Agence d'État Bulletin Officiel de l'État






Contenu non disponible en français
 Este texto consolidado es de carácter informativo y no tiene valor jurídico.
Este texto consolidado es de carácter informativo y no tiene valor jurídico.Incluye la corrección de errores publicada en BOE núm. 306, de 22 de diciembre de 2000. Ref. BOE-A-2000-23507.
[Bloque 2: #preambulo]
La Ley 14/1986, de 25 de abril, General de Sanidad, atribuye a la Administración del Estado, en su artículo 40.5, la reglamentación, autorización y registro u homologación, según proceda, de aquellos productos y artículos sanitarios que, al afectar al ser humano, puedan suponer un riesgo para la salud de las personas. El artículo 110 de esta misma Ley encomienda a la Administración sanitaria del Estado valorar la seguridad, eficacia y eficiencia de las tecnologías relevantes para la salud y la asistencia sanitaria.
Por su parte, la Ley 25/1990, de 20 de diciembre, del Medicamento, en su artículo 1.3, declara tener por objeto, entre otros, la regulación de los principios, normas, criterios y exigencias básicas sobre la eficacia, seguridad y calidad de los productos sanitarios, a los que define en su artículo 8.12, ya que, como señala su exposición de motivos, han de alcanzar idénticos fines que aquellos que la Ley pretende para los medicamentos. Dicha Ley faculta al Gobierno, en su disposición adicional tercera, apartado 2, para determinar aquellos productos sanitarios que hayan de ser autorizados, homologados o certificados por el Estado, en razón de su especial riesgo o trascendencia para la salud.
Ambas Leyes son desarrolladas por el presente Real Decreto.
La regulación de los productos sanitarios de diagnóstico «in vitro» se ha llevado a cabo en la Unión Europea mediante la Directiva 98/79/CE, de 27 de octubre, sobre productos sanitarios para diagnóstico «in vitro», siendo sus objetivos fundamentales la eliminación de obstáculos al comercio intracomunitario, garantizando la libre circulación de los productos en las mejores condiciones de seguridad y el ofrecer a pacientes, usuarios y otras personas un nivel elevado de protección sanitaria, así como que los productos presenten las cualidades de funcionamiento que les haya asignado inicialmente el fabricante.
La Directiva 98/79/CE se trata de una Directiva específica con arreglo al apartado 2 del artículo 2 de la Directiva 89/336/CEE.
Esta regulación comunitaria se incorpora al ordenamiento jurídico nacional mediante el presente Real Decreto, dando así cumplimiento a la obligación establecida en el artículo 22.1 de la citada Directiva.
Por otra parte y con el fin de garantizar la coherencia general en la regulación sobre productos sanitarios, el presente Real Decreto modifica también el Real Decreto 414/1996, de 1 de marzo, sobre productos sanitarios, de acuerdo con las modificaciones que la Directiva 98/79/CE ha introducido en la Directiva 93/42/CEE, cuyo contenido fue incorporado al ordenamiento jurídico interno mediante el citado Real Decreto 414/1996.
Asimismo, se han introducido en este Real Decreto modificaciones al Real Decreto 634/1993, de 3 de mayo, sobre productos sanitarios implantables activos en lo que afecta a medidas nacionales que permiten llevar a cabo las funciones propias de la Administración sanitaria.
Aquellos productos sanitarios de cuya utilización se deriva un mayor riesgo se han incluido en el anexo II del presente Real Decreto, que, a su vez, se subdivide en dos grupos: A y B.
La evaluación de la conformidad de estos productos con los requisitos esenciales que les son de aplicación debe llevarse a cabo por los denominados organismos notificados, que son organismos designados por las Administraciones nacionales y notificados a la Comisión, la cual les atribuye un número de identificación que es publicado en el «Diario Oficial de las Comunidades Europeas».
En el resto de los productos, la conformidad con los requisitos esenciales es llevada a cabo por el fabricante bajo su exclusiva responsabilidad.
Existen diferentes procedimientos para llevar a cabo la evaluación de la conformidad que se recogen en los anexos III a VII.
La citada Directiva 98/79/CE señala que para demostrar la conformidad de los productos sanitarios de diagnóstico «in vitro» con los requisitos esenciales y para hacer posible el control de conformidad, resulta de utilidad la referencia a las normas armonizadas elaboradas por el Comité Europeo de Normalización (CEN) y el Comité Europeo de Normalización Electrotécnica (CENELEC) y, en determinados casos, a las denominadas especificaciones técnicas comunes.
Dichas especificaciones técnicas comunes son elaboradas por las autoridades públicas y podrán ser utilizadas en la evaluación, incluida la reevaluación, del funcionamiento de determinados productos empleados principalmente en la evaluación de seguridad del abastecimiento de sangre y de las donaciones de órganos.
El presente Real Decreto, en cuya elaboración han sido oídos los sectores afectados y el Consejo Interterritorial del Sistema Nacional de Salud, se dicta al amparo de lo dispuesto en el artículo 149.1.10.ª y 16.ª de la Constitución Española, y en virtud de lo establecido en los artículos 40,5 y 6; 95, 100 y 110 de la Ley 14/1986, de 25 de abril, General de Sanidad, y en los artículos 1.3, 2.1, 2.2, 8.12, disposición adicional tercera y disposición final de la Ley 25/1990, de 20 de diciembre, del Medicamento.
En su virtud, a propuesta de la Ministra de Sanidad y Consumo, previa aprobación del Ministro de Administraciones Públicas, de acuerdo con el Consejo de Estado y previa deliberación del Consejo de Ministros en su reunión del día 29 de septiembre de 2000,
DISPONGO:
[Bloque 3: #ci]
[Bloque 4: #a1]
1. El presente Real Decreto establece las condiciones que deben reunir los productos sanitarios para diagnóstico «in vitro» y sus accesorios para su comercialización, puesta en servicio y utilización, así como los procedimientos de evaluación de la conformidad que les son de aplicación.
Igualmente, se determinan las condiciones para su suministro con fines de evaluación del funcionamiento.
Los accesorios de los productos sanitarios para diagnóstico «in vitro» recibirán un tratamiento idéntico a estos últimos. Tanto los productos sanitarios para diagnóstico «in vitro» como los accesorios se denominarán en lo sucesivo «los productos».
2. A efectos del presente Real Decreto, los calibradores y materiales de control engloban todo tipo de sustancia, material o artículo concebido por su fabricante para establecer relaciones de medición o verificar las características de funcionamiento de un producto con respecto al uso para el cual está destinado.
3. El presente Real Decreto no afecta a la obligación de dispensación con receta médica de aquellos productos para los que así lo disponga la legislación vigente.
[Bloque 5: #a2]
1. El presente Real Decreto no se aplicará a los productos destinados a ser utilizados exclusivamente en una institución sanitaria o en locales situados en las inmediaciones directas de ésta y que son fabricados en la misma institución, siempre que no se cedan a otra entidad jurídica.
2. Quedan fuera del ámbito de aplicación del presente Real Decreto los materiales de referencia certificados a nivel internacional y los materiales utilizados en sistemas de evaluación externa de la calidad.
3. La presente disposición no afectará a la aplicación de las disposiciones contenidas en las siguientes normas: Real Decreto 1836/1999, de 3 de diciembre, por el que se aprueba el Reglamento de Instalaciones Nucleares y Radiactivas; Real Decreto 18/1991, de 30 de diciembre, sobre instalación y utilización de aparatos de rayos X con fines de diagnóstico médico; Real Decreto 53/1992, de 24 de enero, por el que se aprueba el Reglamento de Protección Sanitaria contra Radiaciones lonizantes, y Real Decreto 1132/1990, de 14 de septiembre, por el que se establecen las medidas fundamentales de protección radiológica de las personas sometidas a examen y tratamiento médico.
Tampoco afectará a la aplicación de las normas de transposición de la Directiva 96/29/EURATOM del Consejo, de 13 de mayo, por la que se establecen las normas básicas relativas a la protección sanitaria de los trabajadores y de la población contra los riesgos que resulten de las radiaciones ionizantes.
[Bloque 6: #a3]
A efectos del presente Real Decreto, se entenderá por:
a) Producto sanitario: Cualquier instrumento, dispositivo, equipo, material u otro artículo, incluidos los programas informáticos necesarios para su buen funcionamiento, destinado por el fabricante a ser utilizado en seres humanos, solo o en asociación con otros, con fines de: Diagnóstico, prevención, control, tratamiento o alivio de una enfermedad; diagnóstico, control, tratamiento, alivio o compensación de una lesión o de una deficiencia; investigación, sustitución o modificación de la anatomía o de un proceso fisiológico; regulación de la concepción, y que no ejerza la acción principal que se desee obtener en el interior o en la superficie del cuerpo humano por medios farmacológicos, inmunológicos ni metabólicos, pero a cuya función puedan contribuir tales medios.
b) Producto sanitario para diagnóstico «in vitro»: Cualquier producto sanitario que consista en un reactivo, producto reactivo, calibrador, material de control, estuche de instrumental y materiales, instrumento, aparato, equipo o sistema, utilizado solo o en asociación con otros, destinado por el fabricante a ser utilizado «in vitro» para el estudio de muestras procedentes del cuerpo humano, incluidas las donaciones de sangre y tejidos, sólo o principalmente con el fin de proporcionar información: Relativa a un estado fisiológico o patológico, o relativa a una anomalía congénita, o para determinar la seguridad y compatibilidad con receptores potenciales, o para supervisar medidas terapéuticas.
Los recipientes para muestras se considerarán productos sanitarios para diagnóstico «in vitro». Por recipientes para muestras se entiende los productos, tanto si en ellos se ha hecho el vacío como si no, destinados específicamente por el fabricante a la contención directa y a la conservación de muestras procedentes del cuerpo humano para un examen diagnóstico «in vitro».
No se considerarán productos sanitarios para el diagnóstico «in vitro» los artículos de uso general en laboratorio, salvo cuando, por sus características, estén destinados específicamente por el fabricante a usarse en exámenes diagnósticos «in vitro».
c) Accesorio: Un artículo que, sin ser un producto sanitario para diagnóstico «in vitro», esté destinado específicamente por su fabricante a ser utilizado de forma conjunta con un producto para que este último pueda utilizarse de conformidad con su finalidad prevista.
A efectos de la presente definición, los productos invasivos destinados a la obtención de muestras y los productos que se coloquen en contacto directo con el cuerpo humano para la obtención de muestras, con arreglo al Real Decreto 414/1996, no se considerarán accesorios de productos sanitarios para diagnóstico «in vitro».
d) Producto para autodiagnóstico: Cualquier producto destinado por el fabricante para poder ser utilizado por profanos a domicilio.
e) Producto para evaluación del funcionamiento: Cualquier producto destinado por el fabricante a ser objeto de uno o más estudios de evaluación de su funcionamiento en laboratorios de análisis médicos o en otros lugares adecuados fuera de sus propias instalaciones.
Los instrumentos, dispositivos, equipos, materiales u otros artículos, incluidos los programas informáticos, destinados a ser utilizados con fines de investigación sin perseguir objetivos sanitarios no se considerarán productos destinados a la evaluación del funcionamiento.
f) Fabricante: La persona física o jurídica responsable del diseño, fabricación, envasado y etiquetado de un producto con vistas a su comercialización en su propio nombre, independientemente de que estas operaciones sean efectuadas por esta misma persona o por un tercero por cuenta de aquélla.
Las obligaciones del presente Real Decreto a que están sujetos los fabricantes se aplicarán, asimismo, a la persona física o jurídica que monte, acondicione, trate, renueve totalmente o etiquete uno o varios productos fabricados previamente o les asigne una finalidad como productos con vistas a la comercialización de los mismos en su propio nombre. El presente párrafo no se aplicará a la persona que, sin ser fabricante con arreglo al párrafo primero, monte o adapte con arreglo a su finalidad prevista productos ya comercializados, para un paciente determinado.
g) Representante autorizado: La persona física o jurídica establecida en la Unión Europea, designada expresamente por el fabricante, que actúe en su lugar y a la que puedan dirigirse las autoridades y organismos en la Unión Europea en lugar de al fabricante por lo que respecta a las obligaciones de éste con arreglo al presente Real Decreto.
h) Finalidad prevista: La utilización a la que se destina el producto según las indicaciones proporcionadas por el fabricante en el etiquetado, las instrucciones de utilización y/o el material publicitario.
i) Comercialización: La primera puesta a disposición, a título oneroso o gratuito, de un producto que no sea un producto para evaluación del funcionamiento con vistas a su distribución o utilización en el mercado comunitario, independientemente de que se trate de un producto nuevo o totalmente renovado.
j) Puesta en servicio: La fase en la que un producto, que está listo para ser utilizado en el mercado comunitario con arreglo a su finalidad prevista, es puesto a disposición del usuario final por primera vez.
k) Producto nuevo: Se considerará que un producto es nuevo si: Durante los tres años anteriores no ha habido de modo permanente en el mercado comunitario ningún producto de este tipo para el analito de que se trate u otro parámetro; o el procedimiento implica una tecnología analítica que, durante los tres años anteriores, no se haya utilizado de modo permanente en el mercado comunitario en relación con un analito determinado u otro parámetro dado.
[Bloque 7: #cii]
[Bloque 8: #a4]
1. Licencias previas de funcionamiento de instalaciones.
a) De acuerdo con el artículo 100 de la Ley 14/1986, de 25 de abril, General de Sanidad, sobre determinadas actividades en relación con los productos sanitarios, la fabricación, agrupación o esterilización de los productos sanitarios para diagnóstico "in vitro" en territorio nacional requerirá licencia previa de funcionamiento de la instalación, otorgada por la Agencia Española de Medicamentos y Productos Sanitarios. De igual forma requerirán licencia previa de establecimientos aquellos locales ubicados en territorio nacional en los que se efectúe la importación de productos sanitarios para diagnóstico "in vitro" desde terceros países para su comercialización o puesta en servicio en territorio comunitario.
b) Para la obtención de estas autorizaciones, las empresas que desarrollen tales actividades lo solicitarán de la Agencia Española de Medicamentos y Productos Sanitarios, la cual estudiará la documentación presentada y notificará su resolución en el plazo de tres meses a contar desde la fecha en que la solicitud y la documentación que la acompaña hayan tenido entrada en el registro de dicho organismo autónomo.
c) La Agencia Española de Medicamentos y Productos Sanitarios solicitará a las áreas de sanidad de las Delegaciones del Gobierno informe sobre las condiciones en que las empresas desarrollan las actividades relacionadas en este apartado, ordenando a estos efectos las inspecciones de las instalaciones que resulten necesarias. La solicitud de dicho informe suspenderá, por un plazo máximo de tres meses, la tramitación del procedimiento según lo previsto en el artículo 42.5.c) de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común. La petición de dicho informe y su recepción les será comunicada a la empresa interesada.
No obstante lo dispuesto en el párrafo anterior, de forma excepcional, por razones de urgencia o cuando la naturaleza de las actividades lo aconseje, el citado informe y la correspondiente inspección, podrán ser realizados por la propia Agencia Española de Medicamentos y Productos Sanitarios.
d) Cuando las empresas desarrollen las actividades de fabricación, agrupación, esterilización o almacenamiento en instalaciones establecidas fuera del territorio español, los informes e inspecciones citados en los párrafos anteriores podrán ser sustituidos por documentación que avale convenientemente las actividades desarrolladas.
e) Para la realización de las actividades señaladas en este apartado, las empresas contarán con un responsable técnico, titulado universitario, cuya titulación acredite una cualificación adecuada en función de los productos que tenga a su cargo, quien ejercerá la supervisión directa de tales actividades.
f) De acuerdo con lo señalado en los artículos 64.5 y 72.7 de la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios, las empresas deberán contar con un seguro, aval o garantía financiera, para responder de los eventuales daños para la salud que puedan ocasionar sus productos.
g) La Agencia Española de Productos Sanitarios procederá a la denegación, suspensión o revocación de las licencias de funcionamiento cuando de la documentación aportada o de los informes de inspección correspondientes no quede garantizado que la empresa dispone de las instalaciones, medios, procedimientos y personal adecuados para desarrollar las respectivas actividades.
2. Los productos sanitarios para diagnóstico «in vitro» y los productos para evaluación de funcionamiento sólo pueden comercializarse y/o ponerse en servicio si cumplen los requisitos establecidos en la presente disposición cuando hayan sido debidamente suministrados, estén correctamente instalados y mantenidos y se utilicen conforme a su finalidad prevista, no comprometiendo la seguridad ni la salud de los pacientes, de los usuarios ni, en su caso, de terceros.
3. No podrán comercializarse en España productos fabricados en países no pertenecientes a la Unión Europea cuyo fabricante no haya designado un representante autorizado establecido en el territorio de la Unión Europea.
4. En el momento de la puesta en servicio en España de los productos, deberán proporcionarse al usuario los datos e informaciones contenidos en los apartados 5.3, 7 y 8 del anexo I, al menos, en la lengua española oficial del Estado, de modo que se garantice el uso seguro y correcto del producto y permitan disponer de forma cierta y objetiva de una información eficaz, veraz y suficiente sobre sus características esenciales.
5. No podrán comercializarse productos cuyo etiquetado o material promocional contenga menciones o distintivos que induzcan a error, atribuya funciones que no posean o proporcione expectativas de éxito asegurado. Tampoco podrá atribuir carácter superfluo a la intervención médica ni menoscabar la utilidad de otros métodos diagnósticos que requieran la intervención profesional
6. Sólo podrán utilizarse en España productos que cumplan las disposiciones del presente Real Decreto y por profesionales cualificados y debidamente adiestrados, dependiendo del producto de que se trate. Los productos deberán utilizarse en las condiciones y según las finalidades previstas por el fabricante de los mismos. Los productos deberán ser adecuadamente mantenidos de forma que se garantice que, durante el período de utilización, conservan la seguridad y prestaciones previstas por su fabricante.
Se modifica el apartado 1 por el art. 3.1 del Real Decreto 1143/2007, de 31 de agosto. Ref. BOE-A-2007-15890.
[Bloque 9: #a5]
1. Los productos deberán diseñarse y fabricarse de forma que, cuando se usen en las condiciones y para las finalidades previstas, no comprometan directa ni indirectamente el estado clínico o la seguridad de los pacientes, la seguridad o la salud de los usuarios o, en su caso, de otras personas, ni la seguridad de los bienes. Cualquier riesgo que pueda asociarse con su uso deberá ser aceptable en relación con los beneficios para el paciente y compatible con un elevado nivel de protección de la salud y la seguridad.
2. Las soluciones adoptadas por el fabricante para el diseño y la construcción de los productos deberán ajustarse a los principios de integración de la seguridad, teniendo en cuenta el estado actual de la técnica.
Al seleccionar las soluciones más adecuadas, el fabricante deberá aplicar los principios siguientes, en el orden que se indica: Eliminar o reducir tanto como se pueda los riesgos (diseño y construcción intrínsecamente seguros); cuando proceda, tomar medidas adecuadas de protección frente a los riesgos que no puedan eliminarse; informar a los usuarios de los riesgos residuales debidos a la incompleta eficacia de las medidas de protección adoptadas.
