Agencia Estatal Boletín Oficial del Estado






Excelentísimos señores:
De conformidad con lo establecido en las Ordenes ministeriales de 30 de noviembre de 1976 («Boletín Oficial del Estado» de 4 de enero de 1977), 31 de enero de 1977 («Boletín Oficial del Estado» de 14 a 27 de julio) y 31 de julio do 1979 («Boletín Oficial del Estado» de 20 y 30 de agosto), por las que se declaraban como oficiales diversos métodos de análisis, se ha continuado ensayando y poniendo a punto nuevos métodos, no sólo de las materias comprendidas en las citadas Ordenes, sino de otras nuevas donde existe necesidad urgente de disponer de métodos de análisis para el control de determinados parámetros analíticos.
En su virtud, y a propuesta de los Ministros de Agricultura y Pesca, de Hacienda, de Administración Territorial, de Defensa, de Trabajo, Sanidad y Seguridad Social, de Industria y Energía, de Economía y Comercio, de Educación y Ciencia, de Obras Públicas y Urbanismo, esta Presidencia del Gobierno dispone:
Se aprueban como oficiales los métodos de análisis de aceites y grasas, aguas, carne y productos cárnicos, fertilizantes, productos fitosanitarios, leche y productos lácteos, piensos y sus primeras materias, productos orgánicos fertilizantes, plantas, suelos, productos derivados de la uva y similares y toma de muestras que se citan respectivamente en los anejos I al XII.
Cuando no existan métodos oficiales para determinados análisis, y hasta que sean estudiados por el grupo de trabajo correspondiente, podrán ser utilizados los adoptados por Organismos nacionales o internacionales de reconocida solvencia.
Quedan derogadas las disposiciones de igual o inferior rango que se opongan a la presente Orden.
La presente disposición entrará en vigor a los treinta días de su publicación en el «Boletín Oficial del Estado».
Lo que se comunica a VV. EE. para su conocimiento y efectos.
Dios guarde a VV. EE.
Madrid, 17 de septiembre de 1981.
RODRIGUEZ INCIARTE
Excmos. Sres. Ministros de Agricultura y Pesca, Hacienda, Administración Territorial, Industria y Energía, Obras Públicas y Urbanismo, Defensa, Trabajo, Sanidad y Seguridad Social, Economía y Comercio y de Educación y Ciencia.
Anejo I. Aceites y grasas
35. Prueba de Vizern.
43. Reconocimiento de ésteres no glicéridos en grasas comestibles por cromatografía en capa fina.
49. Acidos docosenoicos.
Anejo II. Aguas
14. Flúor.
15 (a). Boro (curcumina).
16. Mercurio.
Anejo III. Carne y productos cárnicos
16. Tiuracilos.
Anejo IV. Fertilizantes
6 (a). Nitrógeno total (método de Kjeldahl, modificado para muestras que no contengan nitratos).
6 (c). Nitrógeno total.
7 (a). Nitrógeno amoniacal (método del óxido de magnesio).
7 (b). Nitrógeno amoniacal (método del formaldehído).
8. Nitrógeno amoniacal y nítrico conjuntamente (método de Devarda).
Anejo V. Fitosanitarios
4 (a). Dimetoato (por ultravioleta).
4 (b). Dimetoato (por infrarrojo).
