.
V0 = volumen en mililitros de ácido clorhídrico utilizado en el ensayo en blanco.
P = peso en gramos de la leche empleada en el análisis.
La diferencia máxima entre dos determinaciones repetidas no debe sobrepasar el 0,005 por 100 de nitrógeno.
2.5.2. Proteínas.
Para expresar el contenido en proteínas de la leche analizada es preciso multiplicar la cantidad de nitrógeno total, obtenida según el método descrito, por un factor de conversión que se fija en 6.38. En consecuencia, el contenido en proteínas viene dado por la fórmula:
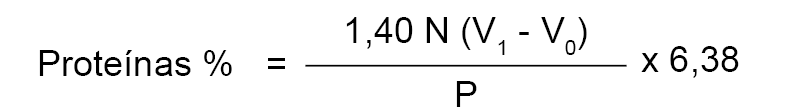
2.6. Referencias.
1. Norma internacional FIL-IDF 20: 1962.
3. Caseína
3.1. Principio.
Se entiende por contenido en caseína de la leche el contenido en proteínas, expresado en porcentaje en peso, obtenidas después de una precipitación a pM 4,6, siguiendo el procedimiento expuesto a continuación, que corresponde a la norma FIL-29: 1964 de la Federación Internacional de Lechería.
Este método es aplicable a las leches no alteradas, natural, certificada, higienizada, esterilizada y a las reconstituidas también, no alteradas posteriormente, de las leches concentrada, evaporada, condensada y en polvo. Asimismo puede aplicarse a las muestras de leche conservadas por adición de formaldehído (1: 2.500).
Se determina la cantidad total en nitrógeno de la leche. A continuación la caseína se precipita con un tampón acético-acetato y se filtra. Se determina luego la cantidad de nitrógeno del filtrado.
La cantidad de caseína se calcula con estas dos determinaciones de nitrógeno, que se realizan por el método Kjeldahl, según el método 2.
3.2. Material y aparatos.
3.2.1. Pipetas de 0, 5, 1 y 10 mililitros.
3.2.2. Probeta graduada de 100 mililitros.
3.2,3. Matraz graduado de 100 mililitros.
3.2.4. Papel filtro, lavado en ácido, velocidad media: De 11 a 12,5 centímetros.
3.2.5. Embudos.
3.3. Reactivos.
3.3.1. Solución de ácido acético al 10 por 100 (en peso por volumen).
3.3.2. Solución de acetato de sodio, 1 N.
3.4. Procedimiento.
3.4.1. Preparación de la muestra.
Antes del análisis poner la muestra a 20 ± 2ºC y mezclar cuidadosamente. Si no se obtiene una dispersión homogénea de la materia grasa, calentar la muestra lentamente a 40ºC; mezclar suavemente y enfriar a 20º ± 2ºC.
3.4.2. Determinación.
Determinar el contenido total en nitrógeno (NT) de la leche utilizando el método Kjeldahl, según el método 2.
Precipitar la caseína de la siguiente manera:
Llevar con pipeta 10 mililitros de leche a un matraz aforado de 100 mililitros. Añadir 75 mililitros de agua a 40ºC; después, un mililitro de la solución de ácido acético. Mezclar suavemente el contenido del matraz y esperar diez minutos, Añadir con pipeta un mililitro de la solución de acetato de sodio. Mezclar de nuevo. Dejar enfriar el contenido del matraz a unos 20ºC, completar hasta 100 mililitros con agua y mezclar invirtiendo lentamente el matraz. Cuando el precipitado de caseína y materia grasa se haya depositado, filtrar por filtro seco y recoger el filtrado en un recipiente seco.
Determinar el contenido en nitrógeno de 50 mililitros del filtrado límpido (nitrógeno no caseínico: NNC), utilizando el método Kjeldahl según el método 2.
3.4.3. Ensayo en blanco.
Además del ensayo en blanco previsto en el método 2, relativo a la determinación del contenido en nitrógeno total de la leche, conviene hacer un ensayo en blanco para comprobar los reactivos que precipitan la caseína, utilizando 50 mililitros de agua, 0,5 mililitros de la solución de ácido acético y 0,5 mililitros de la solución de acetato de sodio.
3.5. Cálculo.
Calcular el porcentaje en peso de NT en la leche y del NNC en el suero límpido con tres cifras significativas. Corregir la cifra obtenida para NNC por el volumen del precipitado, multiplicando el valor obtenido por 0,994. Calcular la cantidad de caseína mediante la fórmula:
Cantidad de caseína % = 6,38 NT – NNO
La aplicación de este factor a todas las leches enteras no entraña error sensible; sin embargo, si se aplica el método a leche desnatada, el factor de corrección utilizado deberá ser 0,998.
La diferencia entre dos determinaciones repetidas no debe sobrepasar el 0,04 por 100 de caseína.
3.6. Referencias.
1. Norma internacional FIL-DIF 29: 1964.
4. Lactosa
4.1. Principio.
Se entiende por contenido en lactosa de la leche el contenido en lactosa monohidratada expresado en porcentaje en peso, determinado por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL-28: 1964 de la Federación Internacional de Lechería.
Este método es aplicable a las leches no alteradas natural, certificada, higienizada, esterilizada y a las reconstituidas, asimismo no alteradas posteriormente, de las leches concentrada, evaporada, condensada y en polvo. También puede aplicarse a las muestras de leche conservadas por adición de formaldehído (1: 2.500).
El contenido en lactosa se determina indirectamente, una vez desproteinizada la leche, por valoración de la cantidad de halógeno reducido al final de la redacción entre lactosa y yoduro potásico-cloramina T.
4.2. Material y aparatos.
4.2.1. Balanza analítica.
4.2.2. Matraces aforados de 100 mililitros.
4.2.3. Pipetas de 5, 10, 20, 25 y 40 mililitros.
4.2.4. Papel filtro (lavado en ácido, velocidad media: 11 a 12,5 centímetros).
4.2.5. Embudos filtrantes.
4.2.6. Matraces Erlenmeyer, de una capacidad de 150 a 200 mililitros.
4.2.7. Matraces Erlenmeyer, de 150 mililitros con cierre esmerilado.
4,2.8. Bureta de 10 mililitros graduada a 0,02 mililitros.
4.3. Reactivos.
4.3.1. Reactivo del ácido tungsténico: Disolver siete gramos de tungstanato de sodio (WO4Na2 . 2H2O) en 870 mililitros de agua; añadir 0,1 mililitro de una solución de ácido ortofosfórico (88 por 100 en peso) y 70 mililitros de una solución de ácido sulfúrico 1 N.
4.3.2. Solución de cloramina T, 0,040 N (5,70 gramos por litro).
4.3.3. Solución de tiosulfato de sodio normalizada a un poco más de 0,040 N.
4.3.4. Solución de yoduro potásico del 10 por 100, recientemente preparada, incolora.
4.3.5. Solución de ácido clorhídrico, 2 N.
4.3.6. Solución de almidón soluble del 1 por 100.
4.4. Procedimiento.
4.4.1. Preparación de la muestra.
Antes del análisis poner la muestra a 20º ± 2ºC y mezclarla con cuidado. Si no se obtiene una dispersión homogénea de la materia grasa, calentar la muestra lentamente a 40 ºC, mezclar suavemente y enfriar a 20º ± 2ºC.