3. Los productos deberán diseñarse y fabricarse de forma que sean adecuados a los fines mencionados en el artículo 3, b) o c), y especificados por el fabricante, teniendo en cuenta el estado generalmente reconocido de la técnica. Deben presentar las prestaciones declaradas por el fabricante, en particular, cuando proceda, por lo que se refiere a sensibilidad analítica, sensibilidad diagnóstica, especificidad analítica, especificidad diagnóstica, exactitud, repetibilidad, reproducibilidad, incluido el control de las interferencias pertinentes conocidas y los límites de detección del método.
La correlación de los valores asignados a los calibradores o a los materiales de control se garantizará mediante procedimientos de medida de referencia disponibles o materiales de referencia disponibles de grado superior.
4. Las características y prestaciones mencionadas en los apartados 1 y 3 del presente artículo no deberán alterarse en grado tal que se ponga en peligro la salud o la seguridad del paciente, del usuario o, en su caso, de otras personas cuando el producto esté sometido a las situaciones que puedan presentarse en las condiciones normales de utilización, durante el período de validez. Lo anterior se aplicará al período de validez que razonablemente pueda esperarse de un producto del tipo correspondiente, teniendo en cuenta la finalidad prevista y el uso previsto del producto.
5. Los productos deberán diseñarse, fabricarse y acondicionarse de forma que sus características y prestaciones durante su uso previsto no se vean afectadas negativamente en las condiciones de almacenamiento y transporte (temperatura, humedad, etc.) indicadas en la información y las instrucciones facilitadas por el fabricante.
6. Los productos deberán cumplir los requisitos esenciales establecidos en el anexo I que les sean aplicables, teniendo en cuenta la finalidad prevista del producto de que se trate.
[Bloque 10: #a6]
1. Sólo podrán comercializarse y ponerse en servicio productos sanitarios para diagnóstico «in vitro» que ostenten el marcado CE. Como excepción, los productos para la evaluación del funcionamiento no ostentarán el marcado CE.
El marcado CE sólo podrá colocarse en productos que hayan demostrado su conformidad con los requisitos esenciales señalados en el artículo 5 y que hayan seguido los procedimientos de evaluación de la conformidad señalados en el artículo 7.
2. El marcado CE de conformidad, indicado en el anexo X, deberá figurar sobre el producto de forma visible, legible e indeleble, siempre que ello resulte apropiado y posible, así como en las instrucciones de utilización. El marcado CE de conformidad deberá figurar también en los envases de venta.
3. El marcado CE irá acompañado del número de identificación del organismo notificado responsable de la aplicación de los procedimientos establecidos en los anexos IV, VI y VII y en el apartado 6 del anexo III.
En el caso de productos para los que el procedimiento de evaluación de la conformidad no requiere la intervención de un organismo notificado, el marcado CE no podrá ir acompañado de ningún número de identificación de un organismo notificado.
4. Queda prohibido poner marcas o inscripciones que puedan inducir a terceros a interpretaciones erróneas en relación con el significado o los gráficos del marcado CE o que menoscaben la significación del mismo. Podrá colocarse sobre el producto, el envase o el prospecto de instrucciones que acompaña al producto cualquier otra marca, siempre que no se reduzca por ello la visibilidad y legibilidad del marcado CE.
5. Tampoco podrá colocarse el marcado CE de conformidad, amparándose en lo previsto en este Real Decreto, en productos no incluidos en el ámbito de aplicación del mismo. En caso de detectarse, tales supuestos serán tratados, a todos los efectos, como «productos no conformes», aplicándose los procedimientos previstos en el presente Real Decreto.
6. Cuando los productos estén sujetos a otras Directivas comunitarias o disposiciones nacionales que hayan transpuesto éstas, relativas a otros aspectos que dispongan también la colocación del marcado CE, este último indicará que los productos cumplen también las disposiciones de las demás Directivas o disposiciones nacionales.
No obstante, si una o más de estas Directivas o disposiciones nacionales que las transpongan permiten que el fabricante, durante un período de transición, escoja qué medidas aplicar, el marcado CE indicará que los productos cumplen solamente lo dispuesto en las Directivas o disposiciones nacionales aplicadas por el fabricante. En este caso, las referencias a estas Directivas, tal como se hayan publicado en el «Diario Oficial de las Comunidades Europeas», deberán indicarse en los documentos, prospectos o manual de instrucciones exigidos por las Directivas, que acompañen a dichos productos.
[Bloque 11: #a7]
1. El fabricante, a efectos de la colocación del marcado CE, deberá optar, de acuerdo con el producto de que se trate, por cualquiera de los siguientes procedimientos de evaluación de la conformidad, que serán, en su caso, desarrollados por alguno de los organismos notificados incluidos en la lista que a tal efecto se publique en el «Diario Oficial de las Comunidades Europeas».
El fabricante podrá dar instrucciones a su representante autorizado para iniciar los procedimientos que disponen los anexos III, V, VI y VIII. Tanto el fabricante como el representante autorizado, en el curso de tales procedimientos, quedan sometidos a las obligaciones que se establecen en los correspondientes anexos.
1. Respecto de los productos enumerados en la lista A del anexo II distintos de los destinados a la evaluación del funcionamiento, el fabricante, para colocar el marcado CE, podrá optar entre:
a) Seguir el procedimiento relativo a la declaración CE de conformidad establecido en el anexo IV (sistema de garantía de calidad total) o
b) Seguir el procedimiento relativo al examen CE de tipo establecido en el anexo V, junto con el procedimiento relativo a la declaración CE de conformidad establecido en el anexo VII (sistema de garantía de calidad de la producción).
2. Respecto de los productos enumerados en la lista B del anexo II distintos de los destinados a la evaluación del funcionamiento, el fabricante, para colocar el marcado CE, podrá optar entre:
a) Seguir el procedimiento relativo a la declaración CE de conformidad establecido en el anexo IV (sistema de garantía de calidad total) o
b) Seguir el procedimiento relativo al examen CE de tipo establecido en el anexo V, junto con: El procedimiento relativo a la verificación CE establecido en el anexo VI o el procedimiento relativo a la declaración CE de conformidad establecido en el anexo VII (sistema de garantía de calidad de la producción).
3. Respecto de los productos para autodiagnóstico, que no sean los contemplados en el anexo II y que no estén destinados a la evaluación del funcionamiento, el fabricante cumplirá los requisitos suplementarios establecidos en el apartado 6 del anexo III y redactará la declaración de conformidad mencionada en este anexo. En vez de aplicar este procedimiento, el fabricante podrá seguir los procedimientos contemplados en el apartado 1.1 y en el apartado 1.2 de este artículo.
4. En el caso de los productos para evaluación del funcionamiento, el fabricante seguirá el procedimiento mencionado en el anexo VIII y redactará la declaración establecida en dicho anexo antes de suministrar tales productos. Asimismo, deberá cumplir lo establecido en el artículo 9.
5. Respecto de los productos que no sean: Los que abarca el anexo II; los productos para evaluación del funcionamiento y los productos para autodiagnóstico, el fabricante, para colocar el marcado CE, seguirá el procedimiento mencionado en el anexo III y redactará la declaración CE de conformidad exigida antes de la comercialización de los productos. La Dirección General de Farmacia y Productos Sanitarios evaluará, en su caso, la documentación señalada en el citado anexo III, a efectos de establecer la conformidad de los productos después de que éstos hayan sido comercializados y/o puestos en servicio.
2. Durante el procedimiento de evaluación de la conformidad de un producto, el fabricante y, en caso de que intervenga, el organismo notificado, tendrán en cuenta los resultados de cualquier operación de evaluación y verificación que se hayan realizado, en su caso, con arreglo al presente Real Decreto en una fase intermedia de fabricación.
3. El fabricante deberá conservar la declaración de conformidad y la documentación técnica citada en los anexos III a VIII, así como las decisiones, informes y certificados procedentes de los organismos notificados, y ponerlos a disposición de las autoridades competentes a efectos de control durante un período de cinco años, contados a partir de la fabricación del último producto. En caso de que el fabricante no esté establecido en la Unión Europea, la obligación de presentar la citada documentación cuando se solicite se aplicará a su representante autorizado.
4. Cuando el procedimiento de evaluación de la conformidad implique la intervención de un organismo notificado, el fabricante o su representante autorizado podrá dirigirse a un organismo de su elección en el marco de las tareas para las que el organismo haya sido notificado.
5. Toda persona física o jurídica que fabrique productos incluidos en el ámbito de aplicación del presente Real Decreto y que, sin ser comercializados, los ponga en servicio y los utilice en el contexto de su actividad profesional deberá colocar el marcado CE a sus productos tras haber aplicado los procedimientos de evaluación de la conformidad establecidos en este artículo que les resulten de aplicación.
[Bloque 12: #a8]
1. Los productos sanitarios que estén provistos del marcado CE y hayan seguido los procedimientos de evaluación de la conformidad señalados en el artículo 7 serán considerados conformes con los requisitos esenciales, salvo indicios razonables en contra.
2. Cuando los productos se ajusten a las normas nacionales correspondientes adoptadas en aplicación de normas armonizadas, que satisfagan determinados requisitos esenciales, se presumirán conformes a los requisitos esenciales de que se trate.
3. A los efectos del apartado anterior, las normas nacionales y las normas armonizadas son aquéllas cuyos números de referencia se hayan publicado en el «Boletín Oficial del Estado» y en el «Diario Oficial de las Comunidades Europeas», respectivamente.
4. Igualmente, se presumirá que los productos diseñados y fabricados con arreglo a las especificaciones técnicas comunes elaboradas para los productos de la lista A del anexo II y, cuando sea necesario, para los productos de la lista B del anexo II, cumplen los requisitos esenciales a que se refiere el artículo 5.
5. Las especificaciones citadas deberán establecer, de manera adecuada, los criterios de evaluación y evaluación de funcionamiento, los criterios de aprobación de lotes, los métodos de referencia y los materiales de referencia.
6. En términos generales, los fabricantes deberán respetar las especificaciones técnicas comunes; si, por razones debidamente justificadas, los fabricantes no cumplieran dichas especificaciones, deberán adoptar soluciones de un nivel, al menos, equivalente a éstas.
7. Cuando se haga referencia en el presente Real Decreto a las normas armonizadas, se considerará que se refieren también a las especificaciones técnicas comunes.
[Bloque 13: #ciii]
[Bloque 14: #a9]
1. Todo fabricante establecido en España que comercialice productos en su propio nombre será incluido en el registro de responsables de la comercialización que existirá en la Dirección General de Farmacia y Productos Sanitarios. Para ello los interesados efectuarán una comunicación a la Comunidad Autónoma donde tenga su domicilio la empresa, en el momento en que se haga efectiva la primera comercialización del producto. La Comunidad Autónoma trasladará inmediatamente la documentación a la Dirección General de Farmacia y Productos Sanitarios.
Las comunicaciones podrán presentarse en cualquiera de los lugares previstos en el artículo 38.4 de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común.
2. La comunicación contendrá, al menos en la lengua española oficial del Estado, los siguientes datos:
a) Datos identificativos de la persona que efectúa la comunicación.
b) Nombre y razón social del fabricante.
c) Dirección del domicilio social del fabricante.
d) Nombre comercial del producto en España y nombres comerciales con los que se comercializa el producto en la Unión Europea en caso de que sean diferentes del primero.
e) Tipo de producto.
f) Características tecnológicas.
g) Finalidad prevista del producto.
h) Indicación de si se trata de un producto nuevo en los términos del artículo 3, k).
3. Cuando un fabricante que comercialice productos en su propio nombre no tenga domicilio social en un Estado miembro de la Unión Europea designará un representante autorizado establecido en la Unión Europea. En el caso de que el representante autorizado fuera una persona física o jurídica establecida en España, ésta deberá seguir el procedimiento indicado en los apartados 1 y 2 del presente artículo.
4. En el caso de los productos nuevos en los términos del artículo 3, k), del presente Real Decreto, la Dirección General de Farmacia y Productos Sanitarios podrá solicitar del fabricante, durante los dos años siguientes a la comunicación a la que se refiere el apartado 1 de este artículo y por motivos justificados, un informe sobre la experiencia adquirida con el producto tras su puesta en el mercado. Dicho informe será remitido a las autoridades de los Estados miembros que lo soliciten.
5. Cualquier modificación de los datos señalados en el apartado 2 de este artículo será comunicada siguiendo el procedimiento establecido en este artículo. También se comunicará el cese de la comercialización de los productos.
[Bloque 15: #a10]
1. Toda persona que comercialice o ponga en servicio productos incluidos en el anexo II del presente Real Decreto o productos para autodiagnóstico deberá dirigir una comunicación a la Dirección General de Farmacia y Productos Sanitarios en el momento en que haga efectiva la primera comercialización o puesta en servicio del producto en España.
En el caso de los productos sanitarios para diagnóstico «in vitro», señalados en el párrafo anterior, que no procedan de terceros países, podrá efectuarse la comunicación al registro que establezca, en su caso, el órgano competente de la Comunidad Autónoma en la que se realice la primera comercialización o puesta en servicio del producto sanitario. El órgano competente de la Comunidad Autónoma trasladará inmediatamente la documentación a la Dirección General de Farmacia y Productos Sanitarios.
Las comunicaciones podrán presentarse en cualquiera de los lugares previstos en el artículo 38.4 de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común.
La Dirección General de Farmacia y Productos Sanitarios mantendrá actualizado un registro de productos del anexo II y productos para autodiagnóstico con todas las comunicaciones a que se refieren los apartados anteriores.
2. La comunicación contendrá, como mínimo, al menos en la lengua española oficial del Estado, los siguientes datos:
a) Datos identificativos de la persona que efectúa la comunicación.
b) Nombre comercial del producto en España y nombres comerciales con los que se comercializa el producto en la Unión Europea en caso de que sean diferentes del primero.
c) Tipo de producto.
d) Finalidad prevista.
e) Parámetros analíticos y/o diagnósticos contemplados en el apartado 3 del artículo 5 del presente Real Decreto.
f) Resultados de la evaluación de funcionamiento.
g) Certificados emitidos por el/los organismo/s notificados que han intervenido en la evaluación de la conformidad a efectos de la colocación del marcado CE.
h) Datos identificativos del fabricante y del lugar de fabricación y de su representante autorizado en la Unión Europea, en su caso.
i) Etiquetado e instrucciones de uso presentados al/certificados por el organismo notificado.
j) Etiquetado e instrucciones de uso con los que se vaya a comercializar el producto en España cuando la versión en la lengua española oficial del Estado no se encuentre incluida en la información señalada en el párrafo i).
En este caso, la versión en lengua española deberá ser una traducción fiel de las presentadas al/certificadas por el organismo notificado
k) Fecha en la que el producto se comercializa o pone en servicio en España.
l) Datos identificativos de los distribuidores en España, caso que no coincidan con la persona señalada en el párrafo a) de este artículo.
3. Cualquier modificación de los datos señalados en el apartado anterior será comunicada siguiendo el procedimiento establecido en el presente artículo. También se comunicará cualquier modificación habida en los certificados, incluida su suspensión o retirada, así como el cese de la comercialización.
[Bloque 16: #a11]
Los registros previstos en los artículos 9 y 10 se mantendrán a disposición de las autoridades competentes de las Comunidades Autónomas.
[Bloque 17: #a12]
Los datos derivados de la aplicación del artículo 9 del presente Real Decreto serán trasladados por la Dirección General de Farmacia y Productos Sanitarios, para su registro, a la base de datos europea descrita en el artículo 12 de la Directiva 98/79/CE, utilizando para su envío un formato normalizado.
[Bloque 18: #civ]
[Bloque 19: #a13]
1. Solamente se venderán y distribuirán productos conformes con el presente Real Decreto y no caducados, tomando como referencia para esto último la fecha indicada en el párrafo e) del apartado 8.4 del anexo I.
2. La distribución y la venta al público estarán sometidas a la vigilancia e inspección de las autoridades sanitarias de la Comunidad Autónoma correspondiente, quien podrá establecer el procedimiento exigido para la autorización de tales actividades.
3. Las personas físicas o jurídicas que se dediquen a la actividad de distribución o de venta al público de productos contemplados en este Real Decreto lo comunicarán previamente a las autoridades sanitarias de la Comunidad Autónoma donde se encuentren establecidas mediante escrito en el que hagan constar:
a) Identificación de los locales de distribución.
b) Tipos de productos que distribuye.
c) Identificación del técnico previsto en el apartado 4 del artículo 14.
Quedan exceptuados de realizar tal comunicación aquellos distribuidores que posean, además, la condición de fabricantes o importadores de los productos, así como las oficinas de farmacia.
4. La venta al público de los productos para autodiagnóstico se realizará exclusivamente a través de las oficinas de farmacia.
5. Queda prohibida la venta ambulante de productos sanitarios para diagnóstico «in vitro».
6. Para la venta al público de los productos de autodiagnóstico se exigirá la correspondiente prescripción. Como excepción, esta prescripción no será necesaria en los productos para el diagnóstico del embarazo y de la fertilidad, así como en los productos de autodiagnóstico para la determinación de la glucemia.
7. Queda prohibida la venta al público por correspondencia o por procedimientos telemáticos de los productos de autodiagnóstico. No obstante, esta modalidad podrá efectuarse por las oficinas de farmacia, con la intervención de un farmacéutico y el asesoramiento correspondiente, para los productos en los que no es necesaria la correspondiente prescripción.
8. Por razones de salud pública, no se pondrán a disposición del público los productos para el diagnóstico genético.
Se añaden los apartados 6 a 8 por la disposición final 2.1 del Real Decreto 1591/2009, de 16 de octubre. Ref. BOE-A-2009-17606.
[Bloque 20: #a14]
1. El distribuidor mantendrá una documentación ordenada de los productos que distribuya o destine para su utilización en territorio nacional.
Esta documentación deberá contener, al menos, los datos siguientes: Nombre comercial del producto, modelo, serie y/o número de lote, fecha de adquisición, fecha de envío o suministro e identificación del cliente.
2. Siempre que le sea requerida, el distribuidor facilitará a las autoridades sanitarias, para el ejercicio de sus respectivas competencias, la documentación que avale la conformidad de los productos con lo dispuesto en el presente Real Decreto. En caso de que el distribuidor no esté en disposición de acceder a esta documentación, deberá ser facilitada por el fabricante.
3. En caso de sospecha o evidencia de riesgo para la salud, el distribuidor ejecutará cualquier medida de restricción o seguimiento de la utilización de los productos que resulte adecuada, así como aquellas que, en su caso, puedan ser determinadas por las autoridades sanitarias.
4. El distribuidor deberá designar un técnico cuya titulación acredite una cualificación adecuada según la naturaleza de los productos de que se trate. Este técnico tendrá directamente a su cargo la ejecución de las actividades contenidas en este artículo y de los procedimientos señalados en los artículos 10 y 20 del presente Real Decreto, cuando corresponda.
Igualmente, se responsabilizará de la información técnico-sanitaria que se suministre sobre los productos comercializados o puestos en servicio en España.
[Bloque 21: #cv]
[Bloque 22: #a15]
Los productos introducidos desde países comunitarios y los importados de terceros países sólo podrán comercializarse y ponerse en servicio en España si cumplen las prescripciones establecidas en el presente Real Decreto.
[Bloque 23: #a16]
Los productos que se fabriquen con destino exclusivo para la exportación a países no comunitarios y no cumplan los requisitos expuestos en la presente disposición deberán ser etiquetados de forma que se diferencien de los destinados al mercado comunitario, con objeto de evitar su utilización en el mismo.