14. Cobre total (por iodometría).
Anejo VI. Leche
19. Sustancias proteicas reductoras.
Anejo VII. Piensos
13. Nitrógeno amoniacal.
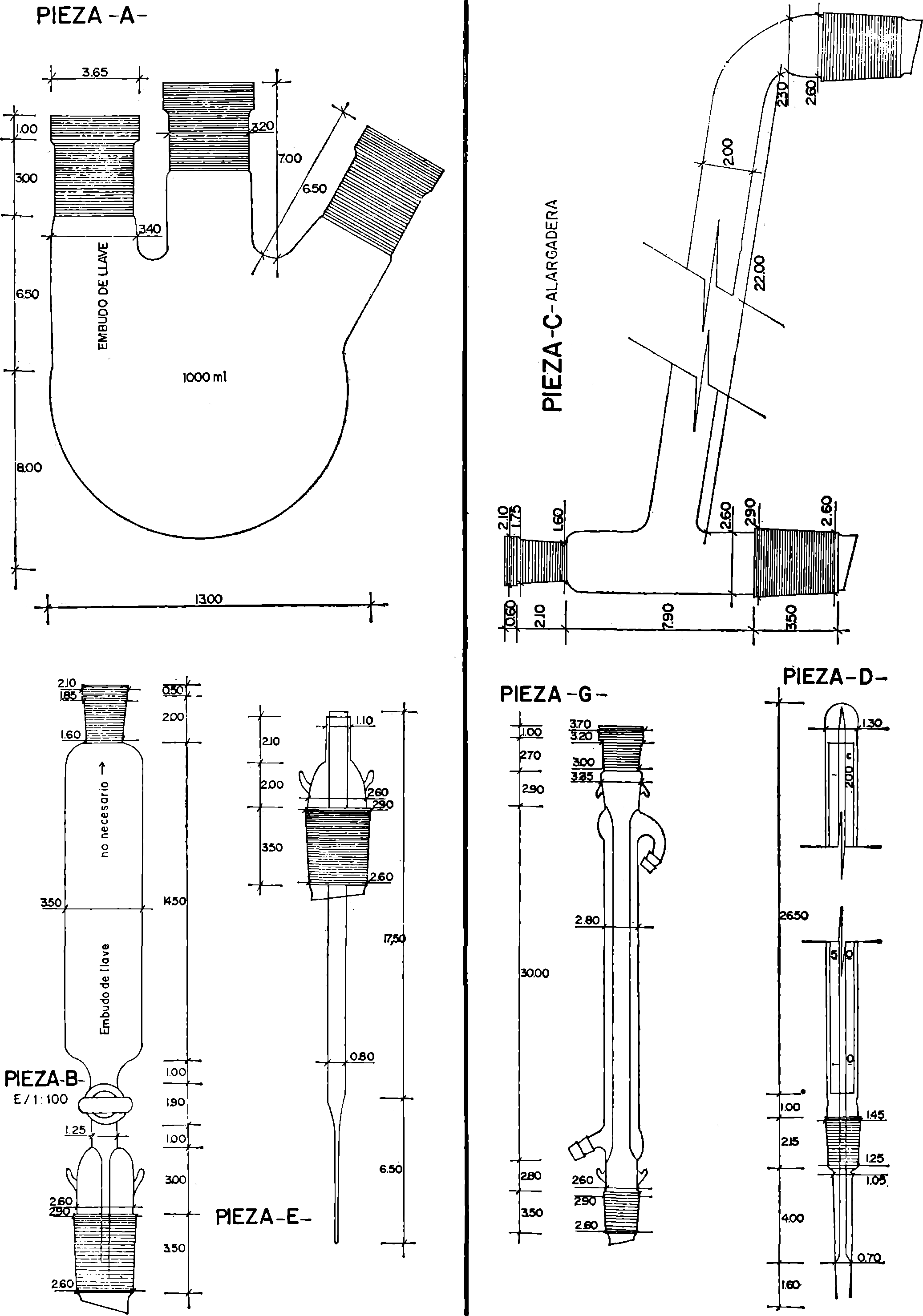
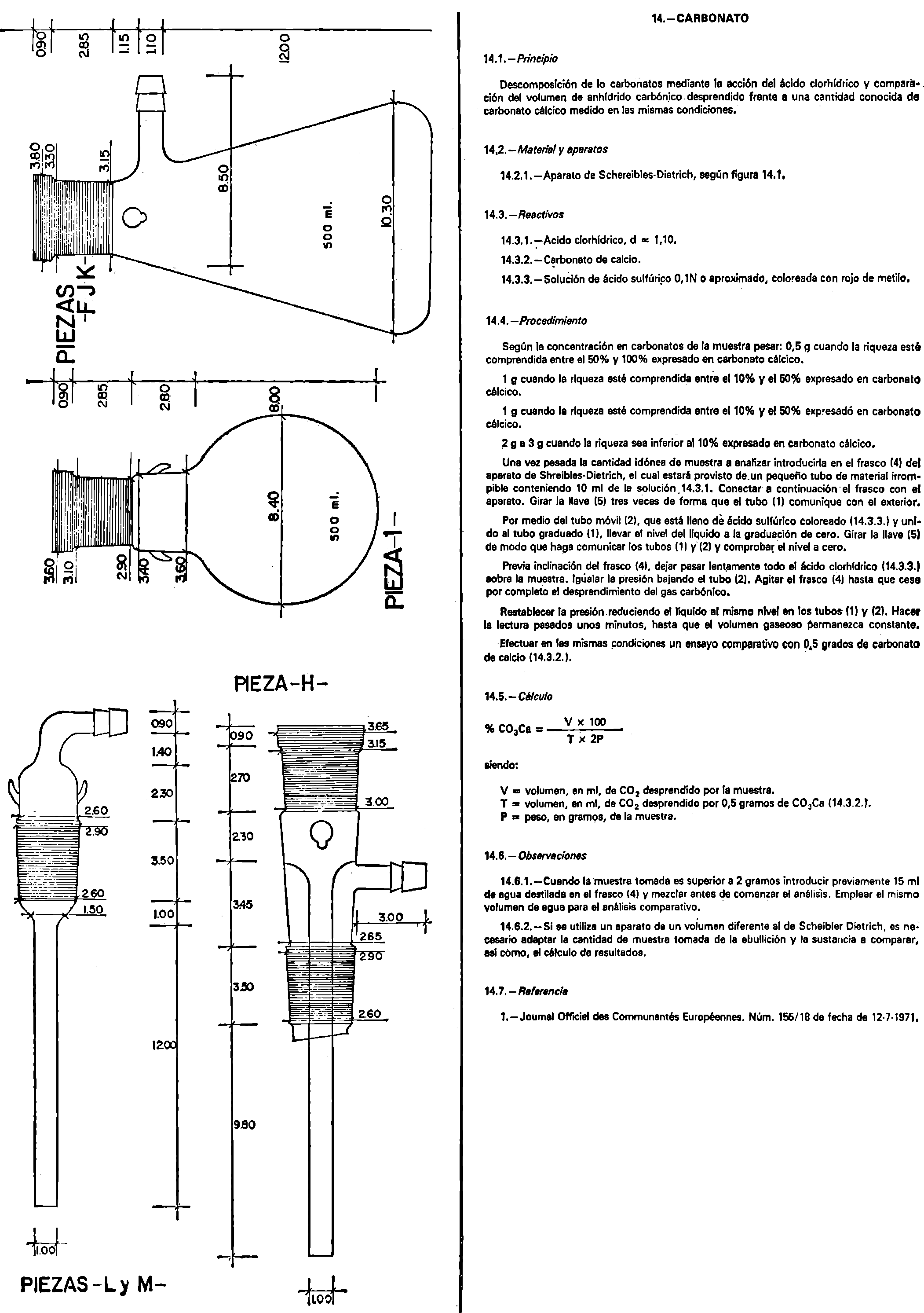
14. Carbonatos
15. Aflatoxina B1.
Anejo VIII. Productos orgánicos fertilizantes
1. Preparación de la muestra.
2. Humedad (pérdida de peso a 105° C).
3 (b). Materia orgánica total (por oxidación).
5. Cenizas.
13. Fósforo total.
16. Potasio soluble en agua (por fotometría de llama).
Anejo IX. Plantas
1. Preparación de la muestra.
2. Nitrógeno.
Anejo X. Suelos
9. Capacidad de cambio catiónico.
11. Acidez valorable de los suelos.
14. Sodio por fotometría de llama.
Anejo XII. Vinos
3 (a). Color.