4.4.2. Determinación.
Llevar con pipeta 10 mililitros de leche a matraz aforado de 100 mililitros. Añadir 25 mililitros de agua, 40 mililitros del reactivo de ácido tungsténico y mezclar suavemente. Completar hasta 100 mililitros con agua, mezclar y dejar que se deposite el precipitado. Filtrar con un filtro seco y recoger en un matraz seco. Con una pipeta tomar 10 mililitros de filtrado y ponerlo en un matraz Erlenmeyer de 150 mililitros provisto de tapón esmerilado. Añadir cinco mililitros de la solución de yoduro potásico y exactamente 20 mililitros de la solución de cloramina T. Mezclar. Tapar el matraz con su tapón, previamente humedecido con un poco de solución de yoduro potásico, y mantenerlo en la oscuridad durante una hora y media a 18º-20ºC. Quitar el tapón, enjuagarlo en el matraz con un poco de agua y añadir cinco mililitros de la solución de ácido clorhídrico. Añadir exactamente 10 mililitros de la solución de tiosulfato de sodio. Valorar con una aproximación de 0,02 mililitros con la solución de tiosulfato. Hacia el final de la valoración, añadir dos o tres gotas de la solución de almidón.
4.4.3. Ensayo en blanco.
Efectuar un ensayo en blanco siguiendo exactamente el método operatorio descrito, pero empleando 10 mililitros de agua en lugar del filtrado.
4.5. Cálculo.
Calcular la diferencia entre los valores de tiosulfato obtenidos para el ensayo en blanco y para el de la leche. Corregir el valor en función del volumen del precipitado, multiplicando por 0,992. Convertir la cifra obtenida en lactosa monohidratada, teniendo en cuenta que un mililitro de solución de tiosulfato 0,040 N corresponde a 0,00720 gramos de lactosa monohidratada. Expresar los resultados en porcentajes de lactosa monohidratada.
La aplicación de este factor 0,992 a todas las leches enteras no ocasiona ningún error sensible; sin embargo, si el método se aplica a la leche desnatada, debe utilizarse 0,996 como factor de corrección.
La diferencia máxima entre dos determinaciones repetidas no debe sobrepasar de 0,05 por 100 de lactosa.
4.6. Observaciones.
La solución de tiosulfato se debe normalizar periódicamente, por ejemplo, por valoración de 10 mililitros de yodato de potasio 0,040 N, a los que se habrá añadido cinco mililitros de la solución de yoduro potásico y cinco mililitros de la solución de ácido clorhídrico.
La concentración de la solución de tiosulfato ha sido elegida de tal forma que la lectura en bureta sea de 2 a 3,5 mililitros para la mayor parte de las muestras de leche y de 9,5 a 9,7 mililitros para los ensayos en blanco.
Cuando se proceda a una serie de análisis se deben preparar los filtrados y los ensayos en blanco hasta la adición de yoduro de potasio inclusive. Finalmente se añade rápidamente la solución de cloramina T a cada matraz y se anota la hora. Después de una hora y media (se admite una tolerancia de una hora veinte minutos a una hora cuarenta minutos) se añade el ácido clorhídrico en todos los matraces y siguiendo el mismo orden. Así se paraliza la reacción y los matraces se pueden valorar por turno.
4.7. Referencias.
1. Norma internacional FIL-IDP 28: 1967.
5 (21). Extracto seco
Leches natural, certificada, higienizada y esterilizada
5 (a).1. Principio.
Se entiende por contenido en extracto seco de las leches natural, certificada, higienizada y esterilizada, el residio, expresado en porcentaje en peso, obtenido después de efectuada la desecación de la leche de que se trate por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL-21: 1962 de la Federación Internacional de Lechería.
Una cantidad conocida de leche se deseca a temperatura constante hasta peso constante. El peso obtenido después de desecar representa el de la materia seca.
5 (a).2. Material y aparatos.
5 (a).2.1. Balanza analítica, de sensibilidad 0,1 miligramo como mínimo.
5 (a).2.2. Desecador provisto de un buen deshidratante (gel de sílice con indicador higrométrico).
5 (a).2.3. Estufa de desecación que permita conseguir una temperatura constante de 102º ± 2ºC.
5 (a).2.4. Cápsulas metálicas planas, en metal inoxidable o de vidrio de dos centímetros de altura aproximadamente y de seis a ocho centímetros de diámetro aproximadamente, con tapas adecuadas.
5 (a).2.5. Baño de agua.
5 (a).3. Procedimiento.
5 (a).3.1. Preparación de la muestra.
Antes del análisis, poner la muestra a 20º ± 2ºC y mezclarla cuidadosamente. Si no se obtiene una buena repartición de la materia grasa, calentar lentamente a 40ºC, mezclarla suavemente y enfriarla a 20º ± 2ºC.
5 (a).3.2. Determinación.
Secar la cápsula y la tapa a 102º ± 2ºC durante treinta minutos. Colocar la cápsula tapada en un desecador, dejarla enfriar a la temperatura ambiente y pesarla. Poner aproximadamente tres mililitros de la muestra en la cápsula, tapar la cápsula con la tapa y pesarla. Poner la cápsula destapada en baño de agua durante treinta minutos. Poner la cápsula y la tapa en una estufa de desecación a 102º ± 2ºC durante dos horas. La tapa se debe poner a un lado de la cápsula. Cubrir la cápsula con la tapa y ponerla en el desecador; dejarla enfriar y pesarla. Ponerla una hora más en la estufa de desecación; dejarla enfriar y pesarla. Repetir la desecación hasta que la diferencia entre dos pesadas consecutivas no sea mayor de 0,5 miligramos.
5 (a).4. Cálculo.

P’ = peso en gramos de la muestra después de la desecación.
P = peso en gramos de la muestra antes de la desecación.
Si a la muestra de la leche se le han adicionado como conservadores sustancias no volátiles, como, por ejemplo, dicromato potásico, se debe corregir la expresión del extracto seco como sigue:
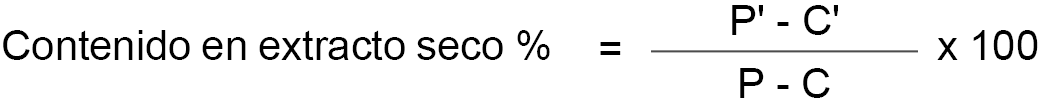
C’ = cantidad de conservante no volátil en la muestra analizada.

En caso de ser dicromato potásico, el contenido porcentual de conservante, se determina según el método 7.
La diferencia entre dos determinaciones repetidas no debe ser mayor de 0,05 por 100 del extracto seco.
5 (a).5. Referencias.
1. Norma Internacional FIL-IDF 21: 1962.
5 (b). Extracto seco
Leches concentrada, evaporada y condensada
5 (b).1. Principio.
Se entiende por contenido en extracto seco de las leches concentrada, evaporada y condensada, el residuo, expresado en porcentaje en peso obtenido después de efectuada la desecación de la leche de que se trate, por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL15: 1961 de la Federación Internacional de Lechería.
El método consiste en la dilución con agua de una cantidad conocida de muestra y la desecación con arena a temperatura constante. El peso obtenido después de desecar representa el de la materia seca.
5 (b).2. Material y aparatos.
5 (b).2.1. Balanza analítica de sensibilidad 0,1 miligramo como mínimo.
5 (b).2.2. Desecador provisto de un buen deshidratante (gel de sílice con indicador higrométrico).
5 (b).2.3. Estufa de desecación que permita obtener una temperatura constante de 98º - 100ºC.
5 (b).2.4. Cápsulas metálicas, planas, en metal inoxidable o de vidrio de 2,5 centímetros de altura aproximadamente y de 7,5 centímetros de diámetro aproximado, con tapas adecuadas.
5 (b).2.5. Arena de cuarzo o arena de mar que pase a través de un tamiz de 10 aberturas por centímetro, pero que no pase a través de un tamiz de 40 aberturas por centímetro y, si es necesario, lavada con ácido clorhídrico concentrado, después con agua y finalmente secada y calcinada.
Para comprobar si la arena es adecuada, desecar una pequeña cantidad a 98º - 100ºC hasta peso constante, añadir agua destilada, desecar de nueva y pesar. No debe haber diferencia entre las dos pesadas.
5 (1).2.6. Varillas cortas de vidrio.
5 (b).2.7. Baño de agua.
5 (b).3. Procedimiento.
5 (b).3.1. Preparación de la muestra.