[Bloque 24: #cvi]
[Bloque 25: #a17]
1. El Ministerio de Sanidad y Consumo designará los organismos que efectuarán los procedimientos recogidos en el artículo 7, así como las tareas específicas asignadas a cada organismo, y lo notificará a la Comisión Europea y a los demás Estados miembros. Tal designación será publicada en el «Boletín Oficial del Estado», junto con el número de identificación asignado por la Comisión Europea y las tareas específicas.
El Ministerio de Sanidad y Consumo realizará las actuaciones necesarias para comprobar la aptitud de los organismos en orden a su designación y llevará a cabo un control continuo para verificar el mantenimiento de estas aptitudes en los organismos designados.
2. Los organismos notificados deberán cumplir los requisitos contemplados en el anexo IX. Se presumirá que los organismos que cumplen los criterios fijados en las normas nacionales que transponen las normas armonizadas correspondientes se ajustan a los citados requisitos. No obstante, el acto de designación resulta independiente de cualquier certificación o acreditación nacional y no queda vinculado por ellas.
3. Cuando se haya designado un organismo y se compruebe que tal organismo ya no satisface los requisitos establecidos en el anexo IX, el Ministerio de Sanidad y Consumo retirará la autorización o limitará su alcance, previo el correspondiente procedimiento administrativo, con audiencia del interesado, e informará de ello a la Comisión Europea y a los demás Estados miembros.
4. En caso de cese de funciones de un organismo notificado, el Ministerio de Sanidad y Consumo adoptará las medidas oportunas para garantizar la continuidad de la gestión de los procedimientos de evaluación de la conformidad.
[Bloque 26: #a18]
1. El organismo notificado comprobará que el producto satisface los requisitos esenciales contemplados en el presente Real Decreto y efectuará las tareas previstas en los procedimientos de evaluación de la conformidad elegidos por los fabricantes.
El organismo notificado emitirá los certificados correspondientes a los procedimientos de evaluación de la conformidad, así como las certificaciones sobre los sistemas de calidad solicitados por los fabricantes de productos sanitarios para diagnóstico "in vitro".
2. La documentación correspondiente a dichos procedimientos de evaluación se redactará, al menos en la lengua española oficial del Estado. No obstante, el organismo notificado podrá aceptar la presentación en otra lengua de documentación científica o especializada que soporte parte de la evaluación de la conformidad.
El organismo notificado podrá exigir cualquier dato o información que juzgue necesario para establecer o mantener el certificado de conformidad, a la vista del procedimiento elegido.
3. En la evaluación de la conformidad de un producto, el organismo notificado tendrá en cuenta cualquier información pertinente relativa a las características y funcionamiento de los productos, incluidos en particular los resultados de todos los ensayos y verificaciones oportunos ya realizados de conformidad con las legislaciones nacionales en vigor el 7 de diciembre de 1998, en cualquier país de la Unión Europea.
4. Las decisiones que tomen los organismos notificados con arreglo a los anexos III, IV y V serán válidas durante un máximo de cinco años y podrán prorrogarse, previa petición realizada en el momento convenido en el contrato firmado por ambas partes, por períodos sucesivos de cinco años como máximo.
5. En caso de que un organismo notificado observe que el fabricante no cumple o que ha dejado de cumplir los requisitos pertinentes del presente Real Decreto, o que no se hubiera debido expedir un certificado, suspenderá o retirará el certificado expedido, teniendo presente el principio de proporcionalidad, a no ser que el fabricante garantice el cumplimiento de tales requisitos mediante la aplicación de medidas correctoras eficaces. En los casos de suspensión o retirada del certificado, o en los casos en que se someta a restricciones, o en los casos en que pudiera requerirse la intervención de la autoridad competente, el organismo notificado informará de los hechos a la Dirección General de Farmacia y Productos Sanitarios trasladando una copia de la decisión correspondiente. La Dirección General de Farmacia y Productos Sanitarios informará a los demás Estados miembros y a la Comisión y mantendrá informadas a las Comunidades Autónomas de los certificados suspendidos o retirados.
6. El organismo notificado informará a los demás organismos notificados y a las autoridades competentes de todos los certificados suspendidos o retirados, así como de los certificados expedidos o denegados, previa petición. Además, previa petición, pondrá a su disposición toda la información adicional pertinente.
7. Previa petición, el organismo notificado facilitará toda la información y documentación pertinentes, incluidos los documentos presupuestarios, necesarios para que el Ministerio de Sanidad y Consumo verifique el cumplimiento de los requisitos del anexo IX.
Se añade un párrafo segundo al apartado 1 y se suprimen el párrafo primero del apartado 2 y el apartado 8 por el art.3.2 del Real Decreto 1143/2007, de 31 de agosto. Ref. BOE-A-2007-15890.
[Bloque 27: #a18bis]
El organismo notificado en sus actuaciones de evaluación de la conformidad y de certificación de sistemas de calidad, seguirá el procedimiento siguiente:
1. El procedimiento se iniciará a solicitud del fabricante o de su representante autorizado en la Unión Europea, conforme lo indicado en el artículo 7.1. Para ello el organismo notificado establecerá los formularios correspondientes adecuados a cada procedimiento, así como la documentación técnica y de calidad a aportar en cada caso.
2. El organismo notificado y el fabricante o su representante autorizado establecido en la Unión Europea fijarán de común acuerdo los plazos para la realización de los procedimientos, teniendo en cuenta la complejidad tecnológica de los productos y de los procesos industriales, el número y la localización geográfica de las instalaciones y el número de productos incluidos en el procedimiento. La fijación de plazo formará parte de la validación de la solicitud y hasta que no se lleve a efecto no quedará admitido a trámite el procedimiento.
3. Si la solicitud o la documentación presentada no se ajustara a lo establecido, el organismo notificado requerirá al interesado para que, en el plazo de 10 días, subsane las deficiencias, indicándole que si así no lo hiciera se le tendrá por desistido de su petición. Este plazo podrá ampliarse a requerimiento del interesado si el organismo notificado considera esta ampliación justificada. Trascurrido el plazo sin que se produzca la subsanación, el organismo notificado comunicará el desistimiento al interesado.
4. El organismo notificado realizará las actuaciones oportunas para establecer o mantener el certificado de conformidad solicitado. En el curso del procedimiento podrá requerir cuantos datos o informaciones considere necesarios para decidir sobre dicha conformidad.
5. El organismo notificado podrá reconocer los informes técnicos sobre productos o sistemas de calidad emitidos por entidades competentes, tanto públicas como privadas, que hayan sido acreditadas previamente por el propio organismo notificado, y que se emitan en el ámbito y las condiciones establecidas en dicha acreditación.
6. La solicitud de los datos o informes suspenderá los plazos acordados hasta tanto obren en poder del organismo notificado. La falta de presentación de dichos datos o informes, transcurridos tres meses de su solicitud, producirá la caducidad del procedimiento y el archivo de las actuaciones, salvo que en ese plazo el organismo notificado haya autorizado, a petición motivada del solicitante, una prórroga de dicho plazo.
7. El organismo notificado comunicará al solicitante la decisión adoptada, remitiéndole los correspondientes certificados o bien desestimando su solicitud. En este último supuesto la notificación contendrá la motivación de la decisión adoptada, así como las vías de alegación que contra la misma procedan. Contra las decisiones adoptadas por el organismo notificado el interesado podrá manifestar su disconformidad, en el plazo de un mes, ante el propio organismo, que, una vez revisadas las alegaciones, comunicará la decisión final al interesado en el mismo plazo.
En caso de persistir el desacuerdo, el interesado podrá manifestar su disconformidad, en el plazo de un mes, ante el Ministro de Sanidad y Consumo, el cual, previa instrucción del oportuno expediente con audiencia del interesado, resolverá en el plazo máximo de tres meses. Contra dicha resolución podrán interponerse los recursos que proceden conforme a lo establecido en los artículos 116 y 117 de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común.
8. Lo previsto en el apartado anterior será también de aplicación en el caso de las decisiones adoptadas por el organismo notificado de acuerdo con lo previsto en el apartado 5 del artículo 18.
Se añade por el art. 3.3 del Real Decreto 1143/2007, de 31 de agosto. Ref. BOE-A-2007-15890.
Texto añadido, publicado el 01/09/2007, en vigor a partir del 01/09/2007.
[Bloque 28: #a19]
1. Los órganos competentes de las Comunidades Autónomas efectuarán inspecciones periódicas para verificar que los productos puestos en el mercado y puestos en servicio son conformes con la presente disposición.
2. La Dirección General de Farmacia y Productos Sanitarios, por sí misma o a través de los servicios de inspección de la Administración General del Estado habilitados a estos fines, podrá ejercer actividades de inspección y control respecto de los productos contemplados en el capítulo V y de los establecimientos en los que se fabriquen, importen o exporten, siempre que estén situados en territorio nacional.
3. El personal al servicio de las Administraciones públicas que desarrolle funciones de inspección procederá según lo establecido en el apartado 3 del artículo 105 de la Ley 25/1990, de 20 de diciembre, del Medicamento, y además podrá solicitar al responsable de la puesta en el mercado información sobre la documentación de los productos comercializados o puestos en servicio en España y de los sometidos a evaluación de funcionamiento.
4. Las autoridades de la Administración General del Estado y de las Comunidades Autónomas competentes se auxiliarán mutuamente a efectos de inspección.
En el seno del Consejo Interterritorial del Sistema Nacional de Salud se adoptarán las medidas adecuadas para favorecer la cooperación y asistencia mutua entre las autoridades sanitarias estatales y autonómicas; asimismo, podrán establecerse programas específicos de control con referencia a la naturaleza, extensión, intensidad y frecuencia de los controles a efectuar.
[Bloque 29: #a20]
1. Cuando, con ocasión de su actividad, los profesionales sanitarios, las autoridades inspectoras, los fabricantes, los responsables de los productos o los centros que realicen programas de evaluación externa de la calidad, adviertan cualquier disfunción, alteración de las características o de las prestaciones de un producto, así como cualquier inadecuación del etiquetado o de las instrucciones de uso que, directa o indirectamente, pueda o haya podido dar lugar a la muerte o al deterioro grave del estado de salud de un paciente, de un usuario o de otras personas, deberán comunicarlo a la Dirección General de Farmacia y Productos Sanitarios, donde dichos datos se evaluarán y registrarán.
Esta comunicación se realizará sin perjuicio de la que, en su caso, sea exigida por la autoridad sanitaria de la Comunidad Autónoma correspondiente. El fabricante o su representante autorizado serán informados de los hechos.
2. Igualmente, el fabricante o cualquier otro responsable del producto notificará la retirada del mercado de un producto ocasionada por razones de carácter técnico o médico en relación con las características o funcionamiento de un producto relacionadas con alguna de las circunstancias señaladas en el apartado anterior.
3. Tras haber procedido a su evaluación, a ser posible conjuntamente con el fabricante, la Dirección General de Farmacia y Productos Sanitarios informará a la Comisión Europea y a los demás Estados miembros de los hechos respecto de los cuales se hayan tomado o se estudie la posibilidad de tomar medidas pertinentes.
[Bloque 30: #a21]
1. Si existieran indicios razonables sobre la no conformidad de un producto, las autoridades sanitarias podrán exigir, motivadamente, del fabricante o del responsable autorizado en la Unión Europea la presentación, en la lengua española oficial del Estado, de cuanta información se considere necesaria para juzgar sobre dicha conformidad.
La negativa a facilitar la documentación señalada podrá considerarse como presunción de no conformidad.
2. Como garantía de la salud y seguridad de las personas, las autoridades sanitarias competentes, cuando consideren que un producto correctamente puesto en servicio, instalado, mantenido y utilizado con arreglo a su finalidad prevista, puede comprometer la salud y/o la seguridad de los pacientes, usuarios o la de terceras personas o la seguridad de los bienes, procederán a adoptar las medidas adecuadas previstas en el capítulo V del Título I de la Ley 14/1986, de 25 de abril, General de Sanidad, y en el capítulo I del Título IX y artículo 110 de la Ley 25/1990, de 20 de diciembre, del Medicamento.
Tales medidas serán previamente puestas en conocimiento del fabricante, salvo que concurran razones de urgencia para su adopción.
3. La Dirección General de Farmacia y Productos Sanitarios deberá ser informada de forma inmediata por la autoridad sanitaria que adoptó la medida indicando las razones que la han motivado.
4. La Dirección General de Farmacia y Productos Sanitarios comunicará inmediatamente a la Comisión Europea las medidas que se hayan adoptado.
5. Cuando un producto no conforme ostente el marcado CE, las autoridades sanitarias competentes adoptarán las medidas apropiadas contra quien ha colocado el marcado e informarán de ellas a la Dirección General de Farmacia y Productos Sanitarios. Dicho centro directivo actuará en consecuencia y lo comunicará a la Comisión Europea y a las autoridades competentes de los demás Estados miembros.
6. La comunicación a la Comisión Europea no será necesaria cuando la falta de conformidad se refiera al incumplimiento de las disposiciones señaladas en los artículos 4.1, 4.4, 9 y 10 del presente Real Decreto.
7. Cuando por iniciativa del fabricante o distribuidor de los productos se acuerden medidas de prevención, alerta o retirada de los productos del mercado, así como la difusión de advertencias relacionadas con productos sanitarios para diagnóstico «in vitro», serán puestas en conocimiento de las autoridades sanitarias competentes antes de su inicio. Dichas autoridades podrán suspender la ejecución de las medidas propuestas, impedirlas o modificarlas por razones justificadas de salud pública.
8. En todos los casos de no conformidad, el fabricante, su representante autorizado o, en su caso, el responsable del producto en España, queda obligado a efectuar las acciones oportunas para que cese la situación de no conformidad, en las condiciones establecidas por la autoridad competente. La persistencia de la no conformidad dará lugar a la prohibición o restricción de la comercialización o puesta en servicio del producto o a su retirada del mercado, siguiéndose el procedimiento establecido en este artículo.
9. Cuando la comprobación de la conformidad de un producto requiera la realización de evaluaciones o ensayos sobre el producto o su documentación técnica, los gastos derivados de tal comprobación serán satisfechos por el fabricante o su responsable, quien facilitará igualmente las muestras necesarias para realizar tal comprobación.
10. Cuando por aplicación del presente Real Decreto se rechace o restrinja la puesta en el mercado y/o la puesta en servicio de un producto, así como cuando se realice la retirada de un producto del mercado, el interesado podrá manifestar su disconformidad ante el órgano superior jerárquico del que dictó la resolución, mediante el correspondiente recurso de alzada en el plazo de un mes, conforme a lo establecido en los artículos 114 y 115 de la Ley de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común.
[Bloque 31: #a22]
Cuando la Dirección General de Farmacia y Productos Sanitarios considere, respecto de un producto o grupo de productos determinado, que para garantizar la protección de la salud de las personas, la seguridad o el cumplimiento de las normas de salud pública con arreglo a lo dispuesto en el artículo 30 del Tratado CE, debe prohibirse, restringirse o someterse a requisitos especiales la disponibilidad de dichos productos, podrá adoptar todas las medidas transitorias necesarias y justificadas, de lo que informará a la Comisión y a los otros Estados miembros indicando los motivos de su decisión.
Por los mismos motivos podrá dictar disposiciones sobre condiciones de utilización de los productos o sobre medidas de seguimiento especial y hacer incluir las advertencias necesarias para evitar riesgos sanitarios en la utilización de los productos.
[Bloque 32: #a23]
Previa petición justificada, la Dirección General de Farmacia y Productos Sanitarios podrá autorizar de forma expresa e individualizada, en interés de la protección de la salud, la comercialización, puesta en servicio y utilización de productos para los que no se hayan satisfecho los procedimientos de evaluación de la conformidad indicados en el artículo 7 del presente Real Decreto.
[Bloque 33: #a24]
Las Administraciones públicas, sin perjuicio de las disposiciones existentes en materia de secreto profesional, velarán porque todas las partes a las que concierne la aplicación del presente Real Decreto mantengan la confidencialidad de cualquier información obtenida en el ejercicio de su función. Ello no afectará a las obligaciones de las autoridades competentes y de los organismos notificados con respecto a la información recíproca, ni a la difusión de advertencias, ni a las obligaciones de información que incumban a las personas afectadas, tanto ante las autoridades como ante los órganos jurisdiccionales.
[Bloque 34: #cvii]
[Bloque 35: #a25]
1. La publicidad dirigida a la promoción de los productos se regirá por los principios generales establecidos en la Ley 34/1988, de 11 de noviembre, General de Publicidad, así como en el artículo 102 de la Ley 14/1986, de 25 de abril, General de Sanidad.
2. Los medios de información y promoción utilizados como soporte, ya sean escritos, audiovisuales o de otra naturaleza, tendrán carácter básicamente científico y estarán dirigidos y se distribuirán con carácter general a profesionales sanitarios.
3. La información se facilitará a través de personas adecuadamente formadas y que posean los conocimientos suficientes para proporcionar orientaciones precisas y completas sobre los productos que promocionan. El contenido de la información incluirá los datos técnicos necesarios para que se pueda juzgar objetivamente sobre la utilidad del producto sanitario para diagnóstico «in vitro».
4. En el caso en que, por la naturaleza del producto, se efectúe publicidad directa al público, se tendrá en cuenta, en particular, lo establecido en el apartado 5 del artículo 4 del presente Real Decreto. Los mensajes publicitarios y/o promocionales dirigidos al público de los productos contemplados en el presente Real Decreto serán objeto de autorización previa por las autoridades sanitarias de las Comunidades Autónomas.
5. Particularmente, los textos de publicidad o promoción deberán reflejar la exactitud de los datos obtenidos con la utilización del producto, así como las limitaciones, restricciones o advertencias necesarias para que los productos alcancen su finalidad prevista.
6. En la publicidad de los productos dirigida al público y efectuada por particulares se prohíbe cualquier mención que haga referencia a una autoridad sanitaria o a recomendaciones que hayan formulado científicos, profesionales de la salud u otras personas que puedan, debido a su notoriedad, incitar a su utilización.
7. De conformidad con el artículo 27 de la Ley 14/1986, de 25 de abril, General de Sanidad, queda prohibida la publicidad de todo producto que, sin ajustarse a lo establecido en este Real Decreto, pretenda realizar alguno de los fines previstos en el artículo 3 b) o 3 c) de este Real Decreto.
8. Queda prohibido efectuar publicidad dirigida al público de los productos de autodiagnóstico, con excepción de los destinados al diagnóstico del embarazo y de la fertilidad. Igualmente queda prohibido efectuar publicidad dirigida al público de los productos para el diagnóstico genético.
Se añade el apartado 8 por la disposición final 2.2 del Real Decreto 1591/2009, de 16 de octubre. Ref. BOE-A-2009-17606.
[Bloque 36: #a26]
1. En el marco de la promoción de los productos sanitarios para diagnóstico «in vitro» queda prohibido otorgar, ofrecer o prometer primas, ventajas pecuniarias o ventajas en especie a los profesionales sanitarios o cualquier otra persona relacionada con la utilización, prescripción o dispensación de los productos así como a sus parientes y personas con las que convivan.
2. Las personas relacionadas en el apartado anterior no podrán solicitar o aceptar ninguno de los incentivos prohibidos.
[Bloque 37: #a27]
1. Las disposiciones del artículo anterior no supondrán un obstáculo para la hospitalidad ofrecida, directa o indirectamente, en el marco de manifestaciones de carácter exclusivamente profesional y científico. Dicha hospitalidad deberá ser siempre moderada en su nivel y subordinada al objetivo principal de la reunión y no podrá ser extensible a personas que no sean profesionales de la salud.
2. Los premios, becas, contribuciones y subvenciones a reuniones, congresos, viajes de estudio y actos similares donados por personas relacionadas con la fabricación, elaboración, distribución y dispensación de productos sanitarios se aplicarán exclusivamente a actividades de índole científica cuando sus destinatarios sean facultativos en ejercicio clínico o las entidades en que se asocian.
En las publicaciones de trabajos y ponencias de reuniones, congresos y actos similares se harán constar los fondos obtenidos para su realización y fuente de financiación. La misma obligación alcanzará al medio de comunicación por cuya vía se hagan públicos y que obtenga fondos por o para su publicación.
3. Lo previsto en este artículo no afectará a los cursos de entrenamiento, necesarios para la correcta utilización de los productos, que sean facilitados a los profesionales por los fabricantes o distribuidores de los mismos.
[Bloque 38: #a28]
En las ferias, exposiciones y demostraciones podrán presentarse productos que no cumplan las disposiciones del presente Real Decreto, siempre que en un cartel suficientemente visible, colocado en los propios productos o junto a ellos, se indique claramente que dichos productos no pueden comercializarse ni ponerse en servicio hasta que se declare su conformidad. Tales demostraciones no podrán nunca implicar la utilización de estos productos sobre muestras procedentes de los participantes.
[Bloque 39: #cviii]
[Bloque 41: #a29]
Tendrán la consideración de infracciones a lo dispuesto en el presente Real Decreto las acciones y omisiones previstas en el artículo 35 de la Ley 14/1986, de 25 de abril, General de Sanidad, y las siguientes específicas:
1. Infracciones leves:
1.ª La presentación en ferias, exposiciones y demostraciones de productos no aptos para la puesta en el mercado o en servicio sin la correspondiente indicación de su no conformidad o imposibilidad de comercialización y/o puesta en servicio.
2.ª Dificultar la labor inspectora mediante cualquier acción u omisión que perturbe o retrase la misma.
3.ª El incumplimiento de los requisitos, obligaciones o prohibiciones establecidos en este Real Decreto que, en razón de los criterios contemplados en este artículo, merezcan la calificación de leves o no proceda su calificación como faltas graves o muy graves.
2. Infracciones graves:
1.ª La fabricación, la agrupación y la esterilización de los productos en territorio nacional sin la licencia sanitaria previa de funcionamiento de la instalación, así como la importación de productos sanitarios sin la licencia previa de establecimiento.
2.ª El incumplimiento del deber de contar con un responsable técnico de acuerdo con los artículos 4.1 y 14.4.
3.ª El uso de cualquier otro marcado que pueda inducir a confusión con el marcado CE o que menoscabe su significación.
4.ª El incumplimiento del deber de comunicación de la puesta en el mercado o puesta en servicio de los productos, así como de las modificaciones de dichas comunicaciones, establecido en el artículo 10.
5.ª El incumplimiento por el responsable técnico de las obligaciones que competen a su cargo.
6.ª La falta de mantenimiento a disposición de las autoridades competentes y por el tiempo señalado de la documentación a que se refiere el artículo 7.3.
7.ª No efectuar la comunicación establecida en el artículo 9 de esta disposición.
8.ª Distribuir y/o vender al público productos sanitarios para diagnóstico «in vitro» en establecimientos que no han sido debidamente comunicados de acuerdo con el artículo 13.3.
9.ª Distribuir, instalar, mantener y utilizar productos sanitarios para diagnóstico «in vitro» sin observar las condiciones exigidas, así como poner a la venta productos sanitarios alterados, en malas condiciones o cuando se haya sobrepasado el plazo de validez.
10. Impedir la actuación de los inspectores, debidamente acreditados, en los centros en que se fabriquen, almacenen, distribuyan o vendan productos sanitarios para diagnóstico «in vitro».
11. El incumplimiento del deber de comunicación previsto en el artículo 20 del presente Real Decreto.
12. El incumplimiento de los requisitos relativos a los textos de publicidad y promoción de los productos sanitarios, así como de las condiciones establecidas en el artículo 25.
13. El ofrecer, otorgar o prometer primas, ventajas pecuniarias o ventajas en especie a los profesionales sanitarios o a cualquier otro cualificado, relacionados con la utilización, prescripción o dispensación de los productos, así como a sus parientes y personas con las que convivan. También el solicitarlas o aceptarlas.
14. La utilización por un profesional de productos sanitarios para diagnóstico «in vitro» en condiciones y para usos distintos a los indicados por el fabricante, o por personal no cualificado o debidamente adiestrado, con riesgo para la salud y seguridad de las personas.
15. La puesta en servicio en España de productos sanitarios para diagnóstico «in vitro» sin haber proporcionado los datos contenidos en los apartados 5.3, 7 y 8 del anexo I, al menos en la lengua española oficial del Estado.
16. El uso indebido del marcado CE en productos no conformes o en los productos señalados en el apartado 5 del artículo 6.
17. La venta al público de productos de autodiagnóstico en establecimientos distintos de las oficinas de farmacia.
3. Infracciones muy graves:
1.ª La puesta en el mercado y/o en servicio de productos sanitarios para diagnóstico «in vitro» que comprometan la salud o la seguridad de los pacientes, usuarios o, en su caso, de terceros.
2.ª La puesta en el mercado y/o en servicio de los productos sanitarios para diagnóstico «in vitro» que no cumplan con los requisitos esenciales que les sean de aplicación, según el presente Real Decreto.
3.ª La puesta en el mercado y/o en servicio de productos sanitarios para diagnóstico «in vitro» que no hayan satisfecho los procedimientos de evaluación de la conformidad o que no hayan efectuado las declaraciones que, en su caso, les resulten de aplicación.
4.ª La instalación y/o mantenimiento inadecuado de productos sanitarios para diagnóstico «in vitro», de forma que comprometan la salud y/o la seguridad de los pacientes, usuarios o, en su caso, de terceros.
5.ª El incumplimiento del deber de ejecución de las medidas y acciones previstas en los artículos 14.3, 20, 21 y 22 del presente Real Decreto.
6.ª La incorrecta ejecución, por el organismo notificado, de las actuaciones que se le encomiendan en el artículo 18 del presente Real Decreto, así como continuar certificando una vez retirada la correspondiente designación.
7.ª La violación del principio de confidencialidad establecido en el artículo 24 del presente Real Decreto.
[Bloque 42: #a30]
1. Las acciones y omisiones constitutivas de infracciones, según lo previsto en el artículo 29 de este Real Decreto, serán objeto de las sanciones administrativas correspondientes, previa instrucción del oportuno procedimiento, sin perjuicio de las responsabilidades civiles, penales o de otro orden que puedan concurrir. El procedimiento para la imposición de sanciones se ajustará a los principios establecidos en el Título IX de la Ley de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común y al Real Decreto 1398/1993, de 4 de agosto, por el que se aprueba el Reglamento para el ejercicio de la potestad sancionadora.
2. Las infracciones a que se refiere el artículo 29 del presente Real Decreto serán sancionadas con multa de acuerdo con la graduación establecida en el artículo 36 de la Ley 14/1986, de 25 de abril, General de Sanidad, y los criterios expresados en el artículo 109 de la Ley 25/1990, de 20 de diciembre, del Medicamento.
3. No tendrá carácter de sanción la clausura o cierre de establecimientos o instalaciones que no cuenten con las previas autorizaciones o registro sanitarios preceptivos, o la suspensión de su funcionamiento hasta tanto se subsanen los defectos, o se cumplan los requisitos exigidos por razones de sanidad, higiene o seguridad.
[Bloque 43: #daprimera]
1. Tienen la condición de norma sanitaria básica, conforme a lo establecido en los artículos 149.1. 16.ª de la Constitución, 1.3, 2.2 y 8.12 y disposición adicional tercera de la Ley 25/1990, de 20 de diciembre, del Medicamento, y 40.5 y 6, 95, 100 y 110 de la Ley 14/1986, de 25 de abril, General de Sanidad, todos los artículos, disposiciones y anexos de este Real Decreto.
2. El artículo 15, en cuanto a la importación de productos, y el artículo 16 del presente Real Decreto se dictan al amparo del artículo 149.1.10.ª y 16.ª de la Constitución.
[Bloque 44: #dasegunda]
Lo previsto en el presente Real Decreto no afectará a los criterios y medidas en materia de financiación de los servicios de salud que se adopten por las autoridades sanitarias competentes.
[Bloque 45: #datercera]
A efectos del presente Real Decreto, la toma, la obtención y la utilización de tejidos, células y sustancias de origen humano se regirán, en materia deontológica, por los principios establecidos en el Convenio del Consejo de Europa para la protección de los derechos humanos y la dignidad del ser humano con respecto a las aplicaciones de la biología y la medicina y por la legislación vigente en dicha materia. En cuanto al diagnóstico, son primordiales tanto la protección de la confidencialidad de la información referente a la vida privada, como el principio de no discriminación basada en las características genéticas familiares de los hombres y de las mujeres.
[Bloque 46: #dacuarta]
La realización de los procedimientos de evaluación de la conformidad, señalados en el artículo 7 de esta disposición, será independiente de la certificación que, en materia de calidad industrial, se señala en la Ley 21/1992, de 16 de julio, de Industria.
[Bloque 47: #daquinta]
A los procedimientos contemplados en los artículos 4.1, 7.1, 10 y 18.1 les serán de aplicación las tasas correspondientes recogidas en el artículo 117 del Título X de la Ley 25/1990, de 20 de diciembre, del Medicamento.
[Bloque 48: #dasexta]
A los productos sanitarios para diagnóstico «in vitro» cuya conformidad haya sido determinada de acuerdo con el presente Real Decreto no les resulta de aplicación el Real Decreto 444/1994, de 11 de marzo, por el que se establecen los procedimientos de evaluación de la conformidad y los requisitos de protección relativos a la compatibilidad electromagnética de equipos, sistemas e instalaciones que transpone la Directiva 89/336/CEE.
[Bloque 49: #daseptima]
1. De acuerdo con el artículo 21 de la Directiva 98/79/CE, de 27 de octubre, se realizan las siguientes modificaciones al Real Decreto 414/1996, de 1 de marzo, por el que se regula los productos sanitarios:
A) Artículo 3. Definiciones:
a) El texto del párrafo c) se sustituye por:
«Producto sanitario para diagnóstico “in vitro”: Cualquier producto sanitario que consista en un reactivo, producto reactivo, calibrador, material de control, estuche de instrumental y materiales, instrumento, aparato, equipo o sistema, utilizado sólo o en asociación con otros, destinado por el fabricante a ser utilizado “in vitro” para el estudio de muestras procedentes del cuerpo humano, incluidas las donaciones de sangre y tejidos, sólo o principalmente con el fin de proporcionar información relativa a un estado fisiológico o patológico, o relativa a una anomalía congénita, o para determinar la seguridad y compatibilidad con receptores potenciales, o para supervisar las medidas terapéuticas.
Los recipientes para muestras se considerarán productos sanitarios para diagnóstico “in vitro”. Por “recipientes para muestras” se entiende los productos, tanto si en ellos se ha hecho el vacío como si no, destinados específicamente por el fabricante a la contención directa y a la conservación de muestras procedentes del cuerpo humano para un examen diagnóstico “in vitro”.
No se considerarán productos sanitarios para diagnóstico “in vitro” los artículos de uso general en laboratorio salvo cuando, por sus características, estén destinados específicamente por el fabricante a usarse en exámenes diagnósticos “in vitro”.»
b) El texto del párrafo i) se sustituye por:
«Puesta en servicio: la fase en la que un producto, que está listo para ser utilizado en el mercado comunitario con arreglo a su finalidad prevista, es puesto a disposición del usuario final por primera vez.»
c) Se añade el párrafo siguiente:
«k) Representante autorizado: cualquier persona física o jurídica establecida en la Unión Europea, designada expresamente por el fabricante, que actúe en su lugar y a la que puedan dirigirse las autoridades y organismos en la Unión Europea en lugar de al fabricante por lo que respecta a las obligaciones de éste con arreglo al presente Real Decreto.»
B) Artículo 5. Condiciones generales:
El texto del apartado 2 se sustituye por:
«Los productos sanitarios y los productos a medida sólo pueden comercializarse y/o ponerse en servicio si cumplen los requisitos establecidos en la presente disposición cuando hayan sido debidamente suministrados, estén correctamente instalados y mantenidos y se utilicen conforme a su finalidad prevista, no comprometiendo la seguridad ni la salud de los pacientes, de los usuarios ni, en su caso, de terceros.»
C) Se añaden los artículos siguientes:
«a) Artículo 15 bis. Base de datos europea.
Los datos reglamentarios derivados de la aplicación del artículo 14 del presente Real Decreto se trasladarán por la Dirección General de Farmacia y Productos Sanitarios, para su registro, a la base de datos europea descrita en el artículo 12 de la Directiva 98/79/CE, utilizando para su envío un formato normalizado.»
«b) Artículo 26 bis. Medidas particulares de seguimiento sanitario.
Cuando la Dirección General de Farmacia y Productos Sanitarios considere, respecto de un producto o un grupo de productos determinado, que para garantizar la protección de la salud de las personas, la seguridad o el cumplimiento de las normas de salud pública con arreglo a lo dispuesto en el artículo 30 del Tratado CE, debe prohibirse, restringirse o someterse a requisitos especiales la disponibilidad de dichos productos, podrá adoptar todas las medidas transitorias necesarias justificadas, de lo que informará a la Comisión y a los otros Estados miembros indicando los motivos de su decisión.
Por los mismos motivos podrá dictar disposiciones sobre condiciones de utilización de los productos o sobre medidas de seguimiento especial y hacer incluir las advertencias necesarias para evitar riesgos sanitarios en la utilización de los productos.»
D) Artículo 23. Actuaciones del organismo notificado:
a) El texto del apartado 5 se sustituye por:
«El organismo notificado informará a los demás organismos notificados y a las autoridades competentes de todos los certificados suspendidos o retirados, así como, previa petición, de los certificados expedidos o denegados. Además, previa petición, pondrá a su disposición toda la información adicional pertinente.»
b) El texto del apartado 6 se sustituye por:
«En caso de que un organismo notificado observe que el fabricante no cumple o que ha dejado de cumplir los requisitos pertinentes del presente Real Decreto, o que no se hubiera debido expedir un certificado, suspenderá o retirará el certificado expedido, teniendo presente el principio de proporcionalidad, a no ser que el fabricante garantice el cumplimiento de tales requisitos mediante la aplicación de medidas correctoras eficaces. En los casos de suspensión o retirada del certificado, o en los casos en que se someta a restricciones, o en los casos en que pudiera requerirse la intervención de la autoridad competente, el organismo notificado informará de los hechos a la Dirección General de Farmacia y Productos Sanitarios trasladando una copia de la decisión correspondiente. La Dirección General de Farmacia y Productos Sanitarios informará a los demás Estados miembros y a la Comisión. La Dirección General de Farmacia y Productos Sanitarios mantendrá informadas a las Comunidades Autónomas de los certificados suspendidos o retirados.»
c) Se añade un nuevo apartado 6 bis:
«Previa petición, el organismo notificado facilitará toda la información y documentación pertinentes, incluidos los documentos presupuestarios, necesarios para que el Ministerio de Sanidad y Consumo verifique el cumplimiento de los requisitos del anexo XI.»
E) Disposición transitoria primera. Vigencia de la legislación anterior sobre productos sanitarios.
El texto de la disposición transitoria primera se sustituye por:
«1. Hasta el 14 de junio de 1998 se permitirá:
a) La comercialización de los productos sanitarios que se ajusten a la reglamentación vigente en España el 31 de diciembre de 1994.
b) La libre comercialización de aquellos productos sanitarios que el 31 de diciembre de 1994 no estuvieran sometidos a ninguna legislación nacional.
2. Hasta el 30 de junio de 2001 se permitirá la puesta en servicio de los productos citados en el párrafo anterior.»
F) Anexo II.
Se suprime el apartado 6.2.
G) Anexo III.
Se suprime el apartado 7.1.
H) Anexo V.
Se suprime el apartado 5.2.
I) Anexo VI.
Se suprime el apartado 5.2.
J) Anexo XI.
Se añade el siguiente párrafo tras la segunda frase del apartado 3:
«Ello presupone la disponibilidad de personal científico suficiente en el seno de la organización, que posea la experiencia adecuada y los conocimientos necesarios para evaluar, desde un punto de vista médico, la funcionalidad y las prestaciones de los productos que le hayan sido notificados, en relación con los requisitos del presente Real Decreto y, en particular, con los requisitos del anexo I.»
2. Además, se modifica el mismo Real Decreto 414/1996, de 1 de marzo, por el que se regulan los productos sanitarios, de la forma siguiente:
A) El artículo 5 se modifica como sigue:
a) En el apartado 1 se incluyen los siguientes párrafos entre el primero y el segundo.
«Para la obtención de estas autorizaciones, las empresas que desarrollen tales actividades las solicitarán de la Dirección General de Farmacia y Productos Sanitarios, la cual estudiará la documentación presentada y notificará su resolución en el plazo de tres meses a contar desde la fecha en que la solicitud y la documentación que la acompaña hayan tenido entrada en cualquiera de los registros del Ministerio de Sanidad y Consumo.
La Dirección General de Farmacia y Productos Sanitarios solicitará a las áreas de sanidad de las Delegaciones del Gobierno informe sobre las condiciones en que las empresas desarrollan las actividades relacionadas en este apartado, ordenando a estos efectos las inspecciones de las instalaciones de dichas empresas que resulten necesarias. Dado que el informe solicitado es determinante del contenido de la resolución que deba adoptarse, suspenderá el transcurso del plazo máximo de tres meses de duración del procedimiento, por el tiempo que medie entre la petición del informe, se comunicará a la empresa interesada, y su recepción, que igualmente le será comunicada, y sin que el plazo de suspensión pueda exceder en ningún caso de tres meses, todo ello de conformidad con lo dispuesto en el artículo 42.5.c) de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común, en la redacción dada al mismo por la Ley 4/1999, de 13 de enero, de modificación de aquélla.
El informe citado en el párrafo anterior no se solicitará cuando las empresas desarrollen las actividades de fabricación, agrupación, esterilización y almacenamiento en instalaciones establecidas fuera del territorio español.»
b) En el apartado 5 se añade la siguiente frase:
«Los productos deberán ser mantenidos adecuadamente de forma que se garantice que, durante su período de utilización, conservan la seguridad y prestaciones previstas por su fabricante.»
B) El apartado 4 del artículo 7 se sustituye por:
«Queda prohibido poner marcas o inscripciones que puedan inducir a terceros a interpretaciones erróneas en relación con el significado o los gráficos del marcado CE o que menoscaben la significación del mismo. Podrá colocarse sobre el producto, el envase o el prospecto de instrucciones que acompaña al producto cualquier otra marca, siempre que no se reduzca por ello la visibilidad y legibilidad del marcado CE.»
C) En el apartado 1.4 del artículo 8 se añade la siguiente frase:
«La Dirección General de Farmacia y Productos Sanitarios evaluará, en su caso, la documentación señalada en el citado anexo, a efectos de establecer la conformidad de los productos después de que éstos hayan sido comercializados y/o puestos en servicio.»
D) El texto del primer párrafo del artículo 14 se sustituye por:
«Todo fabricante establecido en España que comercialice productos de las clases I y IIa, así como productos a medida, será incluido en el registro de responsables de la comercialización que funcionará en la Dirección General de Farmacia y Productos Sanitarios, donde constará su denominación social y la relación de los productos que comercialice o ponga en servicio, especificando tipo de producto, fabricante, clase y nombre comercial. Esta obligación se hará extensiva al representante autorizado en la Unión Europea, responsable de la comercialización o importador de productos fabricados fuera de la Unión Europea cuando se encuentren establecidos en España.»
E) El artículo 16 se modifica como sigue:
a) El texto del apartado 2 se sustituye por:
«La distribución y venta al público estarán sometidas a la vigilancia e inspección de las autoridades sanitarias de la Comunidad Autónoma correspondiente, quien podrá establecer el procedimiento exigido para la autorización de tales actividades.»
b) El texto del último párrafo del apartado 3 se sustituye por:
«Quedan exceptuadas de realizar tal declaración de actividad las oficinas de farmacia, salvo que realicen las actividades contempladas en el apartado 1 del artículo 18.»
F) El artículo 19 se modifica como sigue:
a) En el apartado 1 se añade un nuevo párrafo:
«En el caso de que las investigaciones clínicas se realicen con productos que ostenten el marcado CE, no les resultarán de aplicación los apartados 3, 4, 5, 6, 7 y 8 del presente artículo, salvo que dichas investigaciones tengan por objeto utilizar los productos en una indicación diferente de la contemplada en el procedimiento pertinente de evaluación de la conformidad. Seguirán siendo aplicables las disposiciones correspondientes del anexo X, con excepción del apartado 2.4.»
b) El apartado 6 queda redactado como sigue:
«No obstante lo dispuesto en el apartado 4, en el caso de productos distintos a los de la clase III o los implantable o invasivos a largo plazo de las clases IIa o IIb, podrán iniciarse las investigaciones si transcurridos quince días desde el acuse de recibo de la Dirección General de Farmacia y Productos Sanitarios, ésta no se hubiere pronunciado, y siempre que ,se cuente con el informe favorable del Comité Ético de Investigación Clínica correspondiente.»
G) La segunda frase del apartado 7 del artículo 23 se sustituye por:
«Contra dicha resolución podrá interponerse directamente recurso contencioso-administrativo o, potestativamente, recurso de reposición en el plazo de un mes, conforme a lo previsto en los artículos 116 y 117 de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común.»
H) El texto del apartado 2 del artículo 24 se sustituye por:
«La Dirección General de Farmacia y Productos Sanitarios, por sí misma o a través de los servicios de inspección de la Administración General del Estado habilitados a estos fines, podrá ejercer actividades de inspección y control respecto de los productos contemplados en el capítulo VI y de los establecimientos en que se fabriquen, importen o exporten, siempre que estén situados en territorio nacional. Asimismo, podrá realizar, sin perjuicio de las competencias de otras Administraciones Sanitarias, la inspección y seguimiento de las investigaciones clínicas de aquellos productos que no ostenten el marcado CE.»
I) El artículo 25 se modifica como sigue:
a) El texto del apartado 3 se sustituye por:
«Tras haber procedido a su evaluación, a ser posible conjuntamente con el fabricante, la Dirección General de Farmacia y Productos Sanitarios informará a la Comisión Europea y a los demás Estados miembros de los hechos respecto de los cuales se hayan tomado o se estudie la posibilidad de tomar medidas pertinentes.»
b) El texto del primer guión del apartado 4 se sustituye por:
«Implantes cardíacos e implantes vasculares del sistema circulatorio central.»
c) Se suprimen el tercer y quinto guiones del apartado 4.
J) El artículo 26 se modifica como sigue:
a) El texto del primer párrafo del apartado 1 se sustituye por:
«Si existieran indicios razonables sobre la conformidad de un producto, las autoridades sanitarias podrán exigir, motivadamente, del fabricante, importador o responsable de la comercialización en la Unión Europea la presentación en la lengua española oficial del Estado de cuanta información se considere necesaria para juzgar sobre dicha conformidad.
La negativa a facilitar la documentación señalada podrá considerarse como presunción de no conformidad.»
b) El texto del primer párrafo del apartado 3 se sustituye por:
«Como garantía de la salud y seguridad de las personas, las autoridades sanitarias competentes, cuando consideren que un producto correctamente puesto en servicio, instalado, mantenido y utilizado con arreglo a su finalidad prevista, puede comprometer la salud y/o la seguridad de los pacientes, usuarios o la de terceras personas procederá a adoptar las medidas adecuadas previstas en el capítulo V del Título I de la Ley 14/1986, de 25 de abril, General de Sanidad, y en el capítulo I del Título IX y artículo 110 de la Ley 25/1990, de 20 de diciembre, del Medicamento.»
c) El texto del apartado 6 se sustituye por:
«Cuando un producto no conforme ostente el marcado CE, las autoridades sanitarias competentes adoptarán las medidas apropiadas contra quien ha colocado el marcado e informará de ellas a la Dirección General de Farmacia y Productos Sanitarios. Dicho centro directivo actuará en consecuencia y lo comunicará a la Comisión Europea y a las autoridades competentes de los demás Estados miembros.»
d) El texto del apartado 10 se sustituye por:
«Cuando la comprobación de la conformidad de un producto requiera la realización de evaluaciones o ensayos sobre el producto o su documentación técnica, los gastos derivados de la comprobación serán satisfechos por el fabricante o su responsable, quien facilitará igualmente las muestras necesarias para realizar tal comprobación.»
e) El texto del apartado 11 se sustituye por:
«Cuando por aplicación del presente Real Decreto se rechace o restrinja la puesta en el mercado y/o la puesta en servicio de un producto, así como cuando se realice la retirada de un producto del mercado, el interesado podrá manifestar su disconformidad ante el órgano superior jerárquico del que dictó la resolución, mediante el correspondiente recurso de alzada en el plazo de un mes, conforme a lo establecido en los artículos 114 y 115 de la Ley de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común.»
K) En el artículo 27 se suprime el segundo párrafo.
L) El texto del artículo 28 se sustituye por:
«Las Administraciones Públicas, sin perjuicio de las disposiciones existentes en materia de secreto profesional, velarán porque todas las partes a las que concierne la aplicación del presente Real Decreto mantengan la confidencialidad de cualquier información obtenida en el ejercicio de su función. Ello no afectará a las obligaciones de las autoridades competentes y de los organismos notificados con respecto a la información recíproca, ni a la difusión de advertencias, ni a las obligaciones de información que incumban a las personas afectadas, tanto ante las autoridades, como ante los órganos jurisdiccionales.»
M) El artículo 29 se modifica como sigue:
a) La segunda frase del apartado 4 se sustituye por:
«Los mensajes publicitarios que se inserten en cualquiera de los medios de comunicación general serán objeto de autorización previa por las autoridades sanitarias de las Comunidades Autónomas.»
b) El texto del apartado 6 se sustituye por:
«En la publicidad de los productos dirigida al público y efectuada por particulares se prohíbe cualquier mención que haga referencia a una autoridad sanitaria o a recomendaciones que hayan formulado científicos, profesionales de la salud u otras personas que puedan, debido a su notoriedad, incitar a su utilización.»
N) En el artículo 32 la última frase se sustituye por:
«Tales demostraciones no podrán nunca implicar la utilización de dichos productos en pacientes.»
Ñ) El artículo 33 se modifica como sigue:
a) En el apartado 2, Infracciones graves, se añaden dos nuevos apartados:
«16. La puesta en servicio en España de productos sanitarios que no incluyan los datos contenidos en los apartados 2.7, 5.4 y 7 del anexo I al menos en la lengua española oficial del Estado.
17. El uso indebido del marcado CE en productos no conformes o en los productos contemplados en el apartado 5 del artículo 7.»
b) El texto del apartado 2.9 se sustituye por:
«Distribuir, instalar, mantener y utilizar productos sanitarios sin observar las condiciones exigidas, así como poner a la venta productos sanitarios alterados, en malas condiciones o cuando se haya sobrepasado el plazo de validez.»
c) En el apartado 3, Infracciones muy graves, se añaden dos nuevos apartados:
«8.ª La comercialización y/o puesta en servicio de productos que comprometan la salud o la seguridad de los pacientes, usuarios o, en su caso, de terceros.
9.ª La instalación y/o mantenimiento inadecuado de productos sanitarios, de forma que comprometan la salud o la seguridad de los pacientes, usuarios o, en su caso, de terceros.»
O) El texto de la disposición adicional cuarta, aplicación de tasas, se sustituye por:
«A los procedimientos contemplados en los artículos 8.1, 12 y 19.3 les serán de aplicación las tasas correspondientes recogidas en el artículo 117 del título X de la Ley 25/1990, de 20 de diciembre, del Medicamento.»
P) En la disposición adicional quinta, Extensión, se sustituye el segundo guión del primer párrafo por:
«Aparatos e instrumental utilizados en el maquillaje permanente, semipermanente o en el tatuaje de la piel mediante técnicas invasivas.»
Q) Disposición transitoria segunda, adaptación de las obligaciones sobre comunicación de la comercialización.
El texto del segundo párrafo de la disposición transitoria segunda se sustituye por:
«A las comunicaciones de los productos a que se hace referencia en el apartado 1.a) de la disposición transitoria primera no les será de aplicación la tasa establecida en la disposición adicional cuarta.»
R) Disposición derogatoria primera.
El texto del último párrafo del apartado 2 de la disposición derogatoria primera se modifica como sigue:
«Todas ellas en cuando se opongan al presente Real Decreto y en el ámbito de los productos incluidos en su campo de aplicación.»
[Bloque 50: #daoctava]
El Real Decreto 634/1993, de 3 de mayo, sobre productos sanitarios implantables activos, se modifica de la forma siguiente:
1. En el artículo 7, el texto del apartado 4 se sustituye por:
«4. Productos sanitarios implantables activos destinados a investigaciones clínicas:
a) La realización de investigaciones clínicas con productos sanitarios implantables activos se ajustará a lo dispuesto en el anexo VII y en los Títulos I y III del Real Decreto 561/1993, de 16 de abril, por el que se establecen los requisitos para la realización de ensayos clínicos con medicamentos, tal y como se recoge en la disposición adicional primera del citado Real Decreto.
b) En el caso de que las investigaciones clínicas se realicen con productos que ostenten el marcado CE, no les resultarán de aplicación los apartados 5, 6, 7, 8 y 9 del presente artículo, salvo que dichas investigaciones tengan por objeto utilizar los productos en una indicación diferente de la contemplada en el procedimiento pertinente de evaluación de la conformidad. Seguirán siendo aplicables las disposiciones correspondientes del anexo VII, con excepción del apartado 2.4.
c) Los productos destinados a investigación clínica sólo podrán ser puestos a disposición de los facultativos si esta investigación cuenta con el dictamen favorable del Comité Ético contemplado en el artículo 64 de la Ley 25/1990, de 20 de diciembre, del Medicamento, y sus disposiciones de desarrollo. Esta previsión se extenderá a las modificaciones de investigaciones clínicas en curso que puedan suponer un aumento de riesgo para los sujetos participantes en el ensayo.»
2. El artículo 19 se modifica como sigue:
a) El texto del apartado 3 se sustituye por:
«Tras haber procedido a su evaluación, a ser posible conjuntamente con el fabricante, la Dirección General de Farmacia y Productos Sanitarios informará a la Comisión Europea y a los demás Estados miembros de los hechos respecto de los cuales se hayan tomado o se estudie la posibilidad de tomar medidas pertinentes.»
b) Se añade un nuevo apartado:
«4. Los implantes activos que se distribuyan en España deberán ir acompañados de una tarjeta de implantación. Esta tarjeta no se exigirá al «software» necesario para el buen funcionamiento de los productos.
La tarjeta de implantación, en triplicado ejemplar, incluirá al menos el nombre y modelo del producto, el número de lote o número de serie, el nombre y dirección del fabricante, el nombre del centro sanitario donde se realizó la implantación y fecha de la misma, así como la identificación del paciente (documento nacional de identidad, número de pasaporte), y será cumplimentada por el hospital tras la implantación.
Uno de los ejemplares permanecerá archivado en la historia clínica del paciente, otro será facilitado al mismo y otro será remitido a la empresa suministradora. En el caso en que se haya dispuesto de un registro nacional de implantes, este ejemplar o copia del mismo será remitido al registro nacional por la empresa suministradora.»
3. El texto del artículo 21 se sustituye por:
«Las autoridades sanitarias, sin perjuicio de las disposiciones existentes en materia de secreto profesional, velarán porque todas las partes a las que concierne la aplicación del presente Real Decreto mantengan la confidencialidad de cualquier información obtenida en el ejercicio de su función. Ello no afectará a las obligaciones de las autoridades competentes y de los organismos notificados con respecto a la información recíproca y a la difusión de advertencias, ni a las obligaciones de información que incumban a las personas afectadas, tanto ante la autoridad sanitaria como ante los órganos jurisdiccionales.»
4. Se incluyen los siguientes artículos:
«Artículo 22 bis. Incentivos.
En el marco de la promoción de los productos sanitarios queda prohibido otorgar, ofrecer o prometer primas, ventajas pecuniarias o ventajas en especie a los profesionales sanitarios o cualquier otro cualificado relacionado con la utilización, prescripción o dispensación de los productos así como a sus parientes o personas con las que convivan.
Las personas relacionadas en el apartado anterior no podrán solicitar o aceptar ninguno de los incentivos prohibidos.
Artículo 22 ter. Patrocinio de reuniones científicas.
1. Las disposiciones del artículo anterior no supondrán un obstáculo para la hospitalidad ofrecida, directa o indirectamente, en el marco de manifestaciones de carácter exclusivamente profesional y científico. Dicha hospitalidad deberá ser siempre moderada en su nivel y subordinada al objetivo principal de la reunión y no podrá ser extensible a personas que no sean profesionales de la salud.
2. Los premios, becas, contribuciones y subvenciones a reuniones, congresos, viajes de estudio y actos similares donados por personas relacionadas con la fabricación, elaboración, distribución y dispensación de productos sanitarios se aplicarán exclusivamente a actividades de índole científica cuando sus destinatarios sean facultativos en ejercicio clínico o las entidades en que se asocian.
En las publicaciones de trabajos y ponencias de reuniones, congresos y actos similares se harán constar los fondos obtenidos para su realización y fuente de financiación. La misma obligación alcanzará al medio de comunicación por cuya vía se hagan públicos y que obtenga fondos por o para su publicación.
3. Lo previsto en este artículo no afectará a los cursos de entrenamiento, necesarios para la correcta utilización de los productos, que sean facilitados a los profesionales por los fabricantes o distribuidores de los mismos.»
5. En el artículo 23 apartado 2, infracciones graves, se añade un nuevo apartado:
«11.º La puesta en servicio en España de productos sanitarios implantables activos que no incluyan los datos contenidos en los apartados 3 y 4 del anexo I al menos en la lengua española oficial del Estado.»
6. En el anexo VII apartado 2, se añade un nuevo apartado 4:
«4. Muestras.
a) Las muestras de productos sanitarios implantables activos para ser utilizadas en ensayos clínicos serán proporcionadas gratuitamente por el promotor. En determinadas situaciones se podrán autorizar otras formas de suministro.
Todas las muestras sobrantes serán devueltas al promotor, una vez finalizado el ensayo.
b) Las etiquetas y manual de instrucciones de las muestras para utilización en ensayos clínicos deberán estar redactadas en la lengua española oficial del Estado y permitirán en cualquier momento la perfecta identificación de los productos.
c) El promotor conservará en el archivo principal del ensayo los protocolos de fabricación y control de los lotes de productos fabricados para el ensayo clínico.
d) Los Servicios de Farmacia del Hospital supervisarán el suministro de las muestras que vayan a ser utilizadas en el ensayo clínico.»
7. El texto de la disposición adicional cuarta. Aplicación de tasa, se sustituye por:
«A los procedimientos contemplados en los artículos 6, 7.5 y 9 del presente Real Decreto les serán de aplicación las tasas recogidas en el artículo 117 del Título X de la Ley 25/1990, de 20 de diciembre, del Medicamento.»
Redactado el apartado 1.4.a) y b) conforme a la corrección de errores publicada en BOE núm. 306, de 22 de diciembre de 2000. Ref. BOE-A-2000-23507.
[Bloque 51: #danovena]
El Ministerio de Sanidad y Consumo anunciará en el «Boletín Oficial del Estado» las listas de los organismos notificados en el marco de la Directiva 98/79/CE, publicadas en el Diario Oficial de las Comunidades Europeas (DOCE).
[Bloque 52: #dadecima]
A efectos del presente Real Decreto será de aplicación lo establecido en el Acuerdo sobre el Espacio Económico Europeo.
[Bloque 53: #daundecima]
De acuerdo con los artículos 31.3 de la Ley 16/2003, de Cohesión y Calidad del Sistema Nacional de Salud y 10.3 del Real Decreto 1087/2003, de 29 de agosto, por el que se establece la estructura orgánica del Ministerio de Sanidad y Consumo, las referencias realizadas en este real decreto a la Dirección General de Farmacia y Productos Sanitarios se entenderá efectuadas a la Agencia Española de Medicamentos y Productos Sanitarios.
Se añade por el art. 3.4 del Real Decreto 1143/2007, de 31 de agosto. Ref. BOE-A-2007-15890.
Texto añadido, publicado el 01/09/2007, en vigor a partir del 01/09/2007.
[Bloque 54: #dtprimera]
1. Hasta el 7 de diciembre de 2003 se permitirá:
a) La comercialización de los productos sanitarios de diagnóstico «in vitro» que se ajusten a las reglamentaciones vigentes en España a 7 de diciembre de 1998.
b) La libre comercialización de aquellos productos sanitarios de diagnóstico «in vitro» que el 7 de diciembre de 1998 no estuvieran sometidos a ninguna legislación nacional.
2. Hasta el 7 de diciembre de 2005 se permitirá la puesta en servicio de los productos citados en el párrafo anterior.
[Bloque 55: #dtsegunda]
Las empresas responsables de productos que contasen con autorización sanitaria de comercialización y se estuvieran comercializando en España a la entrada en vigor del presente Real Decreto, deberán cumplir lo establecido en el artículo 10 en el momento de iniciar su comercialización con marcado CE.
A las comunicaciones de los productos a que se hace referencia en el apartado 1, a) de la disposición transitoria primera no les será de aplicación la tasa establecida en la disposición adicional quinta.
[Bloque 56: #dttercera]
Hasta el 7 de diciembre de 2003 se permitirá la fabricación e importación de los productos sanitarios para diagnóstico «in vitro» en las condiciones establecidas por las reglamentaciones vigentes en España el 7 de diciembre de 1998. Igualmente se permitirá hasta esta misma fecha la libre fabricación e importación de aquellos productos sanitarios que el 7 de diciembre de 1998 no tuvieran fijadas reglamentariamente condiciones para efectuar tales actividades.
[Bloque 57: #dtcuarta]
Se fija el 1 de enero de 2002 como fecha límite para que los establecimientos de distribución efectúen la comunicación señalada en el apartado 1 del artículo 14.
[Bloque 58: #ddunica]
1. Queda derogado el artículo 1.b) del Real Decreto 908/1978, de 14 de abril, sobre control sanitario y homologación de material e instrumental médico, terapéutico y correctivo.
2. A partir del 7 de diciembre de 2003, quedan derogadas las siguientes disposiciones:
a) Resolución de 20 de marzo de 1987, de la Subsecretaría, por la que se establece el procedimiento y la documentación necesaria para obtener la autorización de los reactivos para realizar pruebas de detección de marcadores de infección por virus humanos de la familia «retroviridae», entre ellas las pruebas de detección de anticuerpos frente a los virus asociados al Síndrome de Inmunodeficiencia Adquirida (SIDA) y las de detección de antígenos correspondientes a los mismos.
b) Resolución de 11 de septiembre de 1989, de la Subsecretaría, por la que se regula la realización de procesos de investigación controlada de reactivos para la detección de marcadores de infección por virus humanos de la familia «retroviridae», entre ellos los asociados al Síndrome de Inmunodeficiencia Adquirida (SIDA).
c) Orden del Ministro de Sanidad y Consumo de 13 de junio de 1983 por la que se regula el material e instrumental médico-quirúrgico estéril para utilizar una sola vez.
Todas ellas en tanto se opongan al presente Real Decreto y en el ámbito de los productos incluidos en su campo de aplicación.
[Bloque 59: #dfprimera]
Se faculta a la Ministra de Sanidad y Consumo para dictar cuantas disposiciones sean necesarias para la aplicación y desarrollo del presente Real Decreto.
[Bloque 60: #dfsegunda]
El presente Real Decreto entrará en vigor el día siguiente al de su publicación en el «Boletín Oficial del Estado».
[Bloque 61: #firma]
Dado en Madrid a 29 de septiembre de 2000.
JUAN CARLOS R.
La Ministra de Sanidad y Consumo,
CELIA VILLALOBOS TALERO
[Bloque 62: #ani]
1. Propiedades físicas y químicas.
1.1 Los productos deberán diseñarse y fabricarse de forma que presenten las características y prestaciones mencionadas en el artículo 5 «Requisitos generales» del presente Real Decreto. Se tendrá especialmente en cuenta la posibilidad de deterioro del funcionamiento analítico debido a la incompatibilidad entre los materiales utilizados y las muestras (tales como tejidos biológicos, células, fluidos corporales y microorganismos) que vayan a utilizarse con ese producto, habida cuenta de su finalidad prevista.
1.2 Los productos deberán diseñarse, fabricarse y envasarse de forma que se reduzcan en lo posible los riesgos derivados de posibles fugas, contaminantes y residuos para las personas que se encargan del transporte, almacenamiento y uso de los productos, habida cuenta de su finalidad prevista.
2 Infección y contaminación microbiana.
2.1 Los productos y los procedimientos de fabricación deberán estar concebidos de forma que se elimine o reduzca al mínimo el riesgo de infección del usuario u otras personas. El diseño deberá permitir una manipulación fácil y, cuando sea necesario, reducir al mínimo la contaminación y las fugas procedentes del producto durante el uso, así como, en el caso de los recipientes para muestras, el riesgo de contaminación de la muestra. Los procesos de fabricación deberán ser apropiados para estos fines.
2.2 Cuando un producto contenga sustancias biológicas, se reducirán al mínimo los riesgos de infección seleccionando donantes y sustancias adecuados y aplicando procedimientos de inactivación, conservación, ensayo y control apropiados y validados.
2.3 Los productos etiquetados como «ESTÉRILES» o de los que se indique que tienen un estado microbiológico especial deberán diseñarse, fabricarse y acondicionarse en un envase apropiado, de acuerdo con procedimientos adecuados que garanticen que los productos se mantengan en el estado microbiológico adecuado indicado en la etiqueta cuando sean puestos en el mercado y en las condiciones de almacenamiento y transporte establecidas por el fabricante, hasta la apertura o deterioro del envase protector.
2.4 Los productos etiquetados como «ESTÉRILES» o de los que se indique que tienen un estado microbiológico especial deberán haberse elaborado mediante un método validado apropiado.
2.5 Los sistemas de envasado de productos distintos de los mencionados en el apartado 2.3 deberán mantener el producto, sin deterioro, en el nivel de limpieza que haya indicado el fabricante y, en caso de que haya que esterilizarlo antes de su uso, reducir al mínimo el riesgo de contaminación microbiana.
Deberán tomarse medidas para reducir tanto como se pueda la contaminación microbiana en la selección y manipulación de las materias primas, la fabricación, el almacenamiento y la distribución en caso de que dicha contaminación pudiera alterar el funcionamiento del producto.
2.6 Los productos que deban ser esterilizados deberán fabricarse en condiciones adecuadamente controladas (por ejemplo, las relativas al medio ambiente).
2.7 Los sistemas de envasado destinados a los productos no estériles deberán ser tales que conserven el producto sin deterioro en el estado de limpieza previsto y, si el producto ha de esterilizarse antes de su utilización, deberán minimizar el riesgo de contaminación microbiana; el sistema de envasado deberá ser adecuado, en función del método de esterilización indicado por el fabricante.
3. Propiedades relativas a la fabricación y al medio ambiente.
3.1 Si el producto está destinado a utilizarse en asociación con otros productos o equipos, todo el conjunto, incluido el sistema de conexión, deberá ser seguro y no alterar las prestaciones indicadas de los productos. Toda restricción de su uso irá indicada en la etiqueta o en las instrucciones de utilización.
3.2 Los productos deberán diseñarse y fabricarse de manera que se reduzcan al mínimo los riesgos relacionados con su uso en conjunción con los materiales, sustancias y gases con los que puedan entrar en contacto durante su uso normal.
3.3 Los productos deberán diseñarse y fabricarse de forma que se eliminen o se reduzcan al mínimo:
— Los riesgos de lesiones derivados de sus características físicas (en particular los aspectos de volumen por presión, dimensionales y, en su caso, ergonómicos),
— Los riesgos relacionados con influencias externas razonablemente previsibles, como campos magnéticos, influencias eléctricas externas, descargas electrostáticas, presión, humedad, temperatura o variaciones de la presión o la aceleración, o la entrada accidental de sustancias en el producto.
Los productos deberán diseñarse y fabricarse de forma que presenten un nivel adecuado de inmunidad intrínseca frente a perturbaciones electromagnéticas que les permita funcionar de acuerdo con el fin para el que han sido previstos.
3.4 Los productos deberán diseñarse y fabricarse de forma que se reduzcan al mínimo los riesgos de incendio o explosión durante el uso normal y en condiciones de primer defecto. Se prestará atención especial a los productos cuya finalidad prevista incluya la exposición a sustancias inflamables o que puedan dar lugar a combustión, o el uso conjunto con dichas sustancias.
3.5 Los productos deberán diseñarse y fabricarse de forma que se facilite la gestión de la eliminación segura de residuos.
3.6 La escala de medición, control o visualización (incluidos el cambio de color y otros indicadores visuales) se diseñará y fabricará de acuerdo con principios ergonómicos, teniendo en cuenta la finalidad prevista del producto.
4 Productos que sean instrumentos o aparatos con función de medición.
4.1 Los productos que sean instrumentos o aparatos con función primaria de medición analítica deberán diseñarse y fabricarse de forma que proporcionen una estabilidad y exactitud de la medición adecuadas dentro de unos márgenes convenientes de exactitud, teniendo en cuenta la finalidad prevista del producto y los métodos y materiales de medición de referencia disponibles y apropiados. El fabricante especificará los márgenes de exactitud.
4.2 Cuando los valores se expresen de forma numérica, se darán en unidades de medida legales acordes con las disposiciones de la Directiva 80/181/CEE, del Consejo, de 20 de diciembre de 1979, relativa a la aproximación de las legislaciones de los Estados miembros sobre las unidades de medida.
5 Protección contra las radiaciones.
5.1 Los productos deberán diseñarse, fabricarse y envasarse de forma que la exposición de los usuarios y otras personas a emisiones de radiación se reduzca al mínimo.
5.2 Cuando los productos estén destinados a emitir radiaciones potencialmente peligrosas, visibles o invisibles, en la medida de lo posible,
— Deberán diseñarse y fabricarse de forma que sus características y la cantidad de las radiaciones emitidas sean controlables o regulables,
— Deberán dotarse de indicadores visuales o de alarmas sonoras de tales emisiones.
5.3 Las instrucciones de utilización de los productos que emitan radiaciones darán información detallada sobre la naturaleza de la radiación emitida, medios de protección del usuario y formas de evitar manipulaciones incorrectas y de eliminar los riesgos derivados de la instalación.
6 Requisitos para productos sanitarios conectados a una fuente de energía o equipados con ella.
6.1 Los productos que lleven sistemas electrónicos programables, incluidos los programas de ordenador, deberán diseñarse de forma que se garantice la repetibilidad, fiabilidad y funcionamiento de estos sistemas de acuerdo con la finalidad prevista.
6.2 Los productos deberán diseñarse y fabricarse de forma que se reduzca al mínimo el riesgo de creación de perturbaciones electromagnéticas que puedan dañar el funcionamiento de otros productos o equipos en el entorno usual.
6.3 Los productos se diseñarán y fabricarán de forma que se evite en la medida de lo posible el riesgo de descargas eléctricas accidentales durante el uso normal y en condiciones de primer defecto, siempre que los productos estén instalados y mantenidos correctamente.
6.4 Protección contra riesgos mecánicos y térmicos.
6.4.1 Los productos deberán diseñarse y fabricarse de forma que el usuario esté protegido contra riesgos mecánicos. Los productos deberán tener la suficiente estabilidad en las condiciones previstas de funcionamiento. Deberán ser capaces de resistir a los esfuerzos inherentes al ámbito de funcionamiento previsto, y mantener dicha resistencia durante el período de duración previsto de los productos, siempre que se respeten los requisitos de mantenimiento y de inspección indicados por el fabricante.
Cuando existan riesgos debidos a la presencia de elementos móviles, riesgos debidos a rotura o desprendimiento o a la fuga de sustancias, deberán incorporarse medidas de protección adecuadas.
Cualquier resguardo u otro medio incluido en el producto para asegurar protección, en particular contra los elementos móviles, deberá ser seguro y no obstaculizar el acceso para la utilización normal del producto, ni restringir el mantenimiento normal del producto indicado por el fabricante.
6.4.2 Los productos deberán diseñarse y fabricarse de forma que se reduzcan al menor nivel posible los riesgos derivados de las vibraciones generadas por los productos, teniendo en cuenta el progreso técnico y los medios de que se disponga para limitar las vibraciones, en particular en su origen, salvo si las vibraciones forman parte de las prestaciones especificadas.
6.4.3 Los productos deberán diseñarse y fabricarse de forma que se reduzcan al menor nivel posible los riesgos derivados del ruido emitido, teniendo en cuenta el progreso técnico y los medios de que se disponga para reducir el ruido, en particular en su origen, salvo si el ruido emitido forma parte de las prestaciones especificadas.
6.4.4 Los terminales y dispositivos de conexión a la fuente de electricidad, gas o energía hidráulica o neumática que el usuario tenga que manipular deberán diseñarse y fabricarse de forma que se reduzcan al mínimo todos los riesgos posibles.
6.4.5 Las partes accesibles de los productos (con exclusión de las partes o zonas destinadas a suministrar calor o a alcanzar determinadas temperaturas) y las partes circundantes no deberán alcanzar temperaturas potencialmente peligrosas en circunstancias de uso normal.
7. Requisitos para productos de autodiagnóstico.
Los productos para autodiagnóstico deberán diseñarse y fabricarse de forma que sus prestaciones se ajusten a la finalidad prevista, habida cuenta de la capacidad y los medios de que dispone el usuario y la influencia que pueden tener las variaciones razonablemente previsibles de la técnica y del entorno del usuario. La información y las instrucciones dadas por el fabricante serán de fácil comprensión y aplicación por el usuario.
7.1 Los productos para autodiagnóstico deberán diseñarse y fabricarse de forma que:
— Esté garantizada la fácil utilización del producto, en todas las fases de su manipulación, por parte del usuario no profesional a que esté destinado, y
— Se reduzca todo lo posible el riesgo de error por parte del usuario en la manipulación del producto y la interpretación de los resultados.
7.2 Los productos para autodiagnóstico deberán incluir, cuando sea razonablemente posible, un control por el usuario, es decir, un procedimiento que permita al usuario verificar que, en el momento en que vaya a utilizarlo, el producto funcionará como es debido.
8. Información facilitada por el fabricante.
8.1 Cada producto deberá ir acompañado de la información necesaria para utilizarlo de forma correcta y segura, en función de la preparación y los conocimientos de los usuarios potenciales, y para identificar al fabricante.
Esta información estará constituida por los datos de la etiqueta y las instrucciones de utilización.
En la medida en que sea factible y adecuado, la información necesaria para utilizar el producto de forma correcta y segura deberá figurar en el propio producto y/o, cuando proceda, en el envase de venta. Si no es posible etiquetar completamente cada unidad, la información figurará en el envase o en las instrucciones de utilización proporcionadas con uno o más productos.
Las instrucciones de utilización deberán acompañar el envase de uno o más productos, o estar incluidas en dicho envase.
En casos debidamente justificados y con carácter excepcional, no serán necesarias estas instrucciones de utilización si el producto puede ser utilizado de forma correcta y segura sin ellas.
8.2 Cuando proceda, la información que deba facilitarse se expresará en forma de símbolos. Los símbolos y colores de identificación utilizados deberán ajustarse a las normas armonizadas. En los sectores en los que no existan normas, se describirán los símbolos y colores utilizados en la documentación proporcionada con el producto.
8.3 En el caso de productos que contengan una sustancia o un preparado que pueda considerarse peligroso, habida cuenta de la naturaleza y cantidad de sus constituyentes y la forma en que se presenten, se aplicarán los requisitos pertinentes en materia de símbolos de peligro y de etiquetado de los Reales Decretos 2216/1985, de 28 de octubre, y 1078/1993, de 2 de julio. Cuando no se disponga de espacio suficiente para consignar toda la información en el producto mismo o en su etiqueta, los símbolos pertinentes de peligro se colocarán en la etiqueta y se recogerá en las instrucciones de utilización el resto de la información requerida por dichas disposiciones.
Serán de aplicación las disposiciones de las reglamentaciones mencionadas relativas a la ficha de datos de seguridad, salvo que las instrucciones de utilización ya proporcionen de forma adecuada toda la información pertinente.
8.4 En la etiqueta constarán los datos siguientes, que podrán ir en forma de símbolos cuando proceda:
a) El nombre o razón social y la dirección del fabricante. En el caso de productos importados en la Comunidad con el fin de distribuirlos en ésta, en la etiqueta, el envase exterior o las instrucciones de utilización constarán además el nombre y la dirección del representante autorizado del fabricante.
b) Los datos estrictamente necesarios para que el usuario identifique el producto y el contenido del envase inequívocamente.
c) Cuando proceda, la palabra «ESTÉRIL» o una indicación de cualquier estado especial microbiológico o de limpieza.
d) El código de lote precedido de la palabra «LOTE», o el número de serie.
e) En caso necesario, una indicación de la fecha antes de la cual deberá utilizarse el producto o parte de él para tener plena seguridad, sin degradación de las prestaciones, expresada en año, mes y, si procede, día.
f) En el caso de productos para la evaluación del funcionamiento, las palabras «sólo para la evaluación del funcionamiento».
g) Cuando proceda, una indicación del uso «in vitro» del producto.
h) Las condiciones específicas de almacenamiento o mantenimiento.
i) En su caso, las instrucciones especiales de manipulación.
j) Advertencias pertinentes o precauciones que deban adoptarse.
k) Si el producto está destinado al autodiagnóstico, se indicará claramente este extremo.
8.5 Si la finalidad prevista del producto no resulta evidente para el usuario, el fabricante deberá indicarla claramente en las instrucciones de utilización y, si procede, en la etiqueta.
8.6 Siempre que sea razonable y factible, los productos y los componentes separados deberán identificarse, en lotes cuando proceda, a fin de permitir todas las medidas oportunas de detección de cualquier riesgo potencial planteado por los productos y los componentes desmontables.
8.7 Las instrucciones de utilización deberán contener, según proceda, los datos siguientes:
a) Los mencionados en el apartado 8.4, con excepción de los párrafos d) y e).
b) La composición del producto reactivo en función de la naturaleza y cantidad o concentración de la sustancia o sustancias activas del reactivo o reactivos o del estuche de instrumental y materiales, así como una declaración, cuando proceda, de que el producto contiene otros componentes que podrían influir en las mediciones.
c) Las condiciones de almacenamiento y el período de validez tras la primera apertura del envase primario, junto con las condiciones de almacenamiento y la estabilidad de los reactivos de trabajo.
d) Las prestaciones mencionadas en el apartado 3 del artículo 5.
e) La indicación de cualquier equipo especial que se necesite, incluida la información necesaria para la identificación de estos equipos especiales con vistas a un uso correcto.
f) El tipo de muestra que vaya a utilizarse, las condiciones especiales de recogida, el tratamiento previo y, si fuere necesario, las condiciones de almacenamiento y las instrucciones relativas a la preparación del paciente.
g) La descripción detallada del procedimiento que deberá seguirse al utilizar el producto.
h) El procedimiento de medición que debe seguirse con el producto, incluidos, en su caso, los datos siguientes:
— El principio del método.
— Las características específicas de funcionamiento del análisis (por ejemplo, sensibilidad, especificidad, exactitud, repetibilidad, reproducibilidad, límites de detección y margen de medida, incluida la información necesaria para el control de las interferencias pertinentes conocidas), las limitaciones del método y la información acerca del uso, por parte del usuario, de procedimientos de medida de referencia y materiales de referencia disponibles.
— Los datos de cualquier otro procedimiento o manipulación necesarios antes de poder utilizar el producto (por ejemplo, reconstitución, incubación, dilución, controles de instrumentos, etc.).
— La indicación de la eventual necesidad de formación especial.
i) El planteamiento matemático en el que se basa el cálculo del resultado analítico.
j) Las medidas que deberán tomarse en caso de cambios en el funcionamiento analítico del producto.
k) La información adecuada para los usuarios sobre:
— El control de calidad interno, con inclusión de métodos específicos de validación.
— La correlación de la calibración del producto.
l) Los intervalos de referencia de las cantidades que se determinan, con inclusión de una descripción de la población de referencia que deba considerarse.
m) Si el producto debe utilizarse en asociación con otros productos o equipos sanitarios, o instalado o conectado con ellos para que funcione tal como exige su finalidad prevista, datos suficientes de sus características para saber cuáles son los productos o equipos adecuados que se deben usar para obtener una asociación apropiada y segura.
n) Toda la información necesaria para verificar si el producto está instalado adecuadamente y puede funcionar de forma correcta y segura, además de datos sobre la naturaleza y frecuencia del mantenimiento y calibración necesarios para garantizar permanentemente el funcionamiento correcto y seguro del producto, e información sobre la eliminación segura de residuos.
o) La información sobre cualquier tratamiento o manipulación adicional que deba realizarse antes de utilizar el producto (por ejemplo, esterilización, montaje final, etc.).
p) Las instrucciones necesarias en caso de deterioro del envase protector y datos sobre los métodos apropiados de reesterilización o descontaminación.
q) Si el producto es reutilizable, información acerca de los procesos adecuados que permitan la reutilización, incluidos limpieza, desinfección, envasado y reesterilización o descontaminación, así como las eventuales restricciones del número de reutilizaciones.
r) Las precauciones que deben tomarse por lo que respecta a la exposición, en condiciones ambientales razonablemente previsibles, a campos magnéticos, influencias eléctricas externas, descargas electrostáticas, presión o variaciones de la presión, aceleración, fuentes térmicas de ignición, etc.
s) Las precauciones que deben tomarse frente a cualquier riesgo específico e inhabitual derivado del uso o la eliminación del producto, incluidas medidas especiales de protección; cuando el producto incluya sustancias de origen humano o animal, se señalará su naturaleza potencialmente infecciosa.
t) Las especificaciones para productos de autodiagnóstico:
— Los resultados deberán expresarse y presentarse de forma que sean fácilmente comprensibles por un profano; deberá proporcionarse al usuario información sobre la actitud que haya que tomar (en caso de resultado positivo, negativo o indeterminado) y sobre la posibilidad de resultados falsos positivos o falsos negativos.
— Podrán omitirse datos específicos siempre que el resto de la información proporcionada por el fabricante sea suficiente para que el usuario pueda utilizar el producto e interpretar los resultados obtenidos con éste.
— La información proporcionada incluirá una indicación clara de que el usuario no debe tomar ninguna decisión de importancia médica sin consultar antes con su médico.
— La información deberá asimismo precisar que, cuando se utilice un producto de autodiagnóstico para controlar una enfermedad existente, el paciente sólo deberá adaptar el tratamiento cuando haya recibido la formación necesaria para ello.
u) La fecha de publicación o de revisión más reciente de las instrucciones de utilización.
[Bloque 63: #anii]
Lista A.
— Reactivos y productos reactivos, incluidos los materiales asociados de calibrado y control, para la determinación de los grupos sanguíneos siguientes: sistema ABO, Rhesus (C, c, D, E, e) y antiKell.
— Reactivos y productos reactivos, incluidos los materiales asociados de calibrado y control, para la detección, confirmación y cuantificación en muestras humanas de marcadores de infección por VIH (VIH 1 y 2), HTLV I y II y de hepatitis B, C y D.
Lista B.
— Reactivos y productos reactivos, incluidos los materiales asociados de calibrado y control, para la determinación de los grupos sanguíneos siguientes: anti-Duffy y anti-Kidd.
— Reactivos y productos reactivos, incluidos los materiales asociados de calibrado y control, para la determinación de anticuerpos antieritrocíticos irregulares.
— Reactivos y productos reactivos, incluidos los materiales asociados de calibrado y control, para la detección y la cuantificación en muestras humanas de las infecciones congénitas siguientes: rubéola y toxoplasmosis.
— Reactivos y productos reactivos, incluidos los materiales asociados de calibrado y control, para diagnosticar la enfermedad hereditaria siguiente: fenilcetonuria.
— Reactivos y productos reactivos, incluidos los materiales asociados de calibrado y control, para la determinación de las infecciones humanas siguientes: citomegalovirus y Chlamydia.
— Reactivos y productos reactivos, incluidos los materiales asociados de calibrado y control, para la determinación de los grupos tisulares HLA siguientes: DR, A, B.
— Reactivos y productos reactivos, incluidos los materiales asociados de calibrado y control, para la determinación del marcador tumoral siguiente: PSA.
— Reactivos y productos reactivos, incluidos los materiales asociados de calibrado y control, así como los programas informáticos destinados específicamente a la evaluación del riesgo de la trisomía en el par 21.
— El siguiente producto para autodiagnóstico, incluidos los materiales asociados de calibrado y control: equipo para la medición de la glucemia.
[Bloque 64: #aniii]
1. La declaración CE de conformidad es el procedimiento mediante el cual el fabricante o su representante autorizado que cumple las obligaciones impuestas por los apartados 2 a 5 y, además, en el caso de los productos para autodiagnóstico, por el apartado 6 asegura y declara que los productos de que se trata cumplen las disposiciones del presente Real Decreto que les son aplicables. El fabricante colocará el marcado CE con arreglo al artículo 6.
2. El fabricante deberá preparar la documentación técnica descrita en el apartado 3 y asegurarse de que el proceso de fabricación se ajusta a los principios de garantía de calidad que se fijan en el apartado 4.
3. La documentación técnica deberá permitir evaluar la conformidad del producto con los requisitos del Real Decreto. Incluirá, en particular:
— Una descripción general del producto, incluidas las variantes previstas.
— La documentación relativa al sistema de calidad.
— Información sobre el diseño, con inclusión de la determinación de las características de los materiales de base, de las características y limitaciones del funcionamiento de los productos, métodos de fabricación y, en el caso de instrumentos, planos del diseño, diagramas de componentes, subconjuntos, circuitos, etc.
— En el caso de productos que contengan tejidos de origen humano o sustancias derivadas de los mismos, información sobre el origen y sobre las condiciones de recogida de dichos materiales.
— Las descripciones y explicaciones necesarias para la comprensión de las características, planos y diagramas mencionados, así como del funcionamiento del producto.
— Resultados del análisis de riesgo, cuando proceda, y lista de las normas a que hace referencia el artículo 8, aplicadas en todo o en parte, y descripción de las soluciones adoptadas para cumplir los requisitos esenciales del Real Decreto si no se han aplicado en su totalidad las normas a que hace referencia el artículo 8.
— En el caso de productos estériles o de productos con un estado microbiológico o de limpieza especial, descripción de los procedimientos utilizados.
— Resultados de los cálculos de diseño y de las inspecciones efectuadas, etc.
— Si el producto debe ser combinado con otro u otros productos para que funcione de la forma prevista, se aportarán pruebas de que cumple los requisitos esenciales al estar combinado con cualquier producto de ese tipo que tenga las características especificadas por el fabricante.
— Informes de los ensayos.
— Datos adecuados de evaluación del funcionamiento, que demuestren el funcionamiento alegado por el fabricante y respaldados por un sistema de medidas de referencia (si se dispone de él) junto con la información sobre los métodos de referencia, los materiales de referencia, los valores conocidos de referencia, la exactitud y las unidades de medida utilizadas; estos datos deberán haberse obtenido a partir de estudios en un entorno clínico u otro entorno adecuado o derivarse de las referencias bibliográficas pertinentes.
— Etiquetas e instrucciones de utilización.
— Resultados de estudios de estabilidad.
4. El fabricante tomará las medidas necesarias para cerciorarse de que el proceso de fabricación se ajusta a los principios de garantía de calidad de forma adecuada en función de los productos fabricados.
El sistema abarcará los aspectos siguientes:
— La estructura organizativa y las responsabilidades.
— Los procesos de fabricación y el control sistemático de calidad de la producción.
— Los medios de control del funcionamiento del sistema de calidad.
5. El fabricante establecerá y mantendrá actualizado un procedimiento sistemático de análisis de la experiencia adquirida con los productos en la fase posterior a la producción, y de puesta en práctica de los medios apropiados para aplicar las medidas correctoras necesarias, habida cuenta de la naturaleza y de los riesgos inherentes al producto. Notificará a las autoridades competentes los siguientes hechos, tan pronto como tenga conocimiento de los mismos:
i) Cualquier funcionamiento defectuoso, fallo o deterioro de las características o del funcionamiento de un producto, así como cualquier deficiencia del etiquetado o de las instrucciones de utilización que, directa o indirectamente, pudiera o hubiera podido dar lugar a la muerte de un paciente o usuario o de otras personas o a un grave deterioro de su estado de salud.
ii) Cualquier razón de carácter técnico o sanitario ligada a las características o al funcionamiento de un producto por las razones expuestas en el anterior inciso i) que haya dado lugar a la retirada sistemática de productos del mismo tipo por parte del fabricante.
6. En el caso de los productos para autodiagnóstico, el fabricante presentará ante un organismo notificado una solicitud de examen del diseño.
6.1 La solicitud deberá permitir comprender el diseño del producto y evaluar su conformidad con los requisitos de diseño del Real Decreto. Incluirá los aspectos siguientes:
— Informes de los ensayos que incluirán, cuando proceda, los resultados de estudios realizados con profanos.
— Datos que muestren la adecuación del producto para ser manipulado habida cuenta de su finalidad prevista de autodiagnóstico.
— La información que se vaya a facilitar junto con el producto, tanto en la etiqueta como en las instrucciones de utilización.
6.2 El organismo notificado examinará la solicitud y, si el diseño es conforme a las disposiciones pertinentes del presente Real Decreto, expedirá al solicitante un certificado de examen CE de diseño. El organismo notificado podrá exigir que se complete la solicitud con otros ensayos o pruebas que permitan la evaluación de la conformidad con los requisitos del Real Decreto relativos al diseño. En el certificado constarán las conclusiones del examen, las condiciones de su validez, los datos necesarios para la identificación del diseño aprobado y, cuando proceda, una descripción de la finalidad prevista del producto.
6.3 El solicitante informará al organismo notificado que haya expedido el certificado de examen CE de diseño de cualquier modificación importante que se haya introducido en el diseño aprobado. Las modificaciones respecto del diseño aprobado deberán recibir una aprobación complementaria del organismo notificado que haya expedido el certificado de examen CE de diseño, siempre que las modificaciones puedan afectar a la conformidad con los requisitos esenciales del Real Decreto o con las condiciones estipuladas para la utilización del producto. Esta aprobación complementaria se hará en forma de apéndice del certificado de examen CE de diseño.
[Bloque 65: #aniv]
1. El fabricante se cerciorará de que se aplique el sistema de calidad aprobado para el diseño, fabricación e inspección final de los productos de que se trate, tal como se especifica en el apartado 3, y estará sujeto a la auditoría a que se refiere el apartado 3.3 y al control que se especifica en el apartado 5. Además, el fabricante deberá seguir, para los productos contemplados en la lista A del anexo II, los procedimientos establecidos en los apartados 4 y 6.
2. La declaración de conformidad es el procedimiento mediante el cual el fabricante que cumple los requisitos impuestos por el apartado 1 asegura y declara que los productos de que se trata cumplen las disposiciones del presente Real Decreto que les son aplicables.
El fabricante colocará el marcado CE de conformidad con el artículo 6 y efectuará una declaración escrita de conformidad relativa a los productos correspondientes.
3. Sistema de calidad:
3.1 El fabricante presentará ante un organismo notificado una solicitud de evaluación de su sistema de calidad.
Dicha solicitud deberá contener:
— El nombre y la dirección del fabricante y de cualquier otro local de fabricación comprendido en el sistema de calidad.
— Información adecuada sobre el producto o categoría de productos objeto del procedimiento.
— Una declaración escrita de que no se ha presentado ante otro organismo notificado ninguna solicitud paralela relativa al mismo sistema de calidad en relación con el producto.
— La documentación relativa al sistema de calidad.
— El compromiso por parte del fabricante de cumplir las obligaciones impuestas por el sistema de calidad aprobado.
— El compromiso por parte del fabricante de velar por que el sistema de calidad aprobado mantenga su adecuación y eficacia.
— El compromiso por parte del fabricante de establecer y mantener actualizado un procedimiento sistemático de revisión de la experiencia adquirida con los productos en la fase posterior a la producción, y de puesta en práctica de los medios apropiados para aplicar las medidas correctoras necesarias y efectuar la notificación a que se refiere el apartado 5 del anexo III.
3.2 La aplicación del sistema de calidad deberá garantizar la conformidad de los productos con las disposiciones del presente Real Decreto que les sean aplicables en todas las fases, desde el diseño hasta la inspección final. Todos los elementos, requisitos y disposiciones adoptados por el fabricante para su sistema de calidad deberán consignarse en una documentación sistemática y ordenada en forma de planes y procedimientos escritos, como programas de calidad, planes de calidad, manuales de calidad y registros de calidad.
En particular, esta documentación deberá contener una descripción adecuada de:
a) Los objetivos de calidad del fabricante.
b) La organización de la empresa y, en particular:
— Las estructuras de organización, las responsabilidades de los directivos y su autoridad organizativa en materia de la calidad del diseño y de la fabricación de los productos.
— Los métodos para controlar el funcionamiento eficaz del sistema de calidad y, en particular, su aptitud para conseguir la calidad deseada en el diseño y en el producto, incluido el control de los productos no conformes;
c) Los procedimientos de control y verificación del diseño de los productos y, en particular:
— Una descripción general del producto, incluidas las variantes previstas.
— Toda la documentación mencionada en los guiones tercero a decimotercero del apartado 3 del anexo III.
— En el caso de productos para autodiagnóstico, la información mencionada en el apartado 6.1 del anexo III.
— Las técnicas utilizadas para controlar y verificar el diseño y los procesos, y las medidas sistemáticas que se vayan a aplicar en la fase de diseño de los productos.
d) Las técnicas de inspección y de garantía de calidad en la fase de fabricación y, en particular:
— Los procesos y procedimientos que se utilizarán, sobre todo en lo relativo a la esterilización.
— Los procedimientos relativos a las compras.
— Los procedimientos de identificación del producto elaborados y actualizados a partir de dibujos, especificaciones u otros documentos pertinentes en todas las fases de fabricación.
e) Los exámenes y ensayos adecuados que se efectuarán antes, durante y después de la fabricación, la frecuencia con que se llevarán a cabo y el equipo de ensayo utilizado; deberá garantizarse la correlación de la calibración.
El fabricante realizará los exámenes y pruebas pertinentes conforme al nivel técnico más avanzado. Los exámenes y pruebas se referirán al procedimiento de fabricación, incluida la caracterización de la materia prima, así como a cada producto o lote de productos particular fabricado.
Por lo que se refiere a los productos mencionados en la lista A del anexo II, el fabricante tendrá presentes los conocimientos más recientes en particular en lo que se refiere a la complejidad y variabilidad biológica de las muestras que vayan a estudiarse con el producto para diagnóstico «in vitro» de que se trate.
3.3 El organismo notificado realizará una auditoría del sistema de calidad para comprobar si cumple los requisitos mencionados en el apartado 3.2. Dará por supuesta la conformidad con dichos requisitos si los sistemas de calidad cumplen las normas armonizadas pertinentes.
El equipo encargado de la evaluación deberá tener experiencia en evaluaciones de la tecnología de que se trate. El procedimiento de evaluación incluirá una inspección de las instalaciones del fabricante y, en casos debidamente justificados, de las instalaciones de los proveedores o subcontratistas del fabricante, a fin de inspeccionar los procesos de fabricación.
La decisión se comunicará al fabricante, y en ella figurarán las conclusiones de la inspección y una evaluación motivada.
3.4 El fabricante informará al organismo notificado que haya aprobado el sistema de calidad de cualquier proyecto de modificación importante de dicho sistema de calidad o de la gama de productos incluidos.
El organismo notificado evaluará las modificaciones propuestas y comprobará si el sistema de calidad así modificado sigue cumpliendo los requisitos mencionados en el punto 3.2. Comunicará al fabricante su decisión, que incluirá las conclusiones de la inspección y una evaluación motivada.
4. Examen del diseño del producto:
4.1 Para los productos contemplados en la lista A del anexo II, el fabricante, además de las obligaciones que le conciernen con arreglo al apartado 3, presentará al organismo notificado una solicitud de examen del expediente de diseño relativo al producto que vaya a fabricar y que forme parte de la categoría mencionada en el apartado 3.1.
4.2 La solicitud describirá el diseño, la fabricación y las prestaciones del producto sanitario de que se trate e incluirá los documentos necesarios, contemplados en el párrafo c) del apartado 3.2, que permitan juzgar su conformidad con los requisitos del presente Real Decreto.
4.3 El organismo notificado examinará la solicitud y, si el producto cumple los requisitos del presente Real Decreto que sean de aplicación, expedirá un certificado de examen CE del diseño al solicitante. El organismo notificado podrá exigir que la solicitud se complete con ensayos o pruebas adicionales, a fin de que pueda evaluarse la conformidad con los requisitos del presente Real Decreto. En el certificado constarán las conclusiones del examen, las condiciones de su validez, los datos necesarios para la identificación del diseño aprobado y, en su caso, una descripción de la finalidad del producto.
4.4 Las modificaciones introducidas en el diseño aprobado deberán recibir una aprobación complementaria del organismo notificado que haya expedido el certificado de examen CE del diseño, cuando tales modificaciones puedan afectar a la conformidad con los requisitos esenciales del presente Real Decreto o con las condiciones estipuladas para la utilización del producto. El solicitante informará al organismo notificado que haya expedido el certificado de examen CE del diseño sobre cualquier modificación que se haya introducido en el diseño aprobado. La aprobación complementaria será concedida en forma de apéndice del certificado de examen CE del diseño.
4.5 El fabricante informará al organismo notificado sin tardanza cuando haya tenido conocimiento de variaciones del agente patógeno y de los marcadores de las infecciones objeto de la prueba, y especialmente las debidas a su complejidad o variabilidad biológica. A ese respecto, el fabricante comunicará al organismo notificado si existe la probabilidad de que esa variación tenga repercusiones en el funcionamiento del producto sanitario para diagnóstico «in vitro» de que se trate.
5. Control:
5.1 La finalidad del control es garantizar el correcto cumplimiento por parte del fabricante de las obligaciones impuestas por el sistema de calidad aprobado.
5.2 El fabricante deberá autorizar al organismo notificado a llevar a cabo todas las inspecciones necesarias y le proporcionará toda la información pertinente, y en particular la siguiente:
— La documentación relativa al sistema de calidad.
— Los datos estipulados en la parte del sistema de calidad relativa al diseño, tales como resultados de análisis, cálculos, ensayos, etc.
— Los datos estipulados en la parte del sistema de calidad relativa a la fabricación, tales como informes de inspección y datos de ensayos, datos de calibración, informes de cualificación del personal implicado, etc.
5.3 El organismo notificado llevará a cabo periódicamente inspecciones y evaluaciones adecuadas para cerciorarse de que el fabricante aplica el sistema de calidad aprobado y entregará al fabricante un informe de evaluación.
5.4 Además, el organismo notificado podrá visitar al fabricante sin previo aviso. Con ocasión de esas visitas, el organismo notificado podrá, en caso necesario, efectuar o solicitar ensayos para verificar el buen funcionamiento del sistema de calidad. Entregará al fabricante un informe de inspección y, si se ha realizado algún ensayo, un informe de éste.
6. Verificación de los productos elaborados enumerados en la lista A del anexo II:
6.1 Cuando se trate de productos contemplados en la lista A del anexo II, el fabricante comunicará al organismo notificado, sin tardanza y una vez concluidos los exámenes o pruebas, los correspondientes protocolos sobre los exámenes de los productos o lotes de productos fabricados. Además, el fabricante pondrá a disposición del organismo notificado las muestras de productos o lotes de productos elaborados de conformidad con condiciones y modalidades acordadas previamente.
6.2 El fabricante podrá poner en el mercado los productos, salvo en caso de que el organismo notificado le comunicara dentro del plazo convenido, que en cualquier caso no superará los treinta días desde la recepción de las muestras, una decisión diferente, que podrá incluir, en particular, condiciones para la validez de los certificados expedidos.
[Bloque 66: #anv]
1. El examen CE de tipo es la parte del procedimiento mediante la cual un organismo notificado comprueba y certifica que una muestra representativa de la producción considerada cumple las disposiciones pertinentes del presente Real Decreto.
2. El fabricante, o su representante autorizado, presentará ante un organismo notificado la solicitud de examen CE de tipo.
La solicitud deberá incluir:
— El nombre y la dirección del fabricante, y el nombre y la dirección del representante autorizado si es éste quien presenta la solicitud.
— La documentación descrita en el apartado 3 necesaria para evaluar la conformidad de la muestra representativa de la producción considerada, denominada en lo sucesivo «tipo», con los requisitos del presente Real Decreto. El solicitante deberá poner un tipo a disposición del organismo notificado. Si fuere necesario el organismo notificado podrá solicitar otras muestras.
— Una declaración escrita de que no se ha presentado ante otro organismo notificado ninguna solicitud referente al mismo tipo.
3. La documentación deberá permitir comprender el diseño, la fabricación y las prestaciones del producto. Deberá incluir, en particular, los elementos siguientes:
— Una descripción general del tipo, incluidas las variantes previstas.
— Toda la documentación mencionada en los guiones tercero a decimotercero del apartado 3 del anexo III.
— En el caso de los productos para autodiagnóstico, la información a que se refiere el apartado 6.1 del anexo III.
4. El organismo notificado:
4.1 Examinará y evaluará la documentación y comprobará que el tipo se ha fabricado de conformidad con dicha documentación; señalará asimismo los elementos que se hayan diseñado de conformidad con las disposiciones aplicables de las normas mencionadas en el artículo 5, así como los elementos cuyo diseño no se base en las disposiciones pertinentes de dichas normas.
4.2 Realizará o hará realizar los controles adecuados y los ensayos necesarios para comprobar si las soluciones adoptadas por el fabricante cumplen los requisitos esenciales del presente Real Decreto, en caso de que no se hayan aplicado las normas mencionadas en el artículo 5; en caso de que el producto deba combinarse con otro u otros productos para que funcione de la forma prevista, se aportarán pruebas de que cumple los requisitos esenciales al estar combinado con cualquier producto de ese tipo que tenga las características especificadas por el fabricante.
4.3 Realizará o hará que se lleven a cabo los controles adecuados y los ensayos necesarios para comprobar si, en caso de que el fabricante haya decidido aplicar las normas pertinentes, éstas se han aplicado realmente.
4.4 Acordará con el solicitante el lugar en que se vayan a realizar los controles y ensayos necesarios.
5. Si el tipo es conforme a las disposiciones del presente Real Decreto, el organismo notificado expedirá al solicitante un certificado de examen CE de tipo. El certificado contendrá el nombre y la dirección del fabricante, las conclusiones del examen, las condiciones de validez y los datos necesarios para la identificación del tipo aprobado. Se adjuntarán al certificado las partes pertinentes de la documentación, y el organismo notificado conservará una copia.
6. El fabricante informará sin tardanza al organismo notificado cuando haya tenido conocimiento de variaciones del agente patógeno y de los marcadores de las infecciones objeto de la prueba, especialmente las debidas a su complejidad o variabilidad biológica. A ese respecto, el fabricante comunicará al organismo notificado si existe la probabilidad de que esa variación tenga repercusiones en el funcionamiento del producto sanitario para diagnóstico «in vitro» de que se trate.
6.1 Las modificaciones del producto aprobado deberán recibir una aprobación complementaria del organismo notificado que haya expedido el certificado de examen CE de tipo, siempre que las modificaciones puedan afectar a la conformidad con los requisitos esenciales del Real Decreto o con las condiciones estipuladas para la utilización del producto. El solicitante informará al organismo notificado que haya concedido el certificado de examen CE de tipo de cualquier modificación importante que se introduzca en el producto aprobado. Esta nueva aprobación deberá tomar la forma de apéndice del certificado de examen CE de tipo inicial.
7. Disposiciones administrativas: Los demás organismos notificados podrán obtener una copia de los certificados de examen CE de tipo y de sus complementos. Los anexos de los certificados se pondrán a disposición de los demás organismos notificados, previa solicitud motivada y una vez que se haya informado al fabricante.
[Bloque 67: #anvi]
1. La verificación CE es el procedimiento por el que el fabricante o su representante autorizado asegura y declara que los productos sometidos al procedimiento establecido en el apartado 4 son conformes al tipo descrito en el certificado de examen CE de tipo y cumplen los requisitos del presente Real Decreto que les son aplicables.
2.1 El fabricante tomará todas las medidas necesarias para que el proceso de fabricación garantice la conformidad de los productos con el tipo descrito en el certificado de examen CE de tipo y con los requisitos del Real Decreto que les sean aplicables. Antes de comenzar la fabricación, el fabricante preparará una documentación en la que se defina el proceso de fabricación, en particular por lo que respecta a la esterilización y a la idoneidad de los materiales de partida, cuando sea necesario, así como los procedimientos de ensayo necesarios de acuerdo con los avances en ese campo. Se aplicarán todas las disposiciones sistemáticas previamente establecidas, con el fin de garantizar la homogeneidad de la producción y la conformidad de los productos con el tipo descrito en el certificado de examen CE de tipo y con los requisitos del presente Real Decreto que les sean aplicables.
2.2 En la medida en que para ciertos aspectos no proceda el control final con arreglo al apartado 6.3, el fabricante, con la aprobación del organismo notificado, fijará métodos adecuados de ensayo, vigilancia y control durante el proceso. En tal caso, en relación con los antedichos procedimientos aprobados serán aplicables de forma análoga las disposiciones del apartado 5 del anexo IV.
3. El fabricante se comprometerá a establecer y mantener actualizado un procedimiento sistemático de revisión de la experiencia adquirida con los productos en la fase posterior a la producción, y de puesta en práctica de los medios apropiados para aplicar las medidas correctoras necesarias y efectuar la notificación a que se refiere el apartado 5 del anexo III.
4. El organismo notificado efectuará los controles y ensayos adecuados teniendo en cuenta lo dispuesto en el apartado 2.2 a fin de verificar la conformidad del producto con los requisitos del Real Decreto, bien mediante control y ensayo de cada producto tal como se especifica en el apartado 5 o bien mediante examen y ensayo de los productos de forma estadística tal como se especifica en el apartado 6, a elección del fabricante. Al realizar la verificación estadística con arreglo al apartado 6, el organismo notificado deberá decidir si han de aplicarse procedimientos estadísticos para la inspección lote por lote o si ha de efectuarse la inspección de un lote aislado. Antes de tomar esta decisión, deberá consultarse al fabricante.
Si la realización de exámenes y ensayos de forma estadística no resulta adecuada, se efectuarán controles y ensayos de forma aleatoria, siempre que este procedimiento, junto con las medidas tomadas con arreglo al apartado 2.2, garantice un nivel de conformidad equivalente.
5. Verificación mediante examen y ensayo de cada producto:
5.1 Se examinará individualmente cada producto y se realizarán los ensayos adecuados definidos en la norma o normas correspondientes mencionadas en el artículo 5, o ensayos equivalentes, con el fin de verificar la conformidad de los productos con el tipo CE descrito en el certificado de examen CE de tipo y con los requisitos del Real Decreto que les sean aplicables.
5.2 El organismo notificado colocará o hará colocar su número de identificación en cada producto aprobado y expedirá un certificado escrito de conformidad relativo a los ensayos efectuados.
6. Verificación estadística:
6.1 El fabricante presentará los productos fabricados en forma de lotes homogéneos.
6.2 Se tomarán una o más muestras aleatorias de cada lote, conforme sea necesario. Los productos que compongan la muestra se examinarán individualmente y se realizarán los ensayos adecuados definidos en la norma o normas pertinentes mencionadas en el artículo 5, o ensayos equivalentes, con el fin de verificar, cuando proceda, la conformidad de los productos con el tipo descrito en el certificado de examen CE de tipo y con los requisitos del Real Decreto que les sean aplicables, con el fin de determinar la aceptación o rechazo del lote.
6.3 El control estadístico de los productos se basará en atributos o variables, lo que conlleva sistemas de muestreo con características operativas que garanticen un alto nivel de seguridad y de prestaciones correspondiente a los conocimientos más modernos. El método de muestreo se determinará con arreglo a las normas armonizadas mencionadas en el artículo 5, habida cuenta de la naturaleza específica de las categorías de productos de que se trate.
6.4 Si un lote es aceptado, el organismo notificado colocará o hará colocar su número de identificación en cada producto y expedirá un certificado escrito de conformidad relativo a los ensayos efectuados. Podrán ser puestos en el mercado todos los productos del lote, excepto los productos de la muestra que no hayan sido conformes.
Si un lote es rechazado, el organismo notificado competente tomará las medidas adecuadas para impedir su puesta en el mercado. En caso de rechazo frecuente de lotes, el organismo notificado podrá suspender la verificación estadística.
El fabricante, bajo la responsabilidad del organismo notificado, podrá colocar el número de identificación del organismo notificado durante el proceso de fabricación.
[Bloque 68: #anvii]
1. El fabricante se cerciorará de que se aplica el sistema de calidad aprobado para la fabricación de los productos considerados, llevará a cabo la inspección final especificada en el punto 3 y estará sometido al control mencionado en el apartado 4.
2. La declaración de conformidad es la parte del procedimiento mediante la cual el fabricante que cumple las obligaciones impuestas por el apartado 1 asegura y declara que los productos considerados son conformes al tipo descrito en el certificado de examen CE de tipo y que se ajustan a las disposiciones del presente Real Decreto que les son aplicables.
El fabricante colocará el marcado CE de conformidad con el artículo 6 y efectuará una declaración escrita de conformidad referida a los productos de que se trate.
3. Sistema de calidad:
3.1 El fabricante presentará ante un organismo notificado una solicitud de evaluación de su sistema de calidad.
Dicha solicitud deberá contener:
— Toda la documentación y los compromisos mencionados en el apartado 3.1 del anexo IV, y
— La documentación técnica relativa a los tipos aprobados y una copia de los certificados de examen CE de tipo.
3.2 La aplicación del sistema de calidad deberá garantizar la conformidad de los productos al tipo descrito en el certificado de examen CE de tipo.
Todos los elementos, requisitos y disposiciones adoptados por el fabricante para su sistema de calidad deberán figurar en una documentación sistemática y ordenada en forma de declaraciones de planes y procedimientos escritos. Esta documentación del sistema de calidad deberá permitir una interpretación uniforme de los planes yprocedimientos de calidad, como programas, proyectos, manuales y registros relativos a la calidad.
Deberá contener, en particular, una descripción adecuada de:
a) Los objetivos de calidad del fabricante.
b) La organización de la empresa y, en particular:
— Las estructuras de organización, las responsabilidades de los directivos y su autoridad organizativa en lo referente a la calidad de la fabricación de los productos.
— Los métodos para controlar el funcionamiento eficaz del sistema de calidad y, en particular, su aptitud para conseguir la calidad deseada en el producto, incluido el control de los productos no conformes.
c) Las técnicas de inspección y de garantía de calidad en la fase de fabricación y, en particular:
— Los procesos y procedimientos que se utilizarán en lo relativo, sobre todo, a la esterilización.
— Los procedimientos de compras.
— Los procedimientos de identificación del producto elaborados y actualizados a partir de dibujos, especificaciones u otros documentos pertinentes en todas las fases de fabricación.
d) Los exámenes y ensayos adecuados que se efectuarán antes, durante y después de la fabricación, la frecuencia con que se llevarán a cabo y el equipo de ensayo utilizado; deberá garantizarse la correlación de la calibración.
3.3 El organismo notificado realizará una auditoría del sistema de calidad para comprobar si cumple los requisitos mencionados en el apartado 3.2. Dará por supuesta la conformidad con dichos requisitos si los sistemas de calidad cumplen las normas armonizadas pertinentes.
El equipo de evaluación deberá tener experiencia previa en evaluaciones de la tecnología de que se trate. El procedimiento de evaluación incluirá una inspección de las instalaciones del fabricante y, en casos debidamente justificados, de las instalaciones de los proveedores o subcontratistas del fabricante, a fin de inspeccionar los procesos de fabricación.
La decisión se comunicará al fabricante. En ella figurarán las conclusiones de la inspección y una evaluación motivada.
3.4 El fabricante informará al organismo notificado que haya aprobado el sistema de calidad de cualquier proyecto de modificación importante de dicho sistema de calidad.
El organismo notificado evaluará las modificaciones propuestas y comprobará si el sistema de calidad así modificado sigue cumpliendo los requisitos mencionados en el apartado 3.2. Comunicará al fabricante su decisión, que incluirá las conclusiones de la inspección y una evaluación motivada.
4. Control: Se aplicarán las disposiciones del apartado 5 del anexo IV.
5. Verificación de los productos elaborados enumerados en la lista A del anexo II:
5.1 Cuando se trate de productos que figuren en la lista A del anexo II, el fabricante comunicará al organismo notificado, sin tardanza y una vez concluidos los exámenes o pruebas, los correspondientes protocolos sobre los exámenes de los productos o lotes de productos fabricados. Además, el fabricante pondrá a disposición del organismo notificado las muestras de productos o lotes de productos elaborados de conformidad con las condiciones y modalidades acordadas previamente.
5.2 El fabricante podrá poner en el mercado los productos, salvo en caso de que el organismo notificado le comunicara dentro del plazo convenido, que en cualquier caso no superará los treinta días desde la recepción de las muestras, una decisión diferente, que podrá incluir, en particular, condiciones para la validez de los certificados expedidos.
[Bloque 69: #anviii]
1. En el caso de los productos para la evaluación del funcionamiento, el fabricante o su representante autorizado redactará la declaración con la información estipulada en el apartado 2 y velará por que se cumplan las disposiciones pertinentes del presente Real Decreto.
2. La declaración contendrá la información siguiente:
— Los datos que permitan identificar el producto en cuestión.
— Un plan de evaluación en el que se indiquen, en particular, el objetivo, la justificación científica, técnica o médica, el alcance de la evaluación y el número de los productos utilizados.
— La relación de laboratorios u otras instituciones que participen en el estudio de evaluación.
— La fecha de inicio y la duración prevista de las evaluaciones y, en el caso de los productos para autodiagnóstico, el lugar y el número de profanos implicados.
— Una declaración por la que se haga constar que el producto de que se trate cumple los requisitos del presente Real Decreto, con independencia de los aspectos cubiertos por la evaluación y los que figuran específicamente en la declaración, y que se han adoptado todas las precauciones para proteger la salud y seguridad del paciente, del usuario o de otras personas.
3. El fabricante se comprometerá también a tener a disposición de las autoridades nacionales competentes la documentación que permita comprender el diseño, la fabricación y el funcionamiento del producto, incluido el funcionamiento esperado, de forma que se pueda evaluar la conformidad con los requisitos del presente Real Decreto. Esta documentación deberá conservarse durante un período de al menos cinco años a partir del fin de la evaluación del funcionamiento.
El fabricante tomará las medidas necesarias para que el proceso de fabricación garantice que los productos fabricados son conformes a la documentación mencionada en el apartado 1.
[Bloque 70: #anix]
1. El organismo notificado, su director y el personal de verificación y evaluación no podrán ser los diseñadores, fabricantes, proveedores, instaladores o usuarios de los productos que inspeccionen ni los representantes autorizados de ninguna de estas personas. No deberán intervenir, ni directamente ni como representantes, en el diseño, construcción, comercialización o mantenimiento de los productos. Esto no excluye en modo alguno la posibilidad de intercambio de información técnica entre el fabricante y el organismo notificado.
2. El organismo notificado y su personal realizarán las operaciones de evaluación y verificación con el más alto grado de integridad profesional y la necesaria competencia en el campo de los productos sanitarios. Deberán estar libres de todo tipo de presiones e incitaciones, especialmente de orden económico, que pudieran influir en su juicio o en los resultados de la inspección, en particular por parte de personas o grupos de personas con intereses en los resultados de las verificaciones.
En caso de que el organismo notificado subcontrate tareas específicas relacionadas con la observación y verificación de hechos, deberá asegurarse previamente de que el subcontratista cumple las disposiciones del Real Decreto. El organismo notificado tendrá a disposición de las autoridades nacionales los documentos relativos a la evaluación de las cualificaciones del subcontratista y del trabajo realizado por éste en el ámbito del presente Real Decreto.
3. El organismo notificado deberá ser capaz de realizar todas las tareas asignadas a estos organismos por uno de los anexos III a VII y para las que haya sido notificado, ya se realicen estas tareas por el organismo mismo o bajo su responsabilidad. En particular, deberá disponer del personal y de los medios necesarios para desempeñar de forma adecuada las actividades técnicas y administrativas inherentes a la realización de las evaluaciones y verificaciones. Ello implica la existencia, en el seno de la organización, de personal científico suficiente que posea la experiencia adecuada y los conocimientos necesarios para evaluar desde el punto de vista biológico y sanitario el carácter funcional y el funcionamiento de los productos para los que haya sido notificado, en relación con los requisitos del presente Real Decreto y, en particular, con los requisitos del anexo I. También deberá tener acceso al equipo necesario para las verificaciones requeridas.
4. El personal encargado del control deberá tener:
— Una sólida formación profesional que abarque todas las operaciones de evaluación y verificación para las que haya sido designado el organismo.
— Un conocimiento satisfactorio de las normas relativas a las inspecciones que realice y una experiencia adecuada de esas inspecciones.
— La aptitud necesaria para redactar los certificados, actas e informes que demuestren que las inspecciones han sido efectuadas.
5. Deberá garantizarse la imparcialidad del personal que realice las inspecciones. La remuneración de este personal no dependerá del número de inspecciones realizadas ni de los resultados de las mismas.
6. El organismo deberá suscribir un seguro de responsabilidad civil, salvo que dicha responsabilidad sea asumida por la Administración estatal o que las inspecciones sean efectuadas directamente por organismos públicos.
7. El personal del organismo notificado estará vinculado por el secreto profesional respecto a cualquier información de la que tenga conocimiento en el desempeño de sus tareas (excepto en relación con las autoridades administrativas competentes del Estado Español) en el ámbito del presente Real Decreto o de cualquier disposición de Derecho interno adoptada en aplicación del mismo.
[Bloque 71: #anx]
El marcado CE de conformidad consistirá en las iniciales «CE» presentadas de la forma siguiente:
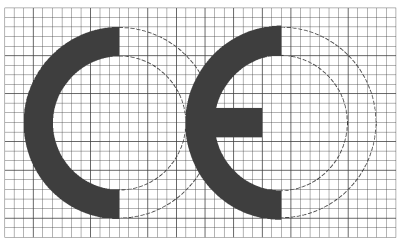
— Si el marcado se reduce o se amplía, se respetarán las proporciones dadas en el dibujo graduado que figura más arriba.
— Los diversos componentes del marcado CE deberán tener, básicamente, la misma dimensión vertical, que no podrá ser inferior a 5 mm. Podrá no exigirse este requisito de dimensión mínima en los productos de pequeño tamaño.
Este documento es de carácter informativo y no tiene valor jurídico.
Ayúdenos a mejorar: puede dirigir sus comentarios y sugerencias a nuestro Servicio de atención al ciudadano
Agence d'État Bulletin Officiel de l'État
Av. Manoteras, 54 - 28050 Madrid