51. Presión del anhídrido carbónico.
52. Antisépticos y antifermentos (método microbiológico).
Vinagre
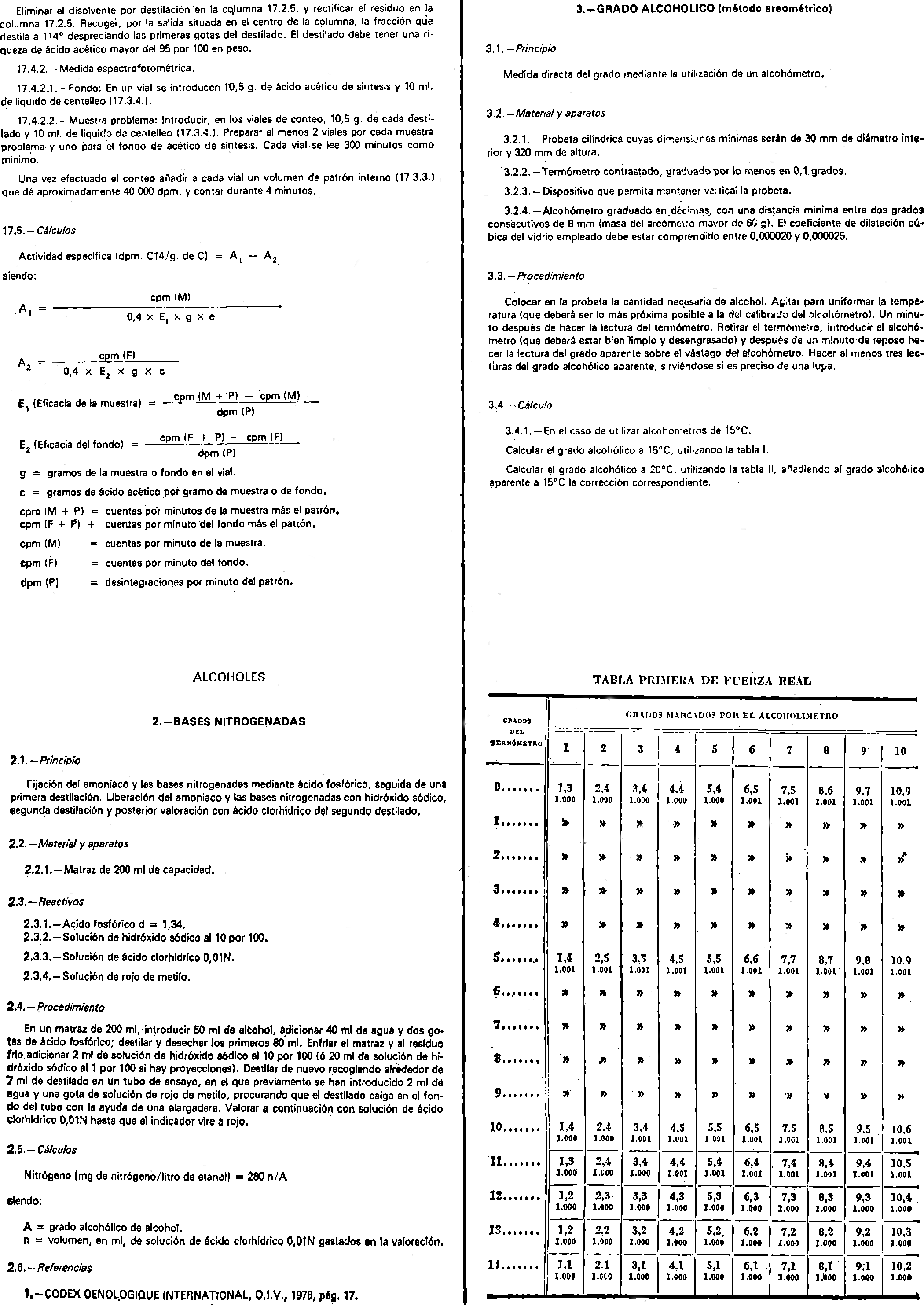
17. Acido acético de síntesis.
Alcoholes
2. Bases nitrogenadas.
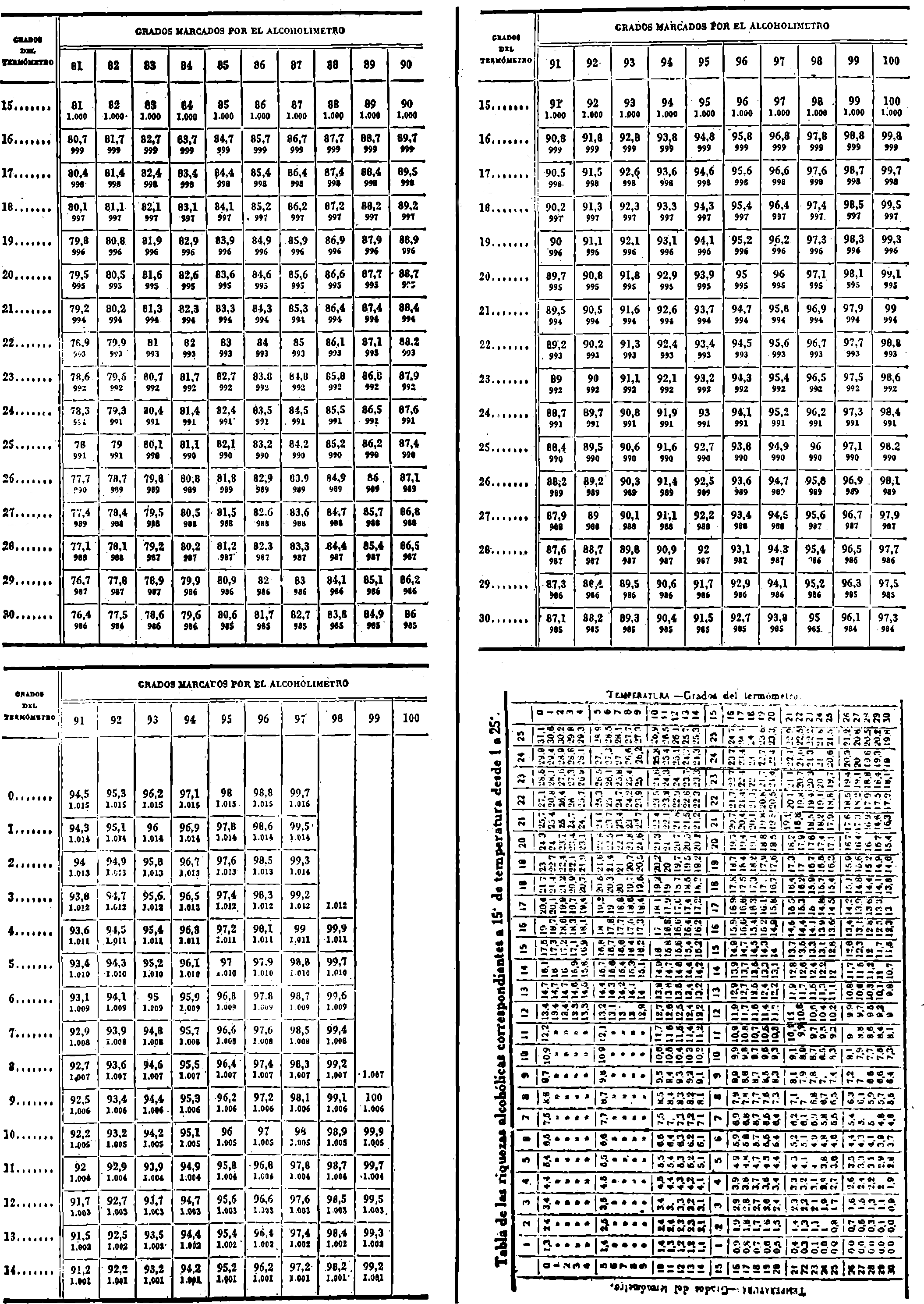