Si se observa una separación parcial más o menos, importante de algunos componentes, por ejemplo, materias proteicas, materia grasa, sales de calcio o lactosa, es necesario mezclar la muestra.
5 (b).3.1.1. Leche concentrada o evaporada.
Abrir el envase al borde de la tapa, verter la leche despacio en otro recipiente y mezclar bien por sucesivos trasvases. La leche o la grasa que se adhiere a la tapa debe reincorporarse a la muestra. Calentar entonces la muestra tapada a una temperatura de 40ºC y mezclar íntimamente removiendo. Dejar enfriar.
5 (b).3.1.2. Leche condensada.
Abrir el bote al borde de la tapa. La leche o grasa adherida a la tapa deberá incorporarse a la muestra. Calentar a 40ºC y mezclar íntimamente removiendo de arriba abajo con una cuchara. Dejar enfriar.
5 (b).3.2. Determinación.
Colocar en la cápsula aproximadamente 25 gramos de arena y una pequeña varilla de vidrio. Secar la cápsula y su contenido, con la tapa abierta, a 98º- 100ºC durante dos horas. Colocar la cápsula tapada en un desecador, dejarla enfriar a la temperatura ambiente y pesar. Arrastrar la arena hacia un borde de la cápsula, pesar exactamente y poner en el espacio libre aproximadamente 1,5 gramos de la muestra. Añadir cinco mililitros de agua destilada, mezclar los líquidos y la arena mediante una varilla de vidrio. Dejar la varilla en la mezcla. Colocar la cápsula en baño de agua hirviendo durante veinte minutos y remover con cuidado la mezcla de vez en cuando. Colocar la cápsula con la varilla y la tapa en una estufa de desecación a 98º-100 ºC durante una hora y treinta minutos. La tapa se debe colocar al lado de la cápsula. Cubrir la cápsula con la tapa y ponerla en el desecador; dejarla enfriar y pesar. Colocarla una hora más en la estufa de desecación; dejarla enfriar y pesar como antes. Repetir la desecación hasta que la diferencia en peso entre dos pesadas sucesivas no exceda de 0,5 miligramos.
5 (b).4. Cálculo.

P’ = peso en gramos de la muestra después de desecar.
P = peso en gramos de la muestra antes de desecar. La diferencia entre dos determinaciones repetidas no debe ser mayor de 0,1 por 100 del extracto seco.
5 (b).5. Referencias.
1. Norma internacional FIL-IDF 15: 1961.
6. Cenizas
6.1. Principio.
Se entiende por contenido en cenizas de la leche el producto resultante de la incineración del extracto seco, expresado en porcentaje en peso, obtenido según el procedimiento descrito a continuación.
El extracto seco se incinera a una temperatura determinada y en una lenta corriente de aire.
6.2. Material y aparatos.
6.2.1. Balanza de sensibilidad 0,1 miligramos como mínimo.
6.2.2. Desecador provisto de un buen deshidratante (gel de sílice con indicador higrométrico).
6.2.3. Estufa de desecación, regulada a 120ºC.
6.2.4. Horno eléctrico con circulación de aire, provisto de un regulador de temperatura.
6.2.5. Cápsula de platino o de un material inalterable en las condiciones del ensayo de aproximadamente 55 milímetros de diámetro y 25 milímetros de altura.
6.2.6. Baño de agua a temperatura de ebullición.
6.3. Procedimiento.
Colocar la cápsula en la estufa de desecación a 102º ± 2ºC durante treinta minutos. Pasarla luego al desecador, dejarla enfriar a la temperatura ambiente y pesar. Pesar exactamente alrededor de 10 gramos de leche en la cápsula. Poner la cápsula en baño de agua hirviendo hasta secado por evaporación (aproximadamente siete horas). Incinerar el extracto seco, procedente de la desecación anterior, por calentamiento durante dos o tres horas en un horno regulado entre 520 y 550ºC (no deben existir en este último partículas carbonosas). Poner a enfriar la cápsula en un desecador. Pesar con una aproximación de 0,5 miligramos.
6.4. Cálculo.
El contenido en cenizas de la leche, expresado en porcentaje en peso, es igual a:
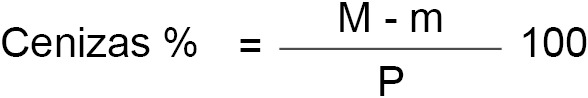
M = peso de la cápsula y de las cenizas después de la incineración y enfriamiento posterior.
m = peso de la cápsula vacía.
P = peso en gramos de la leche empleada en la determinación de las cenizas.
6.5. Observaciones.
El peso de las cenizas es variable según las condiciones de Incineración. La técnica descrita anteriormente proporciona los resultados más constantes: las diferencias no pasan generalmente de un 2 por 100 por término medio, y el 95 por 100 por lo menos de los cloruros se encuentran en las cenizas.
Cuando la temperatura del horno se haya elevado ligeramente por encima de 550ºC, determinar los cloruros y corregir en consecuencia el peso de las cenizas.
Si a la muestra se le ha añadido dicromato potásico, es necesario efectuar dos correcciones para obtener un valor aproximado de las cenizas de la leche inicial, operando como sigue:
1.º Determinar el dicromato potásico y restar su peso del de las cenizas.
2.º Determinar los cloruros en las cenizas, expresarles en CINa y añadir al peso de las cenizas la diferencia entre los cloruros totales de la leche y los cloruros restantes.
7. Dicromato potásico
7.1. Principio.
La determinación se realiza sobre las cenizas de la leche, por reducción de los cromatos mediante una solución de sulfato ferroamónico (sal de Mohr) (SO4) Fe (NE4)2 . 6H2O, cuyo exceso se valora con permanganato potásico.
7.2. Reactivos.
7.2.1. Solución acuosa de sal de Mohr de ocho gramos por litro, para valorar en el momento de su empleo (esta solución se estabiliza por adición de 12 mililitros de ácido sulfúrico por litro).
7.2.2. Solución valorada de permanganato potásico 0,02 N, en la cual un mililitro corresponda a 0,00098 gramos de Cr2 O7 K2.
7.2.3. Ácido sulfúrico (d = 1,84).
7.3. Procedimiento.
Agotar las cenizas por lavados sucesivos con agua destilada; después, con una solución acuosa de ácido sulfúrico al 5 por 100 en volumen; obtener un total de 25 a 30 mililitros de líquido. Recogerlo en un vaso para valorar. Añadir alrededor de cinco mililitros de ácido sulfúrico y 20 mililitros exactamente medidos de la solución sulfúrica de sal de Mohr; la reducción del ácido crómico es inmediata. Valorar. el exceso de sal de Mohr con la solución valorada de permanganato potásico hasta coloración rosa permanente.
Por otra parte, valorar en las mismas condiciones 20 mililitros de la misma solución sulfúrica de sal de Mohr mediante la solución valorada de permanganato potásico.
7.4. Cálculo.
El contenido de la leche en dicromato potásico, expresado en porcentaje en peso es igual a:
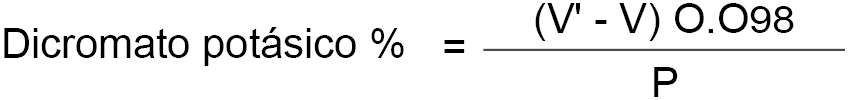
V = volumen en mililitros de permanganato potásico empleados en la valoración del exceso de sal de Mehr.
V’ = volumen en mililitros de permanganato potásico empleados en la prueba en blanco.
P = peso en gramos de la leche empleada en la determinación de las cenizas.
8 (a). Acidez
Leches natural, certificada, higienizada y esterilizada
8 (a).1. Principio.
Se entiende por acidez en las leches natural, certificada, higienizada y esterilizada, el contenido aparente en ácidos, expresado en gramos de ácido láctico por 100 mililitros de leche, determinado por el procedimiento expuesto a continuación, que corresponde al descrito en la norma UNE 34.100 del Instituto de Racionalización del Trabajo.
Un determinado volumen de leche se valora en solución de sosa, empleando solución alcohólica de fenolftaleína y luego se expresa el resultado en peso de ácido láctico, mediante la correspondiente transformación.
8 (a).2. Material y aparatos.
Bureta graduada cada 0,05 mililitros, o cada 0,1 mallares que permita apreciar la semidivisión.
8 (a).3. Reactivos.
8 (a).3.1. Solución 0,111 N (N/9) ó 0,1 N (N/10) de hidróxido sódico (NaOH).
8 (a).3.2. Indicador: 0,5 mililitros de una solución alcohólica de fenolftaleína al 1 por 100.
8 (a).4. Procedimiento.
8 (a).4.1. Preparación de la muestra.
Antes del análisis noper la muestra a 20º ± 2ºC y mezclarla cuidadosamente. Si no se obtiene una dispersión homogénea do la materia grasa, calentar lentamente la muestra a 40ºC, mezclar nuevamente y enfriarla de nuevo a 20º ± 2ºC.
8 (a).4.2. Determinación.
Se determina volumétricamente operando sobre 10 mililitros de leche con solución 0,111 N (N/9) de sosa (NaOH) o sobre 9 mililitros de leche con solución 0,1 N (N/10). Como indicador se emplean 0,5 mililitros de solución alcohólica de fenolftaleína al 1 por 100. (Dar por terminada la valoración cuando aparece una coloración rosa fácilmente perceptible por comparación con un testigo tomado de la misma leche. Dicha coloración desaparece progresivamente, pero se considera obtenido el viraje cuando el tinte rosa persiste durante unos diez segundos.
8 (a).5. Expresión de los resultados.
Los resultados se expresan en peso de ácido láctico por 100 mililitros de leche, dividiendo por 10 el número de mililitros empleados de solución de sosa.
Si a la muestra de leche se le ha adicionado dicromato potásico es preciso tener en cuenta la acidez debida a dicho conservador. En el caso de leches no alteradas se puede considerar que un gramo de dicromato potásico aumenta la acidez en las mismas proporciones que 0,6 gramos de ácido láctico.
8 (a).6. Referencias.
1. Instituto Nacional de Racionalización del Trabajo. Una norma española 34.100.
8 (b). Acidez
Leche en polvo
8 (b).1. Principio.
Se entiende por acidez en la leche en polvo el contenido aparente en ácidos expresado en gramos de ácido láctico por 100 gramos de leche en polvo determinado por el procedimiento expuesto a continuación, que corresponde al descrito en la norma UNE-34.101 del Instituto Nacional de Racionalización del Trabajo.
El método es aplicable a las leches en polvo entera y desnatada.
Una determinada cantidad pesada de leche en polvo disuelta en agua se valora con una solución de sosa empleando fenolftaleína como indicador hasta igualarla en color con otra cantidad igual de leche en polvo disuelta y Mezclada con acetato de rosanilina.
8 (b).2. Material y aparatos.
8 (b).2.1. Cápsulas de porcelana blanca de aproximadamente 100 mililitros de capacidad.
8 (b).3. Reactivos.
8 (b).3.1. Solución 0.111 N (N/9) o 0,1 N (N/10) de hidróxido sódico (NaOH) exento de carbonato.
8 (b).3.2. Solución indicadora de fenolftaleína: Se disuelve un gramo de fenolftaleína en 110 mililitros de alcohol etílico (95 a 96 por 100 en volumen) se añade solución N/9 o N/10 de hidróxido sódico, hasta que una gota dé una débil coloración rosa y se completa hasta 200 mililitros con agua destilada.
8 (b).3.3. Solución concentrada de acetato de rosanilina: Se disuelve 0,12 gramos de acetato de rosanilina en 50 mililitros de alcohol etílico (95 a 96 por 100 en volumen) que contenga 0,5 mililitros de ácido acético glacial y se completa hasta 100 mililitros con alcohol etílico.
8 (b).3.4. Solución diluida de acetato de rosanilina: Se diluye un mililitro de solución concentrada de acetato de rosanilina hasta 500 mililitros con una mezcla, a partes iguales en volumen, de agua destinada y alcohol etílico (95 a 96 por 100 en volumen).
Las soluciones de acetato de rosanilina se conservan en la oscuridad en frascos herméticamente cerrados con tapones de caucho.
8 (b).4. Procedimiento.
Se toman dos cápsulas de porcelana blanca de una capacidad aproximada de 100 mililitros, pesándose en cada una de ellas un gramo de leche en polvo. A una y otra cápsula se añaden 10 mililitros de agua hirviente, se agita con la extremidad aplastada de una varilla de vidrio hasta obtener un líquido perfectamente homogéneo y se deja enfriar durante diez minutos. A una de las cápsulas se le añade un mililitro de solución diluida de acetato de rosanilina y se mezcla. A la otra cápsula se le agrega un mililitro de solución de fenolftaleína y, gota a gota y agitando enérgicamente todo el tiempo, solución valorada de hidróxido sódico hasta obtener una coloración igual a la primera. El tiempo empleado en la valoración no ha de exceder de veinte segundos y ésta se ha de efectuar con una luz difusa.
8 (b).5. Cálculo.
El número de mililitros consumidos de solución 0,111 N (N/9) representa la acidez, expresada en gramos, de ácido láctico por 100 gramos de leche en polvo.
Si se opera con solución 0,1 N (N/10), el número de mililitros multiplicado por 0,9 da el mismo porcentaje de acidez.
8 (b).6. Referencias.
1. Instituto Nacional de Racionalización del Trabajo. Una norma española 34.101.
9. Sacarosa
Determinación polariniétrica en la leche condensada
9.1. Principio.
Se entiende por contenido en sacarosa de la leche condensada el contenido en sacarosa no transformada, expresado en porcentaje en peso, determinado por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL35: 1966 de la Federación Internacional de Lechería.
Este método es aplicable a la leche condensada, entera o desnatada, de composición normal, preparada a partir de leche y sacarosa únicamente que no contenga sacarosa transformada.
El método se basa en el principio de inversión de Clerget: Un tratamiento suave con un ácido hidroliza completamente la sacarosa. La lactosa y los otros azúcares prácticamente no se hidrolizan. La cantidad de sacarosa se deduce del cambio del poder rotatorio de la solución.
Se prepara un filtrado límpido de la muestra, sin mutarrotación debida a la lactosa, por tratamiento de la solución con amoníaco seguido de neutralización y clarificación por adiciones sucesivas de soluciones de acetato de cinc y de ferrocianuro potásico.
En una parte del filtrado la sacarosa se hidroliza en las condiciones especiales que corresponden a este tipo de operación.
Partiendo de los poderes rotatorios del filtrado, antes y después de la inversión, se calcula la cantidad de sacarosa.
9.2. Material y aparatos.
9.2.1. Balanza analítica de sensibilidad 10 miligramos como mínimo.
9.2.2. Vasos de precipitados de 100 mililitros en vidrio.
9.2.3. Matraces graduados, de 200 y 50 mililitros.
9.2.4. Pipetas de 40 mililitros.
9.2.5. Probetas graduadas de 25 mililitros.
9.2.6. Pipetas graduadas de 10 mililitros.
9.2.7. Embudos filtrantes de diámetro entre 8 y 10 centímetros y filtros (plegados) de 15 centímetros de diámetro.
9.2.8. Tubo de polarímetro de 2 decímetros de longitud, exactamente calibrado.
9.2.9. Polarímetro o sacarímetro.
9.2.9.1. Polarímetro con luz de sodio o con luz verde de mercurio (lámpara de vapor de mercurio con prisma o pantalla Wratten número 77 A), permitiendo lecturas con una precisión por lo menos igual a 0,05 grados de ángulo.
9.2.9.2. Sacarímetro con escala internacional de azúcar utilizando luz blanca que pasa a través de un filtro de 15 milímetros de una solución al 8 por 100 de dicromato potásico, o bien luz de sodio, y permitiendo la lectura con una precisión por lo menos igual a 0,1 grados de la escala sacarimétrica internacional.
9.2.10. Baño de agua a 60º ± 1ºC.
9.3. Reactivos.
9.3.1. Solución de acetato de cinc, 2,0 N: Disolver 21,9 gramos de acetato de cinc cristalizado (C2H3O2), Zn. 2H2O y 3 mililitros de ácido acético glacial en agua destilada y completar hasta 100 mililitros.
9.3.2. Solución de ferrocianuro potásico, 1,0 N: Disolver 10,6 gramos de ferrocianuro potásico cristalizado [Fe(CN)6]K4 . 3H2O en agua destilada y completar hasta 100 mililitros.
9.3.3. Solución de ácido clorhídrico, 6,35 ± 0,20 N (20-22 por 100).
9.3.4. Solución diluida de amoniaco, 2,0 ± 0,2 N (3,5 por 100).
9.3.5. Solución diluida de ácido acético, 2,0 ± 0,2 N (12 por 100).
9.4. Procedimiento.
9.4.1. Preparación de la muestra.
Para muestras de productos recientemente preparados en los que no se pueda prever separación alguna apreciable de los componentes. Abrir el recipiente, introducir en él el producto adherido a la tapa y mediante movimientos de arriba abajo, con ayuda de una cuchara, conseguir que se mezclen íntimamente las capas superiores así como el contenido del fondo del recipiente. Trasvasar el contenido de un bote a un frasco provisto de tapón bien adaptado.
Para muestras de productos más antiguos y muestras en las que se pueda prever una separación de componentes, calentar en baño de agua, aproximadamente a 40ºC, hasta que la muestra casi haya alcanzado esta temperatura, abrir el recipiente y operar de la misma manera que arriba. En el caso de un bote, trasvasar el contenido a un frasco, raspar el producto que se haya adherido a las paredes y continuar la mezcla hasta que toda la masa sea homogénea. Cerrar el frasco con una tapadera que se adapte perfectamente. Dejar enfriar.
9.4.2. Comprobación del método.
Proceder como en 9.4.3, utilizando una mezcla de 100 gramos de leche o 110 gramos de leche desnatada y de 18,00 gramos de sacarosa pura, que corresponde a 40,00 gramos de una leche concentrada conteniendo el 45 por 100 de sacarosa. Calcular la cantidad de sacarosa como en 9.5.1, utilizando en la fórmula I), para W, F y P, la cantidad de leche pesada y la riqueza en materia grasa y proteínas de esta leche, y en la fórmula II), pala W, la cifra de 40,00. La media de los valores encontrados no debe diferir de dicho valor (45 por 100) en más del 0,1 por 100.
9.4.3. Determinación.
En un vaso de 100 mililitros pesar aproximadamente 40 gramos de la muestra bien mezclada con una aproximación de 10 miligramos, añadir 50 mililitros de agua destilada caliente (80º - 90ºC) y mezclar cuidadosamente. Trasvasar cuantitativamente la mezcla a un matraz aforado de 200 mililitros, enjuagar el vaso con cantidades sucesivas de agua destilada a 60ºC hasta que el volumen total sea de 120 a 150 mililitros. Mezclar y enfriar a temperatura ambiente. Añadir 5 mililitros de la solución de amoníaco diluida. Mezclar de nuevo y dejar reposar durante quince minutos. Neutralizar el amoníaco añadiendo una cantidad equivalente de la solución diluida de ácido acético. Determinar previamente la cantidad exacta de mililitros por valoración de la solución de amoníaco diluida empleando el azul de bromotimol como indicador. Mezclar. Añadir, mezclando suavemente por rotación del matraz Inclinado, 12,5 mililitros de solución de acetato de cinc. De la misma forma que para la solución de acetato, añadir 12,5 mililitros de solución de ferrocianuro potásico. Poner el contenido del matraz a 20ºC y añadir agua destilada (a 20ºC) hasta alcanzar el enrase de 200 mililitros.
Hasta este momento todas las adiciones de agua o reactivos deberán haberse efectuado de tal manera que se haya evitado la formación de burbujas de aire y por esta misma razón todas las mezclas se habrán realizado por rotación del matraz y no por agitación violenta. Si se observa la presencia de burbujas de aire antes de alcanzar los 200 mililitros se pueden eliminar aplicando al matraz una bomba de vacío e imprimiéndolo un movimiento de rotación. Tapar el matraz con un tapón seco y mezclar íntimamente sacudiendo con energía. Dejar reposar durante algunos minutos, filtrar a continuación por un papel filtro seco. Despreciar los primeros 25 mililitros del filtrado.
9.4.3.1. Polarización directa.
Determinar la rotación óptica del filtrado a 20º ± 2ºC.
9.4.3.2. Inversión.
Introducir con la pipeta en un matraz graduado de 50 mililitros, 40 mililitros del filtrado obtenido de la manera indicada antes: Añadir 6,0 mililitros de ácido clorhídrico 6,35 N. Poner el matraz en baño de agua a 60ºC durante quince minutos, sumergiéndolo hasta el nacimiento del cuello. Mezclar por rotación durante los cinco primeros minutos, al final de los cuales el contenido deberá haber alcanzado la temperatura del baño. Enfriar a 20ºC y completar hasta 50 mililitros con agua destilada a 20ºC, mezclar y dejar reposar una hora a esta temperatura.
9.4.3.3. Polarización después de inversión.
Determinar el poder rotatorio de la solución invertida a 20º ± 2ºC (Cuando la temperatura del liquido en el tubo de polarización difiera en más de 0,2ºC de 20ºC durante la medida, se debe aplicar la corrección de la temperatura indicada en el apartado 9.5.2).
9.5. Cálculo.
9.5.1. Riqueza en sacarosa.
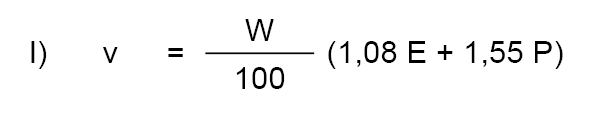
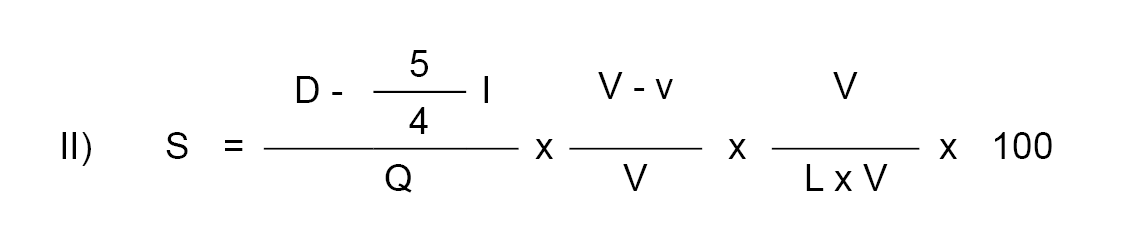
S = Cantidad de sacarosa.
W = Peso de la muestra expresado en gramos.
P = Porcentaje de proteínas (N x 6,38) de la muestra.
F = Porcentaje de materia grasa de la muestra.
V = Volumen en mililitros de la muestra diluida antes de filtrar.
D = Lectura polarimétrica directa (polarización antes de la inversión).
I = Lectura polarimétrica después de la inversión.
L = Longitud en decímetros del tubo del polarímetro.
Q = Factor de inversión cuyos valores se indican más adelante.
Pesando exactamente 40,00 gramos de leche condensada y utilizando un polarímetro con luz de sodio provisto de escala en grados de ángulo y un tubo de 2 decímetros de longitud, el contenido en sacarosa de las leches condensadas normales (C = 9) a 20,0º ± 0,1ºC se puede calcular con ayuda de la siguiente fórmula:
S = (D-5/4 I) (2,833-0,00612 F-0,00878 P).
Si la medida de la polarización después de la inversión se efectúa a una temperatura diferente de 20ºC, las cifras obtenidas se deben multiplicar por
9.5.2. Valores del factor de inversión Q.
Las fórmulas siguientes dan valores precisos de Q para diversas clases de luz con correcciones, si es necesario, para la concentración y la temperatura.
Luz de sodio y polarímetro con escala en grados de ángulo.
Q = 0,8825 + 0,0006 (C-9) – 0,0033 (T-20).
Luz verde de mercurio y polarímetro con escala en grados de ángulo:
Q = 1,0392 + 0,0007 (C-9) – 0,0039 (T-20).
Luz blanca con filtro de dicromato y sacarímetro con escala sacarimétrica:
Q = 2,549 + 0,0017 (C-9) – 0,0095 (T-20).
En las fórmulas precedentes:
C = Porcentaje de azúcares totales en la solución invertida, según la lectura polarimétrica.
T = Temperatura de la solución invertida en la lectura del polarímetro.
El porcentaje de azúcares totales C en la solución invertida se puede calcular a partir de la lectura directa y de la variación después de la inversión según el método habitual, utilizando los valores usuales de, rotación específica de sacarosa de lactosa y de sacarosa invertida. La corrección 0,0006 (C-9), etcétera, no es exacta más que cuando C es aproximadamente 9; para leche concentrada normal esta corrección se puede despreciar, por ser C próximo a 9.
Variaciones en la temperatura de 20ºC influyen escasamente en la lectura de la polarización directa. Por el contrario, diferencias de más de 0,2ºC, en la lectura de polarización después de la inversión necesitan una corrección. La corrección – 0,003 (T-20), etc., no es exacta más que para temperaturas comprendidas entre 18ºC y 22ºC.
La diferencia entre los resultados de dos determinaciones efectuadas simultáneamente o una inmediatamente después de otra por el mismo analista no debe ser mayor de 0,3 gramos de sacarosa para 100 gramos de leche condensada.
9.6. Referencias.
1. Norma Internacional FIL-IDF 35: 1966.
10. Humedad
Leche en polvo
10.1. Principio.
Se entiende por humedad de la leche en polvo el contenido en agua libre, es decir, la pérdida de peso, expresado en porcentaje en peso, determinado por el procedimiento expuesto a continuación, que corresponde al descrito en la norma FIL-26: 1964 de la Federación Internacional de Lechería.
Este método es aplicable a las leches en polvo, entera y desnatada.
El agua, contenida en la leche en polvo, se elimina por calentamiento de la muestra en una estufa de desecación, a una temperatura de 102º ± 2ºC, hasta peso constante.
10.2. Material y aparatos.
10.2.1. Balanza analítica, de sensibilidad 0,1 miligramos como mínimo.
10.2.2. Cápsulas apropiadas, preferentemente en aluminio, níquel, acero inoxidable o vidrio. Las cápsulas deberán estar provistas de tapas que se adapten convenientemente, pero que se puedan quitar con facilidad. Las dimensiones más convenientes son: diámetro aproximado 50 milímetros: profundidad aproximada 25 milímetros.
10.2.3. Un desecador provisto de gel de sílice con indicador de humedad.
10.2.4. Una estufa de desecación bien ventilada, provista de termostato, y regulada a 102º ± 2ºC. Es importante que la temperatura sea uniforme en toda la estufa.
10.2.5. Frascos provistos de tapones herméticos para el mezclado de la leche en polvo.
10.3. Procedimiento.
10.3.1. Preparación de la muestra.
Trasvasar toda la muestra de la leche en polvo a un frasco seco y tapado, de un volumen igual al doble aproximadamente del de la muestra, mezclar íntimamente por rotación y agitación. (En el caso de que no se pueda obtener una homogeneización completa por este procedimiento, extraer, con objeto de realizar determinaciones paralelas, dos muestras en dos puntos, lo más distantes posibles el uno del otro.)
10.3.2. Determinación.
Colocar la cápsula destapada y la correspondiente tapa en la estufa de desecación durante una hora, a la temperatura de 102º ± 2ºC. Colocar la tapa sobre la cápsula y pasarla de la estufa al desecador. A continuación enfriar a la temperatura ambiente y pesar. Introducir aproximadamente un gramo de leche en polvo en la cápsula, tapar la cápsula y pesar rápidamente con la mayor exactitud posible. Destapar la cápsula y colocarla con su tapa, en la estufa a una temperatura de 102º ± 2ºC durante dos horas. Volver a colocar la tapa, poner la cápsula en el desecador, dejar enfriar a temperatura ambiente y pesar rápidamente con la mayor exactitud posible.
Calentar la cápsula abierta y su tapa a 102º ± 2ºC en la estufa durante una hora suplementaria, volver a tapar y dejar enfriar en el desecador a temperatura ambiente; pesar de nuevo. Repetir la operación hasta que las pesadas sucesivas no difieran en más de 0,0005 gramos. La desecación se. acaba aproximadamente en dos horas.
10.4. Cálculo.
Calcular la humedad mediante la fórmula siguiente:
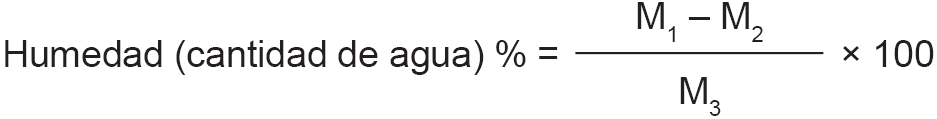
M1 = la masa inicial, en gramos, de la cápsula y su tapa más la leche en polvo utilizada para el análisis.
M2 = la masa final, en gramos, de la cápsula y su tapa más la leche en polvo.
M3 = la masa, en gramos, de la leche en polvo utilizada para el análisis.
La diferencia entre dos determinaciones repetidas no debe sobrepasar el 0,06 por 100 de agua.
10.5. Referencias.
1. Norma Internacional FIL-IDF 26: 1964.
11. Índice de solubilidad
Leche en polvo
11.1. Principio.
Se entiende por índice de solubilidad de la leche en polvo la cantidad de sedimento, expresada en volumen, determinada por el procedimiento expuesto a continuación, que corresponde al descrito en la norma UNE-34.101 del Instituto Nacional de Racionalización del Trabajo.
Este método es aplicable a la leche en polvo, entera o desnatada.
11.2. Material y aparados.
11.2.1. Centrífuga con soportes para la colocación de los tubos centrífugos cónicos. La velocidad requerida varía, como a continuación se indica, con el diámetro.
|
Diámetro |
Revoluciones por minuto |
Diámetro |
Revoluciones por minuto |
|---|---|---|---|
|
250 milímetros |
1.083 |
450 milímetros |
807 |
|
300 milímetros |
988 |
500 milímetros |
766 |
|
350 milímetros |
916 |
550 milímetros |
730 |
|
400 milímetros |
856 |
600 milímetros |
700 |
El «Diámetro» es la distancia entre los fondos internos de dos soportes opuestos, medida a través del centro de rotación de la cabeza de la centrífuga estando los soportes extendidos horizontalmente.
11.2.2. Tubos de centrífuga cónicos, graduados como se indica a continuación:
De 0 a 1,0 ml en divisiones de 0,1 ml.
De 1,0 a 2,0 ml en divisiones de 0,2 ml.
De 2,0 a 10,0 ml en divisiones de 0,5 ml.
De 10,0 a 20,0 ml en divisiones de 1,0 ml.
y a 50 ml una marca que quede, por lo menos, a 13 milímetros del borde del tubo.
11.2.3. Mezclador eléctrico.
11.2.4. Tubo sifón de vidrio.
11.3. Procedimiento.
Se añaden 10 ó 13 gramos de la muestra, según se trate de leche desnatada o completa, respectivamente, a 100 mililitros de agua destilada, a la temperatura de 24ºC, en el vaso especial del mezclador que seguidamente se pone en éste para agitar durante noventa segundos exactos. Si hay necesidad inmediata de volver a emplear el vaso del mezclador, puede verterse la solución total en un vaso de precipitación apropiado. Se deja reposar la solución hasta que la espuma se separe lo suficiente, para poder quitarla completamente con una cuchara. El periodo de reposo después de la mezcla no ha de exceder de quince minutos.
Después de separada la espuma se mezcla completamente con una cuchara durante cinco segundos y, seguidamente, se llena un tubo cónico hasta la marca de 50 mililitros. Se centrifuga durante cinco minutos a las revoluciones por minuto requeridas, según la tabla del apartado 11.2.1. A continuación se sifona el líquido que sobrenada, hasta dos mililitros del nivel del sedimento, teniendo cuidado de no remover éste. Se vierten en el tubo 25 mililitros de agua destilada a 24ºC y se agita para dispersar el sedimento, desalojándole si fuera necesario con un alambre. Se llena con agua destilada, 24ºC, hasta la marca de 50 mililitros se invierte y se agita para la perfecta mezcla del contenido, y se centrifuga de nuevo durante cinco minutos a la velocidad requerida. Se mantiene el tubo en posición vertical, de modo que el nivel superior del sedimento quede a nivel del ojo; se refieren los mililitros de sedimento a la división de la escala más próxima. La lectura se efectúa fácilmente, cuando se observa el tubo, colocado delante de una luz fuerte.
Cuando se opere con leches en polvo parcialmente desnatadas, las cantidades que se han de añadir a los 100 mililitros de agua destilada son, según sus porcentajes grasos, las siguientes:
Hasta el 4 %: 10,0 gramos.
Desde el 4,1 al 9 %: 10,5 gramos.
Desde el 9,1 al 13 %; 11,0 gramos.
Desde el 13,1 al 17 %: 11,5 gramos.
Desde el 17,1 al 21 %: 12,0 gramos.
Desde el 21,1 al 24 %: 12,5 gramos.
Más del 24 %: 13,0 gramos.
11.4. Referencias.
1. Instituto Nacional de Racionalización del Trabajo. Una Norma Española 34.101.
12. Calcio
12.1. Principio.
Se entiende por contenido en calcio de la leche, la cantidad total de calcio expresada en porcentaje en peso, determinada por el procedimiento expuesto a continuación, que corresponde a la norma FIL-36: 1966 de la Federación Internacional de Lechería.
Este método se aplica a todas las leches líquidas normales, así como a las leches reconstituidas por dilución o disolución de leches concentradas o de leches desecadas.
El calcio total se lleva a disolución por precipitación de las materias proteicas con ácido tricloroacético. El calcio contenido en el filtrado es precipitado bajo forma de exalato cálcico, que se separa por centrifugación y se valora con permanganato potásico.
12.2. Material y aparatos.
12.2.1. Balanza analítica.
12.2.2. Matraz aforado de 50 mililitros.
12.2.3. Pipeta para leche de 20 mililitros.
12.2.4. Papel filtro sin cenizas para filtración lenta.
12.2.5. Centrífuga que pueda desarrollar una aceleración centrífuga de 1.400 g (g = aceleración de la gravedad).
12.2.6. Tubos de centrífuga cilíndricos con fondo redondo de 30 mililitros aproximadamente, marcados a 20 mililitros.
12.2.7. Pipetas de dos y cinco mililitros.
12.2.8. Dispositivo de sifón por succión, provisto de un tubo capilar.
12.2.9. Baño de agua, a temperatura de ebullición.
12.2.10. Bureta graduada en 1/50 milímetros.
12.3. Reactivos.
12.3.1. Ácido tricloroacético: solución acuosa al 20 por 100 (en peso por volumen).
12.3.2. Ácido tricloroacético: solución acuosa al 12 por 100 (en peso por volumen).
12.3.3. Oxalato amónico: solución acuosa saturada en frío.
12.3.4. Rojo de metilo: solución al 0,05 por 100 (en peso por volumen) en alcohol etílico del 96 por 100 (en volumen por volumen).
12.3.5. Ácido acético: solución acuosa al 20 por 100 (en volumen por volumen).
12.3.6. Amoníaco: solución acuosa obtenida mezclando volúmenes iguales de amoníaco al 25 por 100 (en peso por peso) y de agua destilada.
12.3.7. Amoníaco: solución acuosa obtenida diluyendo dos mililitros de amoníaco al 25 por 100 (en peso por peso) hasta 100 mililitros, con agua destilada.
12.3.8. Ácido sulfúrico: solución acuosa obtenida añadiendo 20 mililitros de ácido sulfúrico del 98 por 10 (en peso por peso) a 80 mililitros de agua destilada.
12.3.9. Permanganato potásico: solución acuosa titulada, 0,02N.
Todos los reactivos deben ser puros para análisis.
12.4. Procedimiento.
12.4.1. Preparación de la muestra.
Antes del análisis, poner la muestra a 20º ± 2ºC y mezclar con cuidado. Si no se obtiene una dispersión homogénea de la materia grasa, calentar lentamente la muestra a 40ºC, mezclar suavemente y enfriar a 20º ± 2ºC.
12.4.2. Defecación de la muestra.
En un matraz aforado de 50 mililitros pesar exactamente alrededor de 20 gramos de leche, con una aproximación de 10 miligramos. Añadir poco a poco mientras se agita una solución acuosa de ácido tricloroacético al 20 por 100 y completar hasta 50 mililitros con este reactivo. Agitar fuertemente durante algunos segundos. Dejar reposar treinta minutos. Filtrar sobre papel filtro sin cenizas. El filtrado obtenido debe ser límpido.
12.4.3. Precipitación del calcio en forma de oxalato y separación del oxalato.
En un tubo de centrífuga, cilíndrico de fondo redondo, introducir cinco mililitros del filtrado límpido; después cinco mililitros de ácido tricloroacético al 12 por 100, dos mililitros de una solución acuosa saturada de oxalato de amonio, dos gotas de solución alcohólica de rojo de metilo y dos mililitros de ácido acético al 20 por 100. Mezclar mediante agitación circular y añadir poco a poco solución de amoníaco (12.3.6) hasta coloración amarillo pálido; después, algunas gotas de ácido acético al 20 por 100 hasta coloración rosa. Dejar reposar cuatro horas a la temperatura ambiente.
Diluir hasta 20 mililitros con agua y centrifugar diez minutos a 1.400 g (g = aceleración de la gravedad). Mediante un dispositivo de succión; decantar el líquido límpido que sobrenada. Lavar las paredes del tubo de centrifugación (sin remover el depósito de oxalato cálcico) con cinco mililitros de solución de amoníaco diluido (12.3.7). Centrifugar cinco minutos a 1.400 g. Decantar el liquido que sobrenada con el dispositivo de succión. Proceder a tres lavados sucesivos.
12.4.4. Valoración del oxalato.
Después de haber extraído con sifón el agua del último lavado, añadir dos mililitros de solución acuosa de ácido sulfúrico y cinco mililitros de agua destilada sobre el precipitado de oxalato de calcio. Poner el tubo en baño de agua hirviendo y, cuando el oxalato esté completamente disuelto, valorar con solución de permanganato potásico 0,02 N, hasta coloración rosa persistente. La temperatura debe permanecer superior a los 60 ºC durante la valoración.
12.4.5. Ensayo en blanco.
Efectuar un ensayo en blanco con todos los reactivos, utilizando 20 mililitros de agua destilada en vez de leche.
12.5. Cálculo.
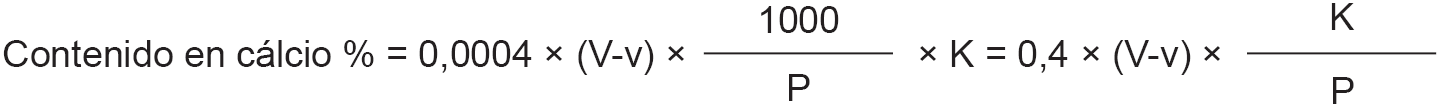
V = volumen en mililitros de MnO4K (0,02 N) gastados en la valoración de la muestra.
v = volumen en mililitros de MnO4K (0,02 N) gastados en el ensayo en blanco.
P = peso en gramos de la muestra inicial (alrededor de 20 gramos).
K = coeficiente de corrección del volumen del precipitado resultante de la precipitación tricloroacética.
Para leche entera (3,5 a 4,5 por 100 de materia grasa): K = 0,972.
Para leche con 3 por 100 de materia grasa: K = 0,976.
Para leche con 2 por 100 de materia grasa: K = 0,980.
Para leche con 1 por 100 de materia grasa: K = 0,985.
Para leche desnatada: K = 0,989.
La diferencia entre los resultados de dos determinaciones efectuadas simultáneamente o una inmediatamente después de otra por el mismo analista, no debe ser mayor de 0,002 gramos de calcio por 100 gramos de leche.
12.6. Referencias.
1. Norma internacional FIL-IDF 36: 1966.
13. Fósforo
13.1. Principio.
Se entiende por contenido en fósforo de la leche la cantidad total de fósforo expresada en porcentaje en peso, determinada por el procedimiento expuesto a continuación, que corresponde a la norma FIL-42: 1967 de la Federación Internacional de Lechería.
Este método se aplica a todas las leches liquidas normales, así como a las leches reconstituidas por dilución o disolución de las leches concentradas o de las leches desecadas.
Las materias orgánicas de la leche se destruyen por mineralización en seco (incineración). El fósforo se determina colorimétricamente, reduciendo el fosfomolibdato de amonio con diaminofenol (amidol) y midiendo la densidad óptica de la solución obtenida.
13.2. Material y aparatos.
13.2.1. Balanza analítica.
13.2.2. Cápsulas de platino o de cuarzo (de 55 mililitros de diámetro aproximadamente) provistas de vidrio de reloj.
13.2.3. Baño de agua.
13.2.4. Pipetas de 1, 2, 5 y 10 mililitros.
13.2.5. Matraces aforados, de 25 mililitros con tapones esmerilados, de 100 y de 1.000 mililitros.
13.2.6. Horno mufla.
13.2.7. Estufa a 105 ± 2ºC.
13.2.8. Espectrofotocolorímetro.
13.3. Reactivos.
13.3.1. Ácido clorhídrico (ClH) 1,0 N.
13.3.2. Ácido perclórico (ClO4H) al 65 por 100 (densidad, 1,61 gramos por mililitro a 20ºC).
13.3.3. Solución de amidol: Disolver en agua destilada un gramo o de amidol (diclorhidrato de 2,4 diaminofenol: (NH2)2 C6H3OH. 2ClH) y 20 gramos de metabisulfito de sodio o pirosulfito de sodio (S2O5Na2). Completar hasta 100 mililitros en un matraz aforado con tapón esmerilado. Preparar cada día una solución nueva.
13.3.4. Solución de molibdato: Disolver en agua destilada 8,3 gramos de molibdato de amonio: Mo7O24 (NH4)6 . 4H2O. Completar hasta 100 mililitros.
13.3.5. Solución acuosa de fosfato monopotásico (a partir de PO4H2K, desecado durante dos horas a 105 ºC), que permite trazar una curva de referencia para la determinación del fósforo: Pesar 4,393 gramos de PO4H2K, disolver en agua destilada y completar hasta 1.000 mililitros (solución A). Diluir 10 mililitros de solución A hasta 1.000 mililitros (solución B). Un mililitro de solución B = 10 microgramos de P.
Todos los reactivos deben ser puros para análisis.
13.4. Procedimiento.
13.4.1. Preparación de la muestra.
Antes del análisis poner la muestra a 20º ± 2ºC y mezclar cuidadosamente. Si no se obtiene una dispersión homogénea de la materia grasa, calentar lentamente la muestra a 40ºC, agitar suavemente y enfriar a 20º ± 2ºC.
13.4.2. Ensayo en blanco.
Efectuar un ensayo en blanco con todos los reactivos, operando como se prescribe en 13.4.3.3, pero reemplazando los cinco mililitros de la dilución por cinco mililitros de agua destilada.
13.4.3. Determinación.
13.4.3.1. Mineralización por vía seca.
En una cápsula de platino o de cuarzo pesar exactamente alrededor de 10 gramos de leche con una aproximación de 10 miligramos. Evaporar, hasta sequedad, al baño de agua hirviendo. Después de la desecación completa, calcinar en la mufla a una temperatura comprendida entre 500ºC y 550ºC hasta la obtención de cenizas blancas.
13.4.3.2. Preparación de la solución de cenizas.
Después de enfriar la cápsula, cubrirla con vidrio de reloj, disolver las cenizas en dos o tres mililitros de ácido clorhídrico normal y diluir con agua destilada. Trasvasar la solución de las cenizas a un matraz aforado de 100 mililitros, lavar el vidrio de reloj y la cápsula, recogiendo las aguas de lavado en el matraz. Completar hasta 100 mililitros con agua destilada, agitar y filtrar. Tomar 10 mililitros del filtrado, introducirlos en un matraz de 100 mililitros. Completar hasta 100 mililitros con agua destilada y agitar.
13.4.3.3. Determinación colorimétrica del fósforo.
Tomar cinco mililitros de la dilución preparada en 13.4.3.2 e introducirlos en un matraz aforado de 25 mililitros.
Añadir sucesivamente dos mililitros de ácido perclórico, dos mililitros de la solución de amidol, un mililitro de la solución de molibdato de amonio. Completar hasta 25 mililitros con agua destilada y mezclar. Esperar cinco minutos y medir la densidad óptica utilizando cubeta de un centímetro en un espectrofotocolorímetro a 750 milimicras. Buscar en la curva patrón la cantidad de fósforo contenida en el matraz de 25 mililitros, expresada en microgramos y correspondiente a la densidad óptica leída.
13.4.3.4. Determinación de la curva patrón.
En cuatro matraces aforados de 25 mililitros introducir 3, 5, 7 y 10 mililitros de la solución B. Las cantidades así introducidas son iguales a 30, 50, 70 y 100 microgramos de fósforo. Proceder a continuación como en 13.4.3.3 situar sobre una gráfica las densidades ópticas obtenidas en función de las cantidades de fósforo presentes en los matreros. Trazar la curva patrón (que es una recta).
13.5. Cálculo.
El contenido en fósforo de la leche expresada en gramos de fósforo por 100 gramos de leche viene dado por la fórmula siguiente:
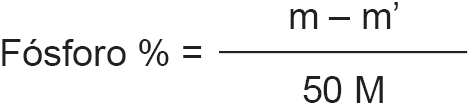
m = masa de fósforo, expresada en microgramos, obtenida según 13.4.3.4 y contenida en el matraz de 25 mililitros con la muestra de aproximadamente 50 miligramos de leche.
m’ = masa de fósforo, expresada en microgramos, obtenida según 13.4.3.4 y contenida en el ensayo en blanco.
M = masa de la leche, expresada en gramos, empleada para la mineralización.
La diferencia entre los resultados de dos determinaciones efectuadas simultáneamente o una inmediatamente después de otra por el mismo analista no debe ser mayor de 0,003 gramos de fósforo por 100 gramos de leche.
13.6. Referencias.
1. Norma internacional FIL-IDF 42: 1967.
Este documento es de carácter informativo y no tiene valor jurídico.
Ayúdenos a mejorar: puede dirigir sus comentarios y sugerencias a nuestro Servicio de atención al ciudadano
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid