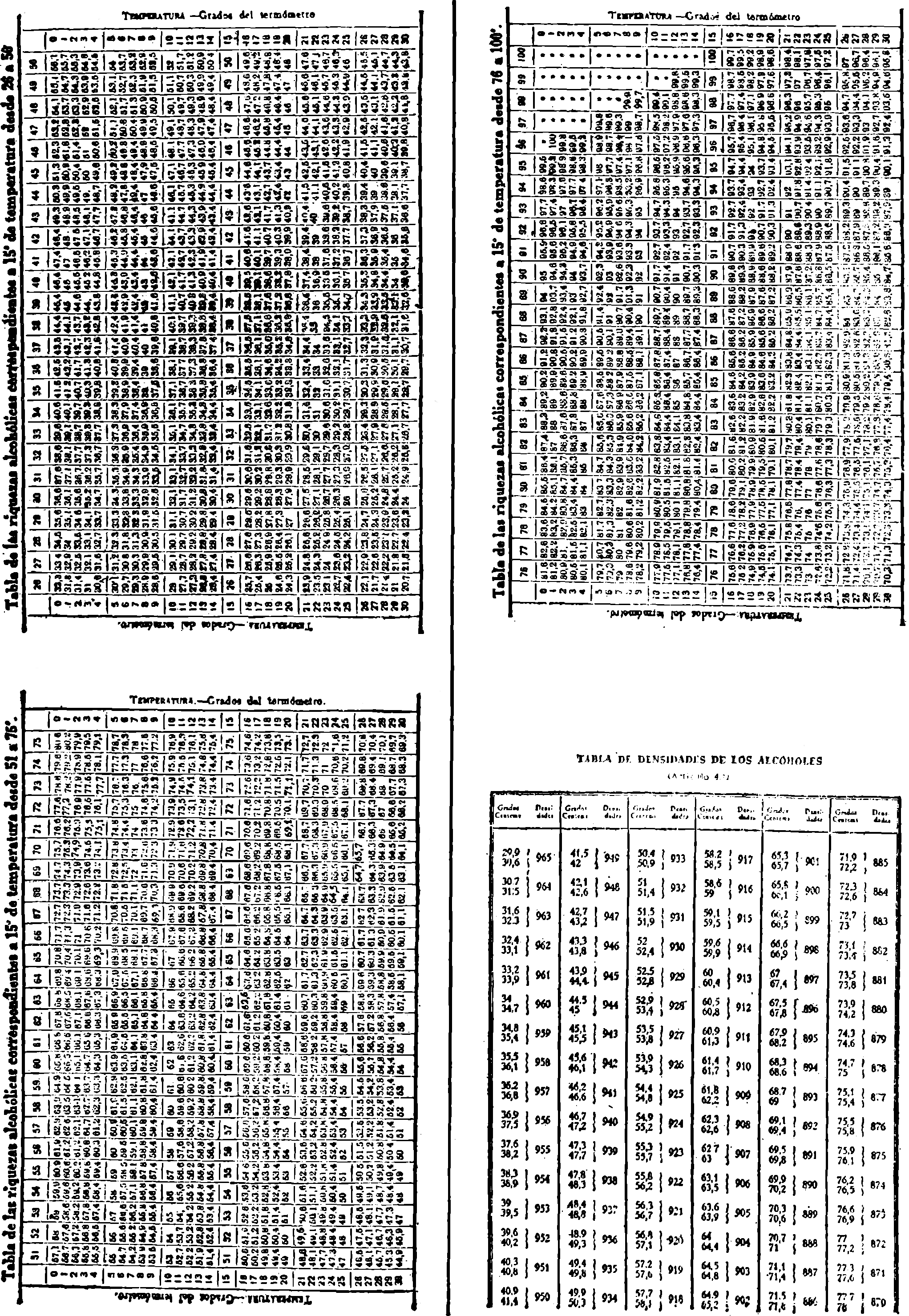
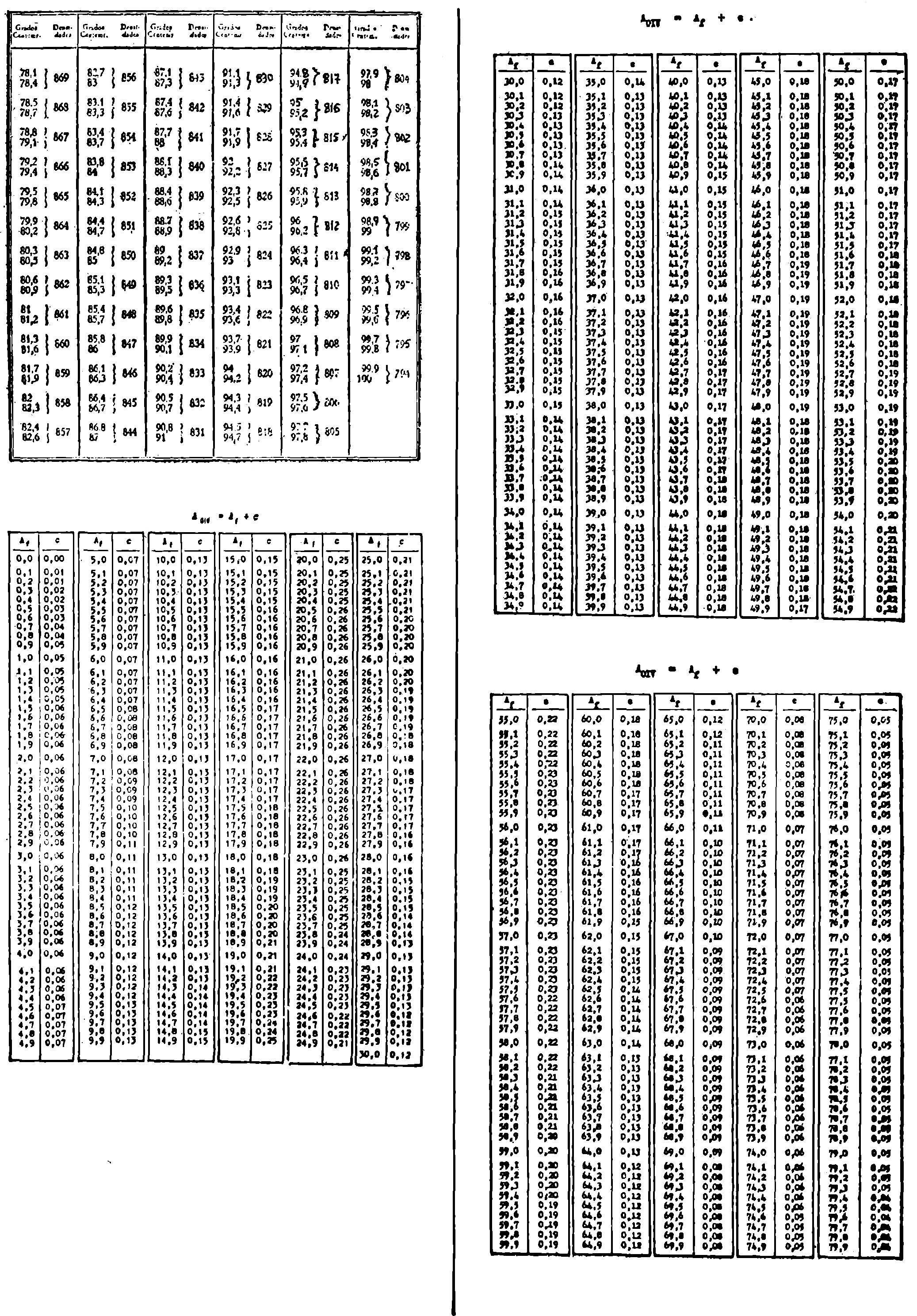
3. Grado alcohólico (método areométrico).
Anejo XII. Toma de muestras
Toma de muestras de suelos.
Anejo A. Toma de muestras de fertilidad.
Anejo B. Toma de muestras para prospecciones edafológicas.
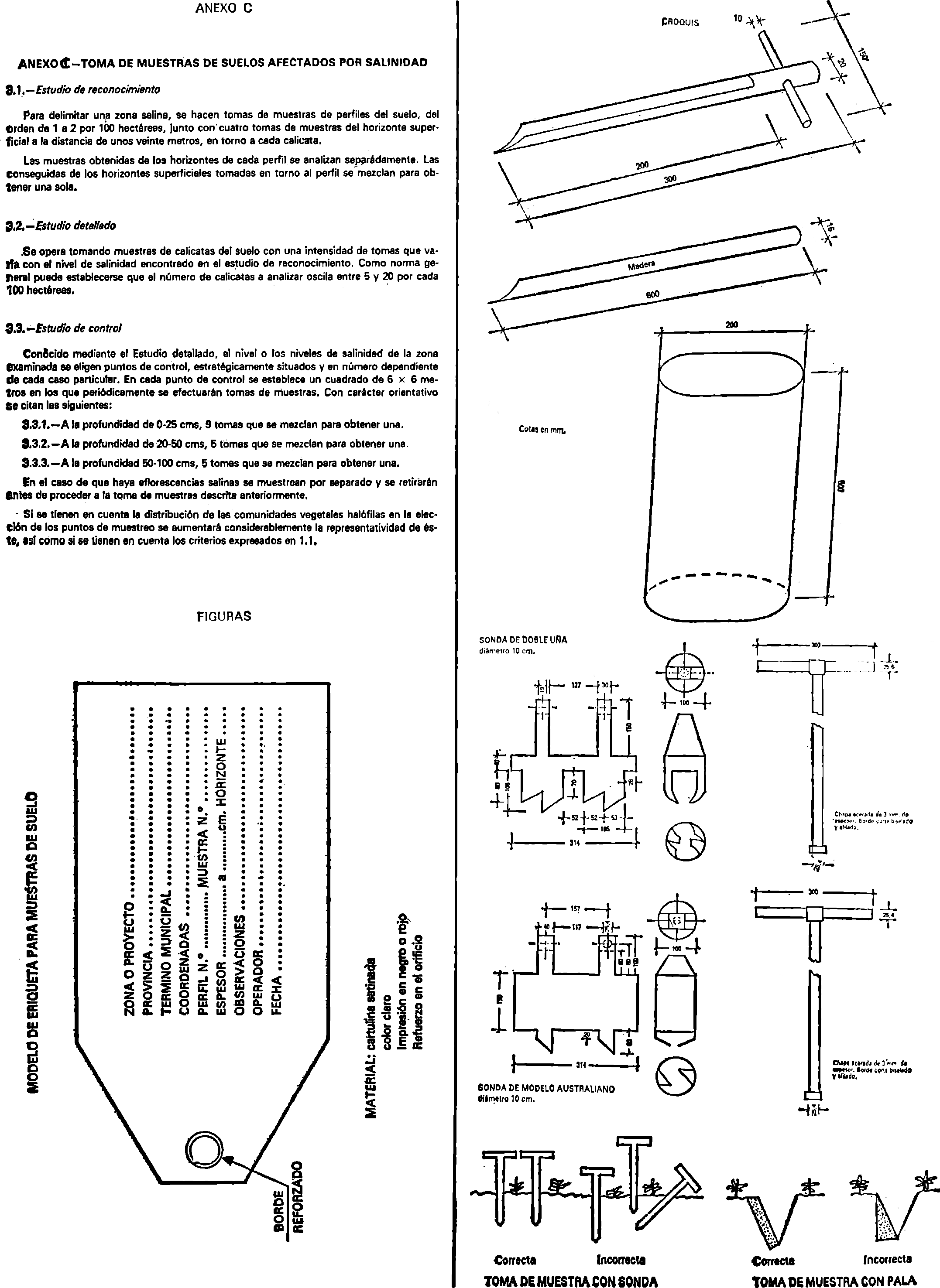
Anejo C. Toma de muestras de suelos afectados por salinidad.
























Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid




