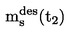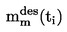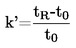Axencia Estatal Boletín Oficial do Estado






Contido non dispoñible en galego
LA COMISIÓN DE LAS COMUNIDADES EUROPEAS,
Visto el Tratado constitutivo de la Comunidad Europea,
Visto el Reglamento (CE) no 1907/2006 del Parlamento Europeo y del Consejo, de 18 de diciembre de 2006, relativo al registro, la evaluación, la autorización y la restricción de las sustancias y preparados químicos (REACH), por el que se crea la Agencia Europea de Sustancias y Preparados Químicos, se modifica la Directiva 1999/45/CE y se derogan el Reglamento (CEE) no 793/93 del Consejo y el Reglamento (CE) no 1488/94 de la Comisión así como la Directiva 76/769/CEE del Consejo y las Directivas 91/155/CEE, 93/67/CEE, 93/105/CE y 2000/21/CE de la Comisión (1), y, en particular, su artículo 13, apartado 3,
Considerando lo siguiente:
(1) De acuerdo con el Reglamento (CE) no 1907/2006, han de adoptarse a nivel comunitario métodos aplicables a los ensayos que sea necesario hacer con sustancias para obtener información sobre las propiedades intrínsecas de dichas sustancias.
(2) La Directiva 67/548/CEE del Consejo, de 27 de junio de 1967, relativa a la aproximación de las disposiciones legales, reglamentarias y administrativas en materia de clasificación, embalaje y etiquetado de las sustancias peligrosas (2), establece en su anexo V métodos de ensayo para la determinación de las propiedades fisicoquímicas, toxicológicas y ecotoxicológicas de las sustancias y preparados. El anexo V de la Directiva 67/548/CEE ha sido suprimido por la Directiva 2006/121/CE del Parlamento Europeo y del Consejo, con efecto a partir del 1 de junio de 2008.
(3) Los métodos de ensayo incluidos en el anexo V de la Directiva 67/548/CEE deben incorporarse al presente Reglamento.
(4) El presente Reglamento no excluye el uso de otros métodos de ensayo, siempre que su uso se ajuste a lo establecido en el artículo 13, apartado 3, del Reglamento (CE) no 1907/2006.
(5) Al elaborar los métodos de ensayo, se han de tener plenamente en cuenta los principios de sustitución, reducción y perfeccionamiento de la experimentación con animales, sobre todo cuando se llegue a disponer de métodos validados adecuados para sustituir, reducir o perfeccionar los ensayos con animales.
(6) Las disposiciones del presente Reglamento se ajustan al dictamen del Comité establecido de conformidad con el artículo 133 del Reglamento (CE) no 1907/2006.
HA ADOPTADO EL PRESENTE REGLAMENTO:
Los métodos de ensayo que deben aplicarse a efectos del Reglamento (CE) no 1907/2006 se recogen en el anexo del presente Reglamento.
La Comisión revisará, cuando proceda, los métodos de ensayo incluidos en el presente Reglamento con el fin de sustituir, reducir o perfeccionar los ensayos con animales vertebrados.
Todas las referencias al anexo V de la Directiva 67/548/CEE se entenderán hechas al presente Reglamento.
El presente Reglamento entrará en vigor el día siguiente al de su publicación en el Diario Oficial de la Unión Europea.
Será aplicable a partir del 1 de junio de 2008.
Hecho en Bruselas, el 30 de mayo de 2008.
Por la Comisión
Stavros DIMAS
Miembro de la Comisión
(1) DO L 396 de 30.12.2006, p. 1; versión corregida en el DO L 136 de 29.5.2007, p. 3.
(2) DO 196 de 16.8.1967, p. 1. Directiva modificada en último lugar por la Directiva 2006/121/CE del Parlamento Europeo y del Consejo (DO L 396 de 30.12.2006, p. 853; versión corregida en el DO L 136 de 29.5.2007, p. 281).
IMÁGENES Y FÓRMULAS OMITIDAS
PARTE A: MÉTODOS PARA LA DETERMINACIÓN DE LAS PROPIEDADES FISICOQUÍMICAS
ÍNDICE
|
A.1. |
PUNTO DE FUSIÓN/CONGELACIÓN |
|
A.2. |
PUNTO DE EBULLICIÓN |
|
A3. |
DENSIDAD RELATIVA |
|
A.4. |
PRESIÓN DE VAPOR |
|
A.5. |
TENSIÓN SUPERFICIAL |
|
A.6. |
HIDROSOLUBILIDAD |
|
A.8. |
COEFICIENTE DE REPARTO |
|
A.9. |
PUNTO DE INFLAMACIÓN |
|
A.10. |
INFLAMABILIDAD (SÓLIDOS) |
|
A.11. |
INFLAMABILIDAD (GASES) |
|
A.12. |
INFLAMABILIDAD (EN CONTACTO CON EL AGUA) |
|
A.13. |
PROPIEDADES PIROFÓRICAS DE SÓLIDOS Y LÍQUIDOS |
|
A.14. |
PROPIEDADES EXPLOSIVAS |
|
A.15. |
TEMPERATURA DE AUTOINFLAMACIÓN (LÍQUIDOS Y GASES) |
|
A.16. |
TEMPERATURA RELATIVA DE AUTOINFLAMACIÓN DE SÓLIDOS |
|
A.17. |
PROPIEDADES COMBURANTES (SÓLIDOS) |
|
A.18. |
PESO MOLECULAR MEDIO EN NÚMERO Y DISTRIBUCIÓN DE LOS PESOS MOLECULARES DE LOS POLÍMEROS |
|
A.19. |
CONTENIDO DE SUSTANCIAS DE BAJO PESO MOLECULAR EN LOS POLÍMEROS |
|
A.20. |
COMPORTAMIENTO DE DISOLUCIÓN/EXTRACCIÓN |
|
A.21. |
PROPIEDADES COMBURENTES (LÍQUIDOS) |
---
A.1. PUNTO DE FUSIÓN/CONGELACIÓN
1. MÉTODO
La mayoría de los métodos descritos se basan en las líneas directrices de la OCDE (1). Los principios fundamentales se dan en las referencias (2) y {3).
1.1. INTRODUCCIÓN
Los métodos y los aparatos descritos a continuación se utilizan para determinar el punto de fusión de los productos químicos, cualquiera que sea su grado de pureza.
La selección del método dependerá de la naturaleza de la sustancia problema. En consecuencia, el factor limitante dependerá de que la sustancia sea fácil, difícilmente o no pulverizable.
En determinadas sustancias será preferible determinar el punto de congelación o de solidificación; las normas para proceder a dichas determinaciones también se recogen en el presente método.
Cuando, debido a propiedades especiales de la sustancia, no pueda medirse convenientemente ninguno de los parámetros mencionados, puede ser adecuado medir el punto de fluidez.
1.2. DEFINICIONES Y UNIDADES
El punto de fusión se define como la temperatura a la que se produce la transición de fase del estado sólido al líquido a presión atmosférica normal; esta temperatura corresponde idealmente a la temperatura de congelación.
Dado que la transición de fase de numerosas sustancias se extiende en una amplia gama de temperaturas, esta se designa muchas veces con el nombre de intervalo de fusión.
Conversión de las unidades (K a oC).
t = T - 273,15
|
t |
= |
temperatura Celsius, grado Celsius ( oC) |
|
T |
= |
temperatura termodinámica, keivin (K) |
1.3. SUSTANCIAS DE REFERENCIA
No es necesario emplear sustancias de referencia cada vez que se estudie una nueva sustancia. Deberán servir, esencialmente, para comprobar el funcionamiento del método de vez en cuando y para comparar con los resultados obtenidos mediante otros métodos.
En la referencia (4) se enumeran determinadas sustancias de calibración.
1.4. PRINCIPIO DEL MÉTODO DE ENSAYO
Se determina la temperatura {o intervalo de temperatura) de transición de fase del estado sólido al liquido o viceversa. En la práctica, las temperaturas del inicio y del final del proceso de fusión/congelación se determinan al calentar/enfriar una muestra de la sustancia problema a presión atmosférica. Se describen cinco tipos de métodos: el método de tubo capilar, el método de superficie caliente, determinación de la temperatura de congelación, métodos de análisis térmico y determinación del punto de fluidez (como el método elaborado para derivados del petróleo).
En algunos casos puede ser conveniente medir la temperatura de congelación en lugar de la temperatura de fusión.
1.4.1. Método de tubo capilar
1.4.1.1. Dispositivos de temperatura de fusión con baño líquido
Introducir en un tubo capilar una pequeña cantidad de sustancia finamente pulverizada y comprimirla firmemente. Calentar dicho tubo al mismo tiempo que un termómetro y ajustar el aumento de temperatura a poco menos de 1 K por minuto, durante la fusión real. Tomar nota de las temperaturas correspondientes al comienzo y al final de la fusión.
1.4.1.2. Dispositivos de temperatura de fusión con bloque metálico
El fundamento es el mismo que el descrito en el punto 1.4.1.1, con la diferencia de que el tubo capilar y el termómetro están colocados en un bloque de metal calentado y se observan a través de aberturas practicadas en este último.
1.4.1.3. Detección fotoeléctrica
Calentar automáticamente en un cilindro metálico la muestra contenida en el tubo capilar. Por una abertura practicada en el cilindro, enviar un rayo de luz a través de la sustancia hacia una célula fotoeléctrica cuidadosamente calibrada. En el momento de la fusión, las propiedades ópticas de la mayor parte de las sustancias se modifican en el sentido de que la opacidad da paso a la transparencia. En consecuencia, la intensidad de la luz que llega a la célula fotoeléctrica aumenta y envía una señal de parada al indicador digital que registra la temperatura del termómetro de resistencia de platino colocado en la cámara de calentamiento. Este método no es aplicable a determinadas sustancias muy coloreadas.
1.4.2. Método de superficie caliente
1.4.2.1. Método de la placa caliente de Kofler
La placa caliente de Kofler se compone de dos piezas de metal de conductividad térmica diferente, que se calientan eléctricamente. Está hecha de manera que el gradiente de temperatura sea casi lineal en toda su longitud. La temperatura de dicha placa puede variar de 283 a 573 K gracias a un dispositivo especial de lectura de la temperatura que tiene un cursor con un índice y una regleta graduada, especialmente concebido para dicha placa. Para determinar un punto de fusión se deposita una fina capa de sustancia directamente sobre la placa caliente. En unos segundos, se forma una fina línea de división entre la fase fluida y la fase sólida. Leer la temperatura a la altura de dicha línea, colocando el índice frente a esta última.
1.4.2.2. Microscopio de fusión
Se utilizan diferentes microscopios de platina caliente para determinar puntos de fusión con cantidades de sustancia muy pequeñas. La temperatura se suele medir con un termopar sensible, pero a veces se usa un termómetro de mercurio. El dispositivo tipo tiene una carcasa de calor que contiene una platina de metal en la que se coloca una lámina de vidrio sobre la que se deposita la muestra. El centro de la platina metálica se atraviesa con un agujero que permite el paso de la luz procedente del espejo de iluminación del microscopio. Al utilizarlo, la carcasa se cierra con una placa de vidrio para impedir la entrada de aire a la zona de la
El calentamiento de la muestra se regula con un reostato. Para realizar mediciones muy precisas se puede utilizar luz polarizada en el análisis de las sustancias ópticamente anisótropas.
1.4.2.3. Método de menisco
Este método se aplica específicamente a las poliamidas.
Se determina la temperatura a la cual se observa, a simple vista, el desplazamiento de un menisco de aceite de silicona, atrapado entre una superficie caliente y un cubreobjetos colocado encima de la muestra de poliamida.
1.4.3. Método de determinación del punto de congelación
Introducir la muestra en un tubo de ensayo especial y colocarlo en un aparato que permita la determinación del punto de congelación. Agitar suavemente la muestra sin interrupción durante el enfriamiento, observando al mismo tiempo la temperatura y registrándola a intervalos adecuados. Cuando varias lecturas indiquen una temperatura constante (previa corrección termométrica), se considera el valor de esta temperatura como el punto de congelación.
Debe evitarse el sobreenfriamiento manteniendo el equilibrio entre las fases sólida y líquida.
1.4.4. Análisis térmico
1.4.4.1. Análisis térmico diferencial (ATD)
Esta técnica registra la diferencia de temperaturas entre ¡a sustancia y un material de referencia en función de la temperatura, cuando ¡a sustancia y el material de referencia se someten al mismo programa de temperatura controlada. Cuando la muestra sufre una transición que suponga un cambio de entalpia, ese cambio se indicará por una desviación endotérmica (fusión) o exotérmica (congelación) de la línea de base del registro de temperatura.
1.4.4.2. Calorimetría diferencial de barrido (CDB)
Esta técnica registra la diferencia de apone energético a una sustancia y a un material de referencia en función de la temperatura, cuando la sustancia y el material de referencia se someten al mismo programa de temperatura controlada. Esta energía es la energía necesaria para establecer una diferencia de temperatura nula entre la sustancia y el material de referencia. Cuando la muestra sufre una transición que suponga un cambio de entalpia, ese cambio se indicará por una desviación endotérmica (fusión) o exotérmica (congelación) de la línea de base del registro del flujo de calor.
1.4.5. Punto de fluidez
Este método, desarrollado para los derivados del petróleo, es adecuado para utilizarse con sustancias oleosas de baja temperatura de fusión.
Tras un calentamiento previo, se va enfriando la muestra a una velocidad específica y se examinan sus características Teológicas a intervalos de 3 K. Se registra como punto de fluidez la temperatura mínima a la que se aprecia movimiento de la sustancia.
1.5. CRITERIOS DE CALIDAD
En el cuadro siguiente se indican las condiciones de aplicación y la precisión de los diferentes métodos de determinación del punto de fusión/intervalo de fusión.
CUADRO: APLICABILIDAD DE LOS MÉTODOS
A. Método de tubo capilar
|
Método de medidas |
Sustancias pulverizablesSí |
Sustancias difícilmente pulverizables |
Gama de temperatura |
Precisión estimada (1) |
Norma existente |
|
Dispositivo de temperatura de fusión con baño liquido |
Sí |
Solo algunas |
De 273 a 573 K |
±0,3 K |
JIS K 0064 |
|
Dispositivo de temperatura de fusión con un bloque metálico |
Sí |
Solo algunas |
De 293 a > 573 K |
±0,5 K |
ISO 1218(E) |
|
Detección fotoeléctrica |
Sí |
Varias con dispositivos de aplicación |
De 253 a 573 K |
±0,5 K |
|
B. Métodos de superficie caliente y de congelación
|
Método de medidas |
Sustancias pulverizables |
Sustancias difícilmente pulverizables |
Gama de temperatura |
Precisión estimada (2) |
Norma existente |
|
Placa caliente Kofler |
Sí |
No |
De 283 a > 573K |
±1,0 K |
ANSI/ASTM D 345176 |
|
Microscopio de fusion |
Sí |
Solo algunas |
De 273 a > 573K |
±0,5 K |
DIN 53736 |
|
Método de menisco |
No |
Específico de las poliamidas |
De 293 a > 573K |
±0,5 K |
ISO 1218 (E) |
|
Métodos de punto de congelación |
Sí |
Sí |
De 223 a 573 K |
±0,5 K |
por ejemplo BS 4695 |
C. Análisis térmico
|
Método de medidas |
Sustancias pulverizables |
Sustancias difícilmente pulverizables |
Gama de temperatura |
Precisión estimada (3) |
Norma existence |
|
Análisis térmico diferenctal |
Sí |
Sí |
De 173 a 1 273 K |
hasta 600 K ±0,5 K hasta 1 273 K ±2,0 K |
ASTME 53776 |
|
Calorimetría diferencial de barrido |
Sí |
Sí |
De 173 a 1 273 K |
hasta 600 K ±0,5 K hasta 1 273 K ±2,0 K |
ASTME 53776 |
D. Punto de fluidez
|
Método de medidas |
Sustancias pulverizables |
Sustancias difícilmente pulverizables |
Gama de temperatura |
Precisión estimada (4) |
Norma existente |
|
Punto de fluidez |
Para derivados del petróleo y sustancias oleosas |
Para derivados del petróleo y sustancias oleosas |
Pe 223 a 323 K |
±3,0 K |
ASTM D 9766 |
1.6. DESCRIPCIÓN DE LOS MÉTODOS
Los procedimientos de casi todos los métodos de ensayo están descritos en normas internacionales y nacionales (véase el apéndice 1).
1.6.1. Métodos de tubo capilar
Cuando la elevación de temperatura es lenta, las sustancias finamente pulverizadas pasarán, normalmente, por las fases de fusión representadas en la figura 1:
Figura I
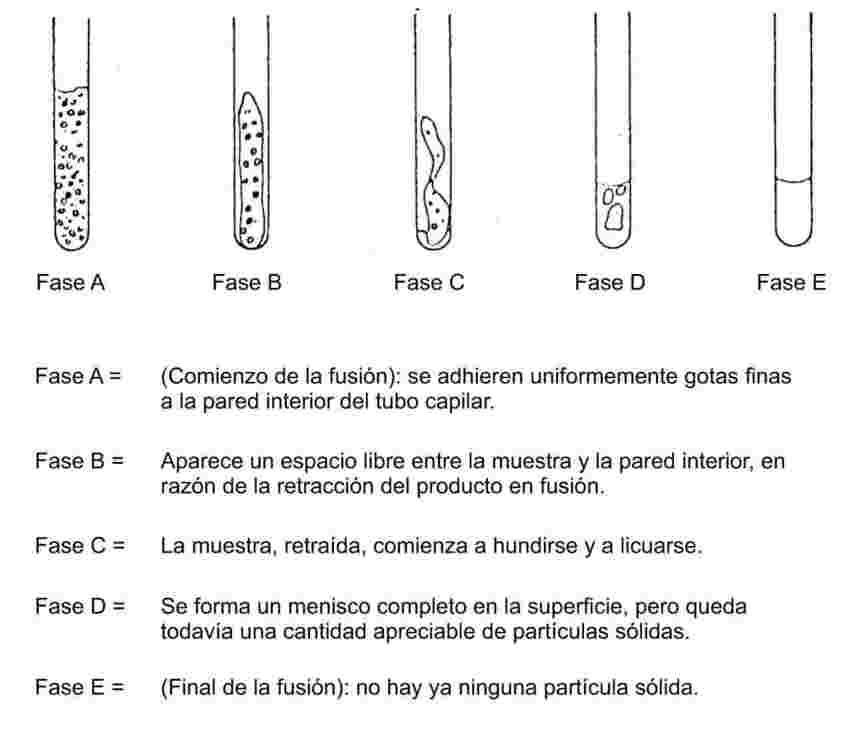
Durante la determinación del punto de fusión, conviene anotar la temperatura al comienzo y al final de la fusión.
1.6.1.1. Dispositivos con baña líquido
La figura 2 representa un tipo de aparato estandarizado de vidrio (JIS K 0064). Todas las dimensiones están expresadas en milímetros.
Figura 2

Baño líquido
El líquido deberá elegirse en función del punto de fusión que deba determinarse; por ejemplo, se utilizará parafina líquida para los puntos de fusión que no pasen de 473 K, aceite de silicona para los puntos de fusión que no pasen de 573 K.
Podrá utilizarse una mezcla de tres partes de ácido sulfúrico y dos partes de sulfato de potasio (en peso) para los puntos de fusión superiores a 523 K. Deben adoptarse precauciones adecuadas cuando se utilice este tipo de mezcla.
Termómetro
Solo podrán utilizarse termómetros que cumplan las exigencias de las normas siguientes o de sus equivalentes:
ASTM E 1-71, DIN 12770, JIS K 8001.
Procedimiento
Pulverizar finamente la sustancia seca en un mortero e introducirla en un tubo capilar cerrado en un extremo. La altura del contenido será de unos 3 mm después de haberlo comprimido bien. Para obtener una muestra uniformemente comprimida, hay que dejar caer el tubo capilar desde una altura de unos 700 mm por el interior de un tubo de vidrio colocado verticalmente sobre un vidrio de reloj.
Colocar el tubo capilar, así llenado, en el baño de tal manera que la parte central del bulbo de mercurio del termómetro esté en contacto con la parte del capilar que contiene la muestra. En general, el tubo capilar se introduce en el aparato en el momento en que el baño está a unos 10 K por debajo del punto de fusión.
Regular el calentamiento del baño de manera que el aumento de la temperatura sea de unos 3 K/minuto. Agitar el líquido. Al llegar a unos 10 K por debajo de la temperatura de fusión esperada, regular el aumento de temperatura a un máximo de 1 K/minuto.
Cálculo
El cálculo del punto de fusión se efectúa mediante la fórmula siguiente;
T = TD+0,00016(TD — TE) n
donde
|
T |
= |
temperatura de fusión corregida, expresada en K |
|
TD |
= |
temperatura leída en el termómetro D, expresada en K |
|
TE |
= |
temperatura leída en el termómetro E, expresada en K |
|
n |
= |
número de graduaciones de la columna de mercurio del termómetro D en el vástago emergente. |
1.6.1.2. Dispositiva con bloque metálico
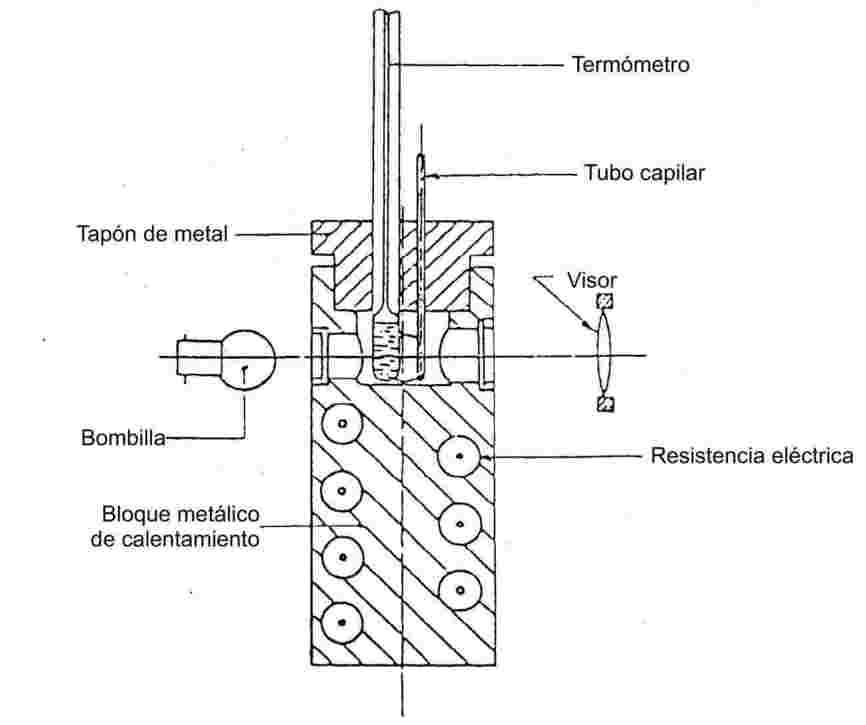
Aparato:
El aparato consta de:
|
— |
un bloque metálico cilíndrico cuya parte superior está vacía y forma un recinto de calentamiento (véase la figura 3), |
|
— |
un tapón metálico atravesado por dos o más agujeros que permitan la introducción de los tubos en el bloque, |
|
— |
un sistema de calentamiento del bloque metálico; por ejemplo, una resistencia eléctrica incorporada al bloque, |
|
— |
un reostato para regular la potencia, si se utiliza calentamiento eléctrico, |
|
— |
cuatro ventanas de vidrio resistente al calor, diametralmente opuestas, en ángulo recto, en las paredes laterales del recinto. Frente a una de dichas ventanas se instalará un visor para observar el tubo capilar. Las otras tres ventanas permitirán iluminar el interior del recinto mediante bombillas, |
|
— |
un tubo capilar, de vidrio resistente al calor, cerrado en un extremo (véase el punto 1.6.1.1). |
Termómetro
Véanse las normas citadas en el punto 1.6.1.1. También podrán utilizarse elementos termoeléctricos de una precisión equivalente.
Figura 3
1.6.1.3. Detección fotoeléctrica
Aparato y procedimiento:
El aparato consiste en un recinto metálico dotado de un sistema de calentamiento automático. Se llenan tres tubos capilares siguiendo las instrucciones del punto 1.6.1.1 y se colocan en el horno.
Se pueden hacer varios aumentos lineales de temperatura para calibrar el aparato. El aumento de temperatura apropiado se regula eléctricamente con una velocidad constante y lineal preseleccionada. Los aparatos registradores indican la temperatura real del horno y la temperatura de la sustancia en los tubos capilares.
1.6.2. Métodos de superficie caliente
1.6.2.1. Placa caliente Kofler
Véase el apéndice.
1.6.2.2. Microscopio de fusión
Véase el apéndice.
1.6.2.3. Método de menisco (poliamidas)
Véase el apéndice.
El aumento de temperatura en la zona del punto de fusión deberá ser inferior a 1 K/min,
1.6.3. Métodos de determinación del punto de congelación
Véase el apéndice.
1.6.4. Análisis térmico
1.6.4.1. Análisis térmico diferencial
Véase el apéndice.
1.6.4.2. Calorimetría diferencial de barrido
Véase el apéndice.
1.6.5. Determinación del punto de fluidez
Véase el apéndice.
2. RESULTADOS
En determinados casos es necesaria la corrección del termómetro,
3. INFORME
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
método utilizado, |
|
— |
especificación precisa de la sustancia (identidad e impurezas) y, en su caso, fase de purificación previa, |
|
— |
estimación de la precisión. |
El punto de fusión indicado en el informe será la media entre dos mediciones, como mínimo, situadas en la zona de la precisión estimada (véase el cuadro).
Si la diferencia entre las temperaturas al comienzo y al final de la fusión se encuentra dentro de los límites de precisión del método, la temperatura leída en la fase final de la fusión se considerará el punto de fusión; de lo contrario, se indicarán las dos temperaturas.
Si la sustancia se descompone o se sublima antes de alcanzar el punto de fusión, se indicará la temperatura a la que se observa el efecto.
Deben indicarse todas las informaciones y observaciones que se consideren útiles para la interpretación de los resultados, en particular lo referente a las impurezas y al estado físico de la sustancia.
4. BIBLIOGRAFÍA
|
(1) |
OCDE, París, 1981, Test Guideline 102, Decision of the Council C (81) 30 Final. |
|
(2) |
IUPAC, B. Le Neindre, B. Vodar, eds. Experimental thermodynamics., Butterworths, Londres, 1975, vol. II, 803-834. |
|
(3) |
R. Weissberger ed.: Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd. ed., Interscience Publ., Nueva York, 1959, Vol I, Part I, Chapter VII. |
|
(4) |
IUPAC, Physicochemical Measurements: Catalogue of Reference Materials from National Laboratories, Pure and Applied Chemistry, 1976, vol. 48, 505-515. |
Apéndice
Para más detalles técnicos, se pueden consultar, por ejemplo, las normas siguientes:
1. Métodos de tubo capilar
1.1. Dispositivos con baño líquido
|
ASTM E 324-69 |
Standard Test Method for Relative Initial and Final Melting Points and the Melting Range of Organic Chemicals |
|
BS 4634 |
Method for the Determination of Melting Point and/or Melting Range |
|
DIN 53181 |
Bestimmung des Schmelzintervalles von Harzen nach Kapillarverfahren |
|
J1S K 00-64 |
Testing Methods for Melting Point of Chemical Products |
1.2. Dispositivos con bloque metálico
|
DIN 53736 |
Visuelle Bestimmung der Schmelztemperatur von teilkristalimen Kunststoffen |
|
ISO 1218 (E) |
Plastics — polyamides — determination of «melting point» |
2. Métodos de superficie caliente
2.1. Placa caliente de Kofler
|
ANSI/ASTM D 3451-76 |
Standard Recommended Practices for Testing Polymeric Powder Coatings |
2.2. Microscopio de fusión
|
DIN 53736 |
Visuelle Bestimmung der Schmelztemperatur von teilkristaliinen Kunststoffen |
2.3. Método de menisco (poliamidas)
|
ISO 1218 (E) |
Plastics-Polyamides-Decermination of «Melting- Point» |
|
ANSI/ASTM D 2133-66 |
Standard Specification for Acetal Resin Injection Moulding and Extrusion Materials |
|
NF T 51050 |
Resines de polyamides. Determination du «point de fusion-. Méthode du ménisque» |
3. Métodos de determinación del punto de congelación
|
BS 4633 |
Method for the Determination of Crystallizing Point |
|
BS 4695 |
Method for Determination of Melting Point of Petroleum Wax (Cooling Curve) |
|
DIN 51421 |
Besrimmung des Gefrierpunktes von Flugkraftsroffen, Ottokraftstoffen und Motorenbenzolen |
|
ISO 2207 |
Cires de pétrole: determination de la temperature de figeage |
|
DIN 53175 |
Besrimmung des Erstarrungspunktes von Fettsauren |
|
NF T 60-114 |
Point de fusion des paraffines |
|
NF T 20-051 |
Méthode de détermination du point de cristalisation (point de congelation) |
|
ISO 1392 |
Method for the determination of the freezing point |
4. Análisis térmico
4.1. Análisis térmico diferencial
|
ASTM E 537-76 |
Standard method for assessing the thermal stability of chemicals by methods of, differential thermal analysis |
|
ASTM E 473-85 |
Standard definitions of terms relating to thermal analysis |
|
ASTM E 472-86 |
Standard practice for reporting thermoanalyrical data |
|
DIN 51005 |
Thermische Analyse, Begriffe |
4.2. Calorimetría diferencial de barrido
|
ASTM E 537-76 |
Standard method for assessing the thermal stability of chemicals by methods of differential thermal analysis |
|
ASTM E 473-85 |
Standard definitions of terms relating to thermal analysis |
|
ASTM E 472-86 |
Standard practice for reporting thermoanalytical data |
|
DIN 51005 |
Thermische Analyse, Begriffe |
5. Determinación del punto de fusión
|
NBN 52014 |
Echantillonnage et analyse des produits du pétrole: Point de trouble et point d'écoulement limite — Monsterneming en ontieding van aardolieproducten: Troebelingspunt en vloeipunt |
|
ASTM D 97-66 |
Standard test method for pour point of petroleum oils |
|
ISO 3016 |
Petroleum oils — Determination of pour point. |
A.2. PUNTO DE EBULLICIÓN
1. MÉTODO
La mayoría de los métodos descritos se basan en las líneas directrices de la OCDE (1). Los principios fundamentales se dan en las referencias (2) y (3).
1.1. INTRODUCCIÓN
Los métodos y dispositivos aquí descritos pueden aplicarse a las sustancias líquidas y de bajo punto de fusión que no sufran reacción química por debajo del punto de ebullición (por ejemplo, autooxidación, redistribución, degradación, etc.). Los métodos son aplicables a las sustancias líquidas puras e impuras.
La importancia dada a la descripción de los métodos basados en la detección fotoeléctrica y en el análisis térmico se debe al hecho de que estos métodos permiten determinar no solo el punto de fusión sino también el punto de ebullición. Además, las medidas pueden efectuarse de manera automática.
El «método dinámico» tiene la ventaja de poder utilizarse igualmente para la determinación de la presión de vapor y hacer innecesaria la corrección de la temperatura de ebullición para llevarla a las condiciones normales de presión (101,325 kPa), ya que durante la medición se puede ajustar la presión normal mediante un manostato.
Observaciones
La influencia de las impurezas en la determinación del punto de ebullición depende mucho de la naturaleza de la impureza. Si en la muestra hay impurezas volátiles, que pudieran afectar a los resultados, puede procederse a purificar la sustancia.
1.2. DEFINICIONES Y UNIDADES
La temperatura de ebullición normal se define como la temperatura en que la presión de vapor de un líquido es 101,325 kPa.
Si la temperatura de ebullición no se mide a la presión atmosférica normal, la dependencia de la presión de vapor respecto a la temperatura puede calcularse cuantitativamente por la ecuación de Clausius-Clapeyron:

donde
|
p |
= |
presión de vapor de la sustancia en pascales |
|
A Hv |
= |
su calor de vaporización en J mol-1 |
|
R |
= |
constante universal de los gases = 8,314 J mol-1 K-1 |
|
T |
= |
temperatura termodinámica, expresada en K. |
La temperatura de ebullición se establece con relación a la presión ambiente en el momento de la medición.
Conversiones
Presión (unidad: kPa)
|
100 kPa |
= |
1 bar = 0,1 MPa (todavía se permite la unidad «bar» pero no es recomendable). |
|
133 Pa |
= |
1 mm Hg 1 Torr (no se permite la unidad «Torr» ni «mm Hg»). |
|
1 atm |
= |
atmósfera normal = 101 325 Pa (no se permite la unidad «atm»). |
Temperatura (unidad: K)
t = T - 273,15
|
t |
: |
temperatura Celsius, grado Celsius ( oC) |
|
T |
: |
temperatura termodinámica, Kelvin (K) |
1.3. SUSTANCIAS DE REFERENCIA
No es necesario utilizar sustancias de referencia cada vez que se estudie una nueva sustancia. Dichas sustancias de referencia sirven esencialmente para comprobar la validez del método de vez en cuando y para poder comparar con los resultados según otros métodos.
En los métodos enumerados en el apéndice figuran algunas sustancias de calibración.
1.4. PRINCIPIO DEL MÉTODO DE ENSAYO
Cinco métodos de determinación del punto de ebullición (o del intervalo de ebullición) se basan en la medición de la temperatura de ebullición; otros dos se basan en el análisis térmico.
1.4.1. Método del ebullómetro
Aunque en un principio los ebullómetros se pensaron para determinar el peso molecular por elevación del punto de ebullición, se prestan también para realizar mediciones exactas del punto de ebullición. En la norma ASTM D 1120-72 (véase el apéndice), se describe un aparato muy sencillo. En dicho aparato, el líquido se calienta en condiciones de equilibrio a la presión atmosférica hasta ebullición.
1.4.2. Método dinámico
Este método se basa en la medición de la temperatura de recondensación del vapor mediante un termómetro adecuado que se coloca en el reflujo durante la ebullición. En este método puede modificarse la presión.
1.4.3. Método de destilación para el punto de ebullición
Este método se basa en la destilación del líquido, la medida de la temperatura de recondensación del vapor y la determinación de la cantidad de destilado.
1.4.4. Método de Siwoloboff
Se calienta una muestra en un tubo de ensayo, que se sumerge en un baño caliente. Se introduce en el tubo de ensayo un capilar cerrado, con una burbuja de aire en su parte inferior.
1.4.5. Detección fotoeléctrica
De acuerdo con el principio de Siwoloboff, la ascensión de las burbujas permite una medición fotoeléctrica automática.
1.4.6. Análisis térmico diferencial
Esta técnica registra la diferencia de temperatura entre la sustancia y un material de referencia en función de la temperatura, cuando la sustancia y el material de referencia se someten al mismo programa de temperatura controlada. Cuando la muestra sufre una transición que implique un cambio de entalpia, ese cambio se indica con una desviación endotérmica (ebullición) respecto a la línea base del registro de temperatura.
1.4.7. Calorimetría diferencial de barrido
Esta técnica registra la diferencia de aporte energético a una sustancia y a un material de referencia en función de la temperatura, cuando la sustancia y el material de referencia se someten al mismo programa de temperatura controlada. Esta energía es la energía necesaria para establecer una diferencia de temperatura nula entre la sustancia y el material de referencia. Cuando la muestra sufre una transición que implique un cambio de entalpia, ese cambio se indica con una desviación endotérmica (ebullición) respecto a la línea base del registro de flujo de calor.
1.5. CRITERIOS DE CALIDAD
La aplicación y la precisión de los diferentes métodos utilizados para determinar el punto de ebullición/intervalo de ebullición se indican en el cuadro 1.
Cuadro 1
Comparación de los métodos
|
Método de medición |
Precisión estimada |
Norma existente |
|
Ebullómerro |
±1,4 K (hasta 373 K) (5) (6) ±2,5 K (hasta 600 K) (5) (6) |
ASTM D 1120-72 (5) |
|
Método dinámico |
±0,5 k (hasta 600 K) (6) |
|
|
Método de destilación (intervalo de ebullición) |
±0,5 k (hasta 600 K) |
ISO/R 918, DIN 53171, BS 4591/71 |
|
Método de Siwoloboff |
± 2 K (hasta 600 K) (6) |
|
|
Detección fotoeléctrica |
±0,3 K (a 373 K) (6) |
|
|
Calorimetría térmica diferencial |
±0,5 K (hasta 600 K) ±2,0 K (hasta 1 273 K) |
ASTM E 537-76 |
|
Calorimetría diferencial de barrido |
±0,5 K (hasta 600 K) ±2,0 K (hasta 1 273 K) |
ASTM E 537-76 |
1.6. DESCRIPCIÓN DE LOS MÉTODOS
En las normas internacionales y nacionales (véase el apéndice) se describen los procedimientos de algunos métodos de ensayo.
1.6.1. Ebullómetro
Véase el apéndice.
1.6.2. Método dinámico
Véase el método de ensayo A.4 para la determinación de la presión de vapor.
Se considera como temperatura de ebullición la registrada a una presión de 101,325 kPa,
1.6.3. Método de destilación (intervalo de ebullición)
Véase el Apéndice,
1.6.4. Método de Siwoloboff
Se introduce la muestra en un tubo de muestra de unos 5 mm de diámetro y se calienta en un aparato apropiado para determinar el punto de fusión (véase la figura 1).
En la figura 1 hay un ejemplo de aparato normalizado apropiado para determinar el punto de fusión y el punto de ebullición (JIS K 0064). Las dimensiones están expresadas en milímetros. El aparato es de vidrio.
Figura 1
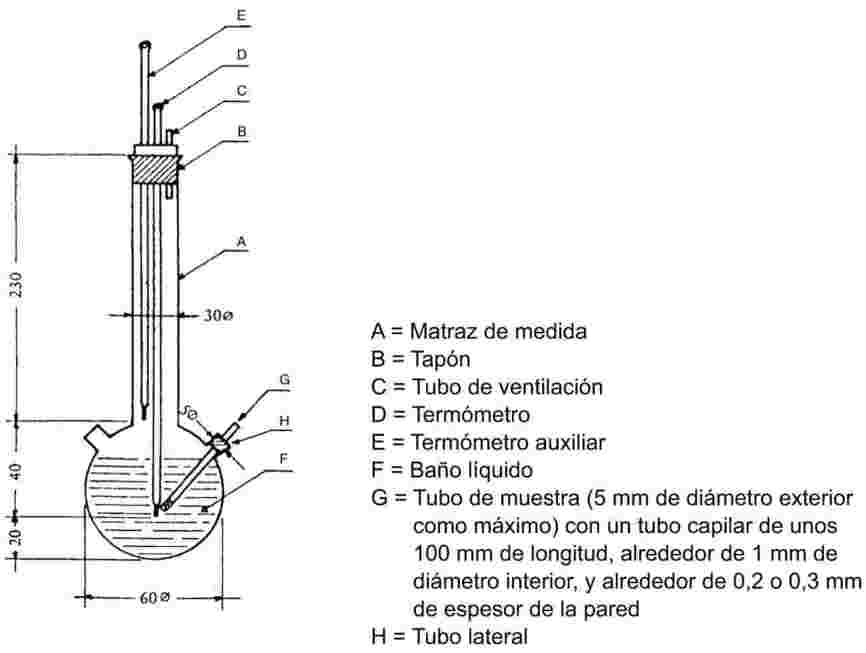
En el tubo de muestra que contiene la sustancia se introduce un tubo capilar (capilar de ebullición), cerrado a aproximadamente 1 cm de su extremo inferior. El nivel hasta el que se añade la sustancia problema es tal que la parte cerrada del capilar quede situada por debajo de la superficie del líquido. El tubo de muestra con el capilar de ebullición debe estar unido al termómetro mediante una cinta elástica o un soporte lateral (véase la figura 2).
|
Figura 2 Método de Siwoloboff |
Figura 3 Método modificado |
|
|
|
El líquido del baño se elige en función de la temperatura de ebullición. Se puede utilizar aceite de silicona para temperaturas de hasta 573 K. La parafina liquida solo puede usarse para temperaturas inferiores a 473 K. Al principio, el calentamiento del baño debe graduarse de manera que se obtenga un aumento de temperatura de 3 K por minuto. Hay que agitar el líquido del baño. A unos 10 K por debajo del punto de ebullición supuesto, se reduce el calentamiento de tal manera que el aumento de la temperatura no llegue a 1 K/minuto. Al acercarse a la temperatura de ebullición, comienzan a salir rápidamente burbujas del capilar de ebullición.
El punto de ebullición se define como la temperatura a la cual, en un enfriamiento momentáneo, se interrumpe el rosario de burbujas y el líquido comienza a elevarse súbitamente por el capilar. La temperatura leída en el termómetro en ese preciso momento corresponde a la temperatura de ebullición de la sustancia de ensayo.
En el método modificado (véase la figura 3), el punto de ebullición se determina en un capilar de punto de fusión. Este último se alarga en una punta fina de unos 2 cm de longitud (a) y se aspira hacia el interior una pequeña cantidad de sustancia. El extremo abierto del fino capilar se cierra por fusión, de forma que quede una pequeña burbuja de aire en el extremo. Al calentarla en el aparato de punto de fusión (b), la burbuja de aire se va dilatando. El punto de ebullición corresponde a la temperatura a la que el tapón de muestra llega al nivel de la superficie del baño de líquido (c).
1.6.5. Detección fotoeléctrica
Se calienta una muestra de la sustancia en un tubo capilar colocado dentro de un bloque metálico de calentamiento.
Por las aberturas practicadas en el bloque, se envía un haz de luz a través de la sustancia hacia una célula fotoeléctrica calibrada en forma precisa.
Durante el aumento de la temperatura de la muestra, van escapando del capilar de ebullición algunas burbujas aisladas de aire. Cuando se alcanza la temperatura de ebullición, el número de burbujas aumenta mucho. La consiguiente modificación de la intensidad luminosa es registrada por la célula, que envía una señal de interrupción al indicador que da la temperatura de un termómetro de resistencia de platino colocado en el bloque.
Este método es especialmente útil, pues permite efectuar determinaciones por debajo de la temperatura ambiente hasta 253,15 K (-20 oC) sin ninguna modificación del aparato. Basta con colocar este en un baño refrigerante
1.6.6. Análisis térmico
1.6.6.1. Análisis térmico diferencial
Véase el apéndice.
1.6.6.2. Calorimetría diferencial de barrido
Véase el apéndice.
2. RESULTADOS
Cuando haya diferencias pequeñas respecto a la presión normal (máxima de ± 5 kPa), las temperaturas del punto de ebullición podrán normalizarse a Tn mediante la ecuación de valor numérico de Sidney Young:
Tn = T + (fT × Δ p)
donde
|
A p |
= |
(101,325 — p) [atención al signo] |
|
p |
= |
medida de la presión, en kPa |
|
fT |
= |
tasa de variación del punto de ebullición con la presión, en K/kPa |
|
T |
= |
valor medido de la temperatura de ebullición, en K |
|
Tn |
= |
valor de la temperatura de ebullición corregido a presión normal, en K. |
Los factores de corrección de la temperatura (fT) y las ecuaciones para su aproximación figuran en las normas internacionales citadas en el texto para numerosas sustancias.
Por ejemplo, el método DIN 53171 presenta las siguientes correcciones aproximadas para disolventes contenidos en las pinturas:
Cuadro 2
Factores de corrección de la temperatura (ft)
|
Temperatura T en K |
Factor de corrección fT en K/kPa |
|
323,15 |
0,26 |
|
348,15 |
0,28 |
|
373,15 |
0,31 |
|
398,15 |
0,33 |
|
423,15 |
0,35 |
|
448,15 |
0,37 |
|
473,15 |
0,39 |
|
498,15 |
0,41 |
|
523,15 |
0,44 |
|
548,15 |
0,45 |
|
573,15 |
0,47 |
3. INFORME
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
método utilizado, |
|
— |
especificación precisa de la sustancia (identidad e impurezas) y, en su caso, fase de purificación previa, |
|
— |
estimación de la precisión. |
El punto de ebullición registrado será la media entre dos mediciones, como mínimo, situadas en la zona de precisión estimada (véase el cuadro 1).
Deberán indicarse los valores medidos de los puntos de ebullición así como su media, y la presión o presiones a que se hayan efectuado las mediciones deberán registrarse en kPa. La presión debe, preferentemente, ser próxima a la presión atmosférica normal.
Deben suministrarse todas las informaciones y observaciones que se consideren útiles para la interpretación de los resultados, en particular lo referente a las impurezas y al estado físico de la sustancia.
4. BIBLIOGRAFÍA
|
(1) |
OCDE, París, 1981, Test Guideline 103 — Decision of the Council C (81) 30 final. |
|
(2) |
IUPAC, B. Le Neindre, B. Vodar, eds. Experimental thermodynamics, Butterworths, Londres 1975, vol. II. |
|
(3) |
R. Weissberger ed.: Technique of organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Interscience Publ., Nueva York, 1959, vol. I, Part I, Chapter VIII. |
Apéndice
Para más detalles técnicos, se pueden consultar las normas siguientes:
1. Ebullómetro
|
ASTM D 1120-72 |
Standard test method for boiling point of engine anti-freezes |
2. Método de destilacion (intervalo de ebullicion)
|
ISO/R 918 |
Test method for distillation (distillation yield and distillation range) |
|
BS 4349/68 |
Method for determination of distillation of petroleum products |
|
BS 4591/'71 |
Method for the determination of distillation characteristics |
|
DIN 53171 |
Losungsmittel fur Anstrichstoffe. Bestimmung des Siedeverlaufes |
|
NF T 20-608 |
Distillation; determination du rendement et de l'intervalle de distillation |
3. Analisis termico diferencial y calorimetria diferencial de barrido
|
ASTM E 537-76 |
Standard method for assessing the thermal stability of chemicals by methods of differential thermal analysis |
|
ASTM E 473-85 |
Standard definitions of terms relating to thermal analysis |
|
ASTM E 472-86 |
Standard practice for reporting thermoanalytical data |
|
DIN 51005 |
Thermische Analyse: Begriffe |
A.3. DENSIDAD RELATTVA
1. MÉTODO
Los métodos descritos se basan en las líneas directrices de la OCDE (1). Los principios fundamentales se dan en la referencia (2).
1.1. INTRODUCCIÓN
Los métodos descritos de determinación de la densidad relativa son aplicables a las sustancias sólidas y líquidas, cualquiera que sea su grado de pureza. En el cuadro 1 se indican los diversos métodos utilizables.
1.2. DEFINICIONES Y UNIDADES
La densidad relativa, D20 4, de los sólidos o líquidos es la relación entre la masa de un volumen de sustancia problema, determinada a 20 oC, y la masa del mismo volumen de agua, determinada a 4 oC. La densidad relativa es un número adimensional.
La densidad, p, de una sustancia es el cociente de su masa m por su volumen v.
En unidades SI, la densidad, p, se expresa en kilogramos por metro cúbico.
1.3. SUSTANCIAS DE REFERENCIA (1) (3)
No es necesario emplear sustancias de referencia cada vez que se estudie una nueva sustancia. Deben utilizarse, esencialmente, para comprobar la validez del método de vez en cuando y para poder comparar con los resultados obtenidos según otros métodos,
1.4. PRINCIPIO DE LOS MÉTODOS
Se aplican cuatro clases de métodos.
1.4.1. Métodos de flotabilidad
1.4.1.1. Areómetro (para líquidos)
Se pueden obtener determinaciones de densidad suficientemente precisas y rápidas con areómetros flotantes, que permiten deducir la densidad de un líquido a partir de la profundidad de inmersión leída en una escala graduada.
1.4.1.2. Balanza hidrostática (para sustancias líquidas y sólidas)
La diferencia entre el peso de una muestra medido en el aire y en un líquido adecuado (como el agua) puede servir para determinar su densidad.
En el caso de los sólidos, la densidad medida solo es representativa de la muestra utilizada en concreto. Para determinar la densidad de un líquido, se pesa un cuerpo, con un volumen v conocido, primero en el aire y luego en el líquido.
1.4.1.3. Método del cuerpo sumergido (para las sustancias líquidas) (4)
En este método, la densidad de un líquido se determina a partir de la diferencia entre los resultados de la pesada del líquido, antes y después de sumergir un cuerpo de volumen conocido en dicho líquido problema.
1.4.2. Métodos picnométricos
Para sólidos o líquidos, se pueden utilizar picnómetros de diversas formas cuyos volúmenes sean conocidos. La densidad se calculará a partir de la diferencia de peso entre el picnómetro lleno y el picnómetro vacío, por una parte, y de su volumen conocido, por la otra.
1.4.3. Picnómetro de comparación de aire (para sólidos)
La densidad de un sólido, cualquiera que sea su forma, se puede medir a la temperatura ambiente mediante un picnómetro de comparación de gases. El volumen de una sustancia en el aire o en un gas inerte se mide en una probeta calibrada de volumen variable. Para el cálculo de la densidad, se efectúa una medida de la masa después de la medida del volumen.
1.4.4. Densímetro oscilante (5) (6) (7)
La densidad de un líquido se puede medir con un densímetro oscilante. Un oscilador mecánico con forma de tubo en U se hace vibrar a la frecuencia de resonancia del oscilador, que depende de su masa. La introducción de una muestra modifica la frecuencia de resonancia del oscilador, el cual debe calibrarse con ayuda de dos sustancias líquidas de densidades conocidas. Las sustancias deben elegirse de tal manera que sus densidades cubran el intervalo de medición.
1.5. CRITERIOS DE CALIDAD
En el cuadro se indica la aplicabilidad de los diferentes métodos que se utilizan para determinar la densidad relativa.
1.6. DESCRIPCIÓN DE LOS MÉTODOS
Las referencias de las normas citadas como ejemplo, las cuales pueden consultarse para obtener detalles técnicos suplementarios, figuran en el apéndice.
Los ensayos deberán realizarse a la temperatura de 20 oC y deben hacerse, al menos, dos medidas.
2. RESULTADOS
Véanse las normas.
3. INFORME
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
método utilizado, |
|
— |
especificación precisa de la sustancia (identidad e impurezas) y, en su caso, fase de purificación previa. |
También se indicarán tanto la densidad relativa, ![]() , según lo definido en el punto 1.2, como el estado físico de la sustancia examinada,
, según lo definido en el punto 1.2, como el estado físico de la sustancia examinada,
Deben suministrarse todas las informaciones y observaciones que sean útiles para la interpretación de los resultados, en particular lo referente a las impurezas y al estado físico de la sustancia.
Cuadro
Aplicabilidad de los métodos
|
Método de medida |
Densidad |
Viscosidad dinámica máxima posible |
Normas existentes |
|||
|
sólido |
líquido |
|||||
|
|
Sí |
5 Pa s |
ISO 387, ISO 649-2, NF T 20-050 |
||
|
|
|
|
|
||
|
Sí |
|
|
ISO 1183 (A) |
||
|
|
Sí |
5 Pa S |
ISO 901 y 758 |
||
|
|
Sí |
20 Pa s |
DIN 53217 |
||
|
|
|
|
ISO 3507 |
||
|
Sí |
|
|
ISO 1183 (B), NF T 20-053 |
||
|
|
Sí |
500 Pa s |
ISO 758 |
||
|
Sí |
|
|
DIN 55990 parte 3, DIN 53243 |
||
|
|
Si |
5 Pa s |
|
4. BIBLIOGRAFÍA
|
(1) |
OCDE, París, 1981, Test Guideline 109 — Decisión of the Council C (81) 30 Final. |
|
(2) |
R. Weissberger ed., Technique of Organic Chemistry. Physical Methods of Organic Chemistry, 3rd ed., Chapter IV, Interscience Publ. Nueva York, 1959, Vol. I, Part 1, |
|
(3) |
IUPAC, Recommended reference materials for realizatíon of physico-chemical properties. — Pure and Applied Chemistry, 1976, Vol. 48, 508. |
|
(4) |
Wagenbreth, H., Die Tauchkugel zur Besrimmung der Dichte von Flüssigkeiten, Technisches Messen tm, 1979, Vol 11, 427-430. |
|
(5) |
Leopold, H., Die digitale Messung von Flüssigkeiten, Elektronik, 1970, Vol. 19, 297-302. |
|
(6) |
Baumgarten, D., Füllmengenkontrolle bei vorgepackten Erzeugnissen — Verfahren zur Dichtebestim-mung bei flüssigen Produkten und ihre praktische Anwendung, Die Pharmazeurische Industrie, 1975, Vol. 37, 717-726. |
|
(7) |
Riemann, J., Der Einsatz der digitalen Dichtemessung im Brauereilaboratorium, Brauwissenscbaft, 1976, Vol. 9, 253-255. |
Apéndice
Para más detalles técnicos, se pueden consultar las normas siguientes:
1. Métodos de flotabilidad
1.1. Areómetro
|
DIN 12790, ISO 387 |
Hydrometer: general instructions |
|
DIN 12791 |
Part I: Density hydrometers; construction, adjustment and use Part II: Density hydxometers; standardised sizes, designation Part III: Use and test |
|
ISO 649-2 |
Laboratory glassware: Density hydrometers for general purpose |
|
NF T 20-050 |
Chemical products for industrial use — Determination of density of liquids — Areometric method |
|
DIN 12793 |
Laboratory glassware: range find hydrometers |
1.2. Balanza hidrostática
Para sustancias sólidas
|
ISO 1183 |
Method A: Methods for determining the density and relative density of plastics excluding cellular plastics |
|
NF T 20-049 |
Chemical products for industrial use — Determination of the density of solids other than powders and cellular products — Hydrostatic balance method |
|
ASTM-D-792 |
Specific gravity and density of plastics by displacement |
|
DIN 53479 |
Testing of plastics and elastomers; determination of density |
Para sustancias líquidas
|
ISO 901 |
ISO 758 |
|
DIN 51757 |
Testing of mineral oils and related materials; determination of density. |
|
ASTM D 941-55, ASTM D 12 96-67 y ASTM D 1481-62 |
|
|
ASTM D 1298 |
Density, specific gravity or API gravity of crude petroleum and liquid petroleum products by hydrometer method |
|
BS 4714 |
Density, specific gravity or API gravity of crude petroleum and liquid petroleum products by hydrometer method |
1.3. Métodos del cuerpo sumergido
|
DIN 53217 |
Testing of paints, varnishes and similar coating materials; determination of density; immersed body method |
2. Métodos picnométricos
2.1. Para sustancias líquidas
|
ISO 3507 |
Pycnometers |
|
ISO 758 |
Liquid chemical products; determination of density at 20 oC |
|
DIN 12797 |
Gay-Lussac pycnometer (for non-volatile liquids which are not too viscous) |
|
DIN 12798 |
Lipkin pyenometer (for liquids with a kinematic viscosity of less than 100,10-6 m2 -l at 15 oC) |
|
DIN 12800 |
Sprengel pycnometer (for liquids as DIN 12798) |
|
DIN 12801 |
Reischauer pycnometer (for liquids with a kinematic viscosity of less than 100,10—6 m2 s-1 at 20 oC, applicable in particular also to hydrocarbons and aqueous solutions as well as to liquids with higher vapour pressure, approxi mately 1 bar at 90 oC) |
|
DIN 12806 |
Hubbard pycnometer (for viscous liquids of all types which do not have a too high vapour pressure, in particular also for paints, varnishes and bitumen) |
|
DIN 12807 |
Bingham pycnometer (for liquids, as in DIN 12801) |
|
DIN 12808 |
Jaulmes pycnometer (in particular for ethanol-water mixture) |
|
DIN 12809 |
Pycnometer with ground-in thermometer and capillary side tube (for liquids which are not too viscous) |
|
DIN 53217 |
Testing of paints, varnishes and similar products; determination of density by pycnometer |
|
DIN 51757 |
Point 1: Testing of mineral oils and related materials; determination of density |
|
ASTM D 297 |
Section 15: Rubber products — chemical analysis |
|
ASTM D 2111 |
Method C: Halogenated organic compounds |
|
BS 4699 |
Method for determination of specific gravity and density of petroleum products (graduated bicapillary pycnometer method) |
|
BS 5903 |
Method for determination of relative density and density of petroleum products by the capillary-stoppered pycnometer method |
|
NF T 20-053 |
Chemical products for industrial use— Determination of density of solids in powder and liquids — Pyknometric method |
2.2. Para sustancias sólidas
|
ISO 1183 |
Method B: Methods for determining the density and relative density of plastics excluding cellular plastics. |
|
NF T 20-053 |
Chemical products for industrial use — Determination of density of solids in powder and liquids — Pyknometric method |
|
DIN 19683 |
Determination of the density of soils |
3. Picnómetro de comparación de aire
|
DIN 55990 |
Part 3: Prüfung von Anstrichstoffen und ähnlichen Beschichtungsstoffen; Pulverlack; Bestimmung der Dichte |
|
DIN 53243 |
Anstrichstoffe; Chlorhaltige Polymere; Prüfung |
A.4. PRESIÓN DE VAPOR
1. MÉTODO
La mayoría de los métodos descritos se basan en las líneas directrices de la OCDE (1). Los principios fundamentales se dan en las referencias (2) y (3).
1.1. INTRODUCCIÓN
Es conveniente disponer de datos previos sobre la estructura, el punto de fusión y el punto de ebullición de la sustancia antes de proceder al ensayo.
No hay ningún método de medida que sea aplicable a toda la gama de presiones de vapor. Por eso se recomiendan varios métodos para medir las presiones de vapor que van de < 10-4 Pa a 105 Pa.
En general, las impurezas influyen sobre la presión de vapor en una medida que depende en gran parte de la naturaleza de la impureza.
Cuando la muestra contenga impurezas volátiles que puedan afectar el resultado, será posible purificar la sustancia. También puede ser adecuado indicar la presión de vapor del material técnico.
Algunos de los métodos descritos aquí utilizan equipos con partes metálicas; esto deberá tenerse en cuenta cuando se estudien sustancias corrosivas.
1.2. DEFINICIONES Y UNIDADES
La presión de vapor de una sustancia es la presión de saturación por encima de la sustancia sólida o líquida. En equilibrio termodinámico, la presión de vapor de una sustancia pura es función únicamente de la temperatura.
La unidad SI de presión que se debe utilizar es el pascal (Pa).
Los factores de conversión de las unidades que solían utilizarse en el pasado, son:
|
1 Torr(- 1 mm Hg) |
= 1,333 × 102 Pa |
|
1 atmósfera |
= 1,013 × 105 Pa |
|
1 bar |
= 105 Pa |
La unidad SI de temperatura es el grado Kelvin (K).
La constante universal molar de los gases R es 8,314 J mol-1 K-1.
La relación de la presión de vapor con la temperatura se refleja en la ecuación de Clausius-Clapeyron;

donde
|
p |
= |
presión de vapor de la sustancia en pascales |
|
A Hv |
= |
calor de vaporización en J mol-J |
|
R |
= |
constante universal molar de los gases en J mol-1 K-1 |
|
T |
= |
temperatura termodinámica en K. |
1.3. SUSTANCIAS DE REFERENCIA
No es necesario emplear sustancias de referencia cada vez que se estudie una nueva sustancia. Dichas sustancias deberán servir, esencialmente, para comprobar de vez en cuando la validez del método y para comparar con los resultados obtenidos según otros métodos.
1.4. PRINCIPIO DE LOS MÉTODOS
Se proponen siete métodos para determinar la presión de vapor, aplicables a diferentes gamas de presiones de vapor. En cada uno de los métodos, la presión de vapor se determina a diferentes temperaturas. En una gama de temperaturas limitada, el logaritmo de la presión de vapor de una sustancia pura es una función lineal de la inversa de la temperatura.
1.4.1. Método dinámico
El método dinámico se basa en la medida de la temperatura de ebullición correspondiente a una presión específica.
Intervalo recomendado:
de 103 hasta 105 Pa.
Este método se ha recomendado también para determinar el punto de ebullición normal y, para este fin, es útil hasta a 600 K.
1.4.2. Método estático
En este procedimiento, la presión de vapor que se establece en un sistema cerrado, en equilibrio termodinámico, se determina a una temperatura específica. Este método es aplicable a sólidos y líquidos que contengan uno o varios componentes.
Intervalo recomendado:
de 10 hasta 105 Pa.
Este método puede utilizarse también en el intervalo de 1 a 10 Pa siempre que se tenga cuidado.
1.4.3. Isoteniscopio
Este método normalizado es también un procedimiento estático, pero no es aplicable generalmente a los sistemas con varios componentes. Se puede encontrar información suplementaria en la norma ASTM D-2879-86.
Intervalo recomendado:
de 100 hasta 105 Pa,
1.4.4. Método de efusión: Balanza de presión de vapor
La cantidad de sustancia que sale por unidad de tiempo de un compartimento a través de una abertura de tamaño conocido, se determina bajo presión reducida para que el retorno de sustancia al compartimento-depósito sea despreciable (por ejemplo, midiendo el impulso que imprime a una balanza sensible un chorro de vapor o, también, midiendo la pérdida de peso del compartimento-depósito).
Intervalo recomendado:
de 10-3 hasta 1 Pa.
1.4.5. Método de efusión: Pérdida de peso o retención de la parte evaporada
El método se basa en la estimación de la masa de sustancia problema que sale por unidad de tiempo de una célula Knudsen (4) en forma de vapor a través de un microorificio en condiciones de ultravacío. La masa de vapor desprendido puede obtenerse o bien determinando la pérdida de masa de la célula o bien condensando el vapor a baja temperatura y determinando la cantidad de sustancia volatilizada mediante análisis cromatográfico. La presión de vapor se calcula según la relación de Hertz-Knudsen.
Intervalo recomendado:
de 10-3 hasta 1 Pa.
1.4.6. Método de saturación de gases
Se envía una corriente de gas portador inerte sobre la sustancia para que aquel se sature del vapor de esta. La medida de la cantidad de sustancia transportada por un volumen conocido de gas portador puede realizarse o bien mediante su recogida en un sifón adecuado o bien mediante una técnica analítica acoplada. Así se puede calcular después la presión de vapor a una temperatura dada.
Intervalo recomendado:
de 10-4 hasta 1 Pa:
Este método puede utilizarse también en el intervalo de 1 a 10 Pa siempre que se tenga cuidado.
1.4.7. Rotor
En el indicador de rotor, el elemento medidor en realidad es una pequeña bola de acero, suspendida en un campo magnético, que gira a gran velocidad. La presión del gas se deduce del frenado de la bola de acero, que depende de la presión.
Intervalo recomendado:
de 10-4 hasta 0,5 Pa.
1.5. CRITERIOS DE CALIDAD
El cuadro siguiente muestra una tabla comparativa de la aplicación, repetibilidad, reproducibilidad, intervalos de medida y normas existentes de los diferentes métodos de determinación de la presión de vapor.
Cuadro
Criterio de calidad
|
Método de medida |
Sustancia |
Repetibililidad estimada (7) |
Reproducibilidad estimada (7) |
Intervalo recomendado |
Norma existente |
|||
|
sólida |
liquida |
|||||||
|
Fusión baja |
Sí |
Hasta el 25 % |
Hasta el 25 % |
De 103 Pa a 2 × 103 Pa |
— |
||
|
|
|
|
1-5 % |
1-5 % |
De 2 x 103 Pa a 105 Pa |
— |
||
|
Sí |
Sí |
5-10 % |
5-10 % |
De 10 Pa a 105 Pa (8) |
NFT 20-048 (5) |
||
|
Sí |
Sí |
5-10 % |
5-10 % |
De 102 Pa a 105 Pa |
ASTM-D 2879-86 |
||
|
Sí |
Sí |
5-20 % |
Hasta el 50 % |
De 10-3 pa a 1 Pa |
NFT 20-047 (6) |
||
|
Sí |
Sí |
10-30 % |
— |
De 10-3 Pa a 1 Pa |
— |
||
|
Sí |
Sí |
10-30 % |
Hasta el 50 % |
De 10-4 Pa a 1 Pa-4 (8) |
— |
||
|
Sí |
Sí |
10-20 % |
— |
De 10-4 Pa a 0,5 Pa |
— |
1.6. DESCRIPCIÓN DE LOS MÉTODOS
1.6.1. Medida dinámica
1.6.1.1. Aparato
El equipo de medida típico consta de un recipiente de ebullición con refrigerante de vidrio o de metal (véase la figura 1), un dispositivo de medida de la temperatura y un dispositivo de regulación y de medida de la presión. El equipo de medida representado en el esquema es de vidrio termorresistente y consta de cinco partes principales:
El tubo ancho con un tramo de doble pared tiene junta esmerilada, refrigerante, recipiente de enfriamiento y orificio de entrada.
El cilindro de vidrio unido a una bomba Cottrell está montado en la sección del tubo donde se efectúa la ebullición y tiene una superficie rugosa de vidrio triturado para evitar las sacudidas durante el proceso de ebullición.
Para medir la temperatura se puede utilizar un termosensor adecuado (por ejemplo, un termopar de camisa o un termómetro de resistencia) introducido en el aparato hasta el punto de medida (n° 5, figura 1) a través de una entrada adecuada (por ejemplo, junta esmerilada macho).
Se hacen las conexiones necesarias con el dispositivo de regulación y de medida de la presión.
El bulbo, que actúa como volumen-tampón, está conectado al aparato de medida a través de un tubo capilar.
Un elemento calefactor (por ejemplo, un calentador de cartucho) introducido en la parte inferior del aparato de vidrio, calienta el recipiente de ebullición. La corriente necesaria se establece y se regula por medio de un termopar.
La bomba de vacío produce el vacío necesario, entre 102 y aproximadamente 102 Pa.
Una válvula adecuada se utiliza para medir aire o nitrógeno con el fin de regular la presión (zona aproximada de medición: de 102 a 105 Pa) y de ventilar el aparato.
La presión se mide con un manómetro.
1.6.1.2. Procedimiento de medida
La presión de vapor se mide determinando el punto de ebullición de la muestra a diversas presiones específicas comprendidas entre 103 Pa y 105 Pa, aproximadamente. El punto de ebullición se alcanza cuando la temperatura se mantiene estable a presión constante. Este procedimiento de medida no es aplicable a las sustancias que formen espuma.
Se introduce la sustancia en el recipiente limpio y seco. Los sólidos no pulverulentos pueden presentar problemas, que a veces se resuelven calentando la camisa de refrigeración. Una vez lleno el recipiente, se cierra el aparato con la pestaña y se extrae el gas de la sustancia. Se regula a la presión más baja prevista y se acciona el sistema de calentamiento, conectando, simultáneamente, el termosensor a un registrador.
Cuando este indique una temperatura de ebullición fija, a presión constante, se habrá alcanzado el equilibrio. Hay que evitar las sacudidas durante la ebullición. Además, debe obtenerse condensación completa en el refrigerante. Cuando se determina la presión de vapor de sólidos de bajo punto de fusión, hay que evitar que se bloquee el condensador.
Una vez registrado el punto de equilibrio, se regulará a una presión más elevada y se irá repitiendo de nuevo el mismo proceso hasta que se alcance una presión de 10-5 Pa (en total, de 5 a 10 medidas). Los puntos de equilibrio deben repetirse a presiones decrecientes para comprobar ¡os resultados.
1.6.2. Medida estática
1.6.2.1. Aparato
El aparato se compone de un recipiente para la muestra, de un sistema de calentamiento y enfriamiento para llevar la muestra a la temperatura deseada, así como de un dispositivo, para medir la temperatura. El aparato también incluye instrumentos para fijar y medir la presión. Las figuras 2a y 2b ilustran los principios básicos del aparato.
El compartimento que contiene la muestra (figura 2a) está unido, por una parte, a una llave de alto vacío y, por la otra, a un tubo en forma de U lleno de un líquido manométrico apropiado. Un extremo del tubo en U se ramifica para unirse a la bomba de vacío, a la bombona de nitrógeno o a la válvula de ventilación y a un manómetro.
En lugar de un tubo en U puede utilizarse un manómetro con indicador de presión (figura 2b).
Para poner la muestra a la temperatura deseada, se sumergen en un baño a temperatura constante ±0,2 K, el compartimento de la muestra con la llave y el tubo en U o el manómetro. La temperatura se mide en la pared exterior del compartimento de la muestra o en el propio compartimento.
Para evacuar los gases del aparato se utilizará una bomba de vacío con purgador previo de enfriamiento.
En el método 2a, la presión de vapor de una sustancia se mide indirectamente con un indicador de cero. Esto tiene en cuenta el hecho de que la densidad del líquido en el tubo en U se altera si la temperatura cambia de forma importante.
Los siguientes líquidos pueden utilizarse como indicadores de cero para el tubo en U, según el intervalo de presiones y el comportamiento químico de la sustancia problema: aceites de silicona y ftalatos. La sustancia problema no debe disolverse de forma apreciable o reaccionar con el líquido del tubo U.
En el manómetro, el mercurio puede utilizarse en la zona desde la presión atmosférica hasta 102 Pa, los aceites de silicona y los ftalatos de 102 Pa a 10 Pa; en cuanto al manómetro de membrana calentable, puede utilizarse hasta a presiones inferiores a 10-1 Pa. También pueden utilizarse por debajo de 102 Pa otros tipos de manómetros.
1.6.2.2. Métodos de medida
Antes de medir, se limpian y se secan completamente todas las partes del aparato cuyo esquema aparece en la figura 2.
Para el método 2a, se llena el tubo en U con el líquido previsto, al que antes de realizar las lecturas se le ha extraído el gas a temperatura elevada.
Después de introducir la sustancia, se cierra el aparato y se enfría suficientemente para desgasificar. La temperatura debe ser suficientemente baja como para garantizar que se ha extraído el aire aunque, en el caso de sistemas multicomponenciales, no debe alterarse la composición del material. En caso necesario, el equilibrio puede alcanzarse más rápidamente por agitación.
La muestra puede subenfriarse, por ejemplo, con nitrógeno líquido (precaución: condensación de aire, líquido de la bomba) o una mezcla de etanol y hielo seco. Para las medidas a baja temperatura, úsese un baño termostatizado conectado a un ultracriomat.
Se abre la llave que hay sobre el recipiente de la muestra y se aplica una succión durante varios minutos para extraer el aire. Se cierra luego la llave y se reduce la temperatura de la muestra hasta el nivel más bajo que se desee. En caso necesario, la operación de desgasificado puede repetirse varias veces.
Al calentar la muestra aumenta la presión de vapor, lo que hace cambiar el equilibrio del líquido del tubo en U. Para compensar este fenómeno, hay que permitir la entrada de nitrógeno o aire en el aparato a través de una llave hasta que el líquido del manómetro esté otra vez a cero. La presión necesaria para conseguir este efecto puede leerse en un manómetro de precisión a temperatura ambiente y corresponde a la presión de vapor de la sustancia a esa temperatura concreta de medida.
El método 2b es similar pero la presión de vapor puede leerse directamente.
La relación de la presión de vapor con la temperatura se determina a pequeños intervalos adecuados (aproximadamente, entre 5 y 10 puntos de medida en total) hasta llegar al máximo previsto. Las lecturas a baja temperatura deben repetirse como comprobación.
Si los valores obtenidos en las lecturas repetidas no coinciden con la curva obtenida al ir aumentando las temperaturas, puede deberse a las causas siguientes:
|
1) |
la muestra sigue conteniendo aire (por ejemplo, materiales de elevada viscosidad) o sustancias de bajo punto de ebullición, que se liberan durante el calentamiento y pueden eliminarse mediante succión tras un mayor subenfriamiento; |
|
2) |
la temperatura de enfriamiento no es bastante baja. En este caso se utiliza nitrógeno líquido como agente de enfriamiento. Si se trata de las causas 1 o 2, hay que repetir las medidas; |
|
3) |
la sustancia sufre una reacción química en el intervalo de temperaturas investigado (por ejemplo, descomposición, polimerización). |
1.6.3. Isoteniscopio
Para una descripción completa de este método, consultar la referencia 7. Los principios del aparato de medida están descritos en la figura 3. Como el método estático descrito en el punto 1.6.2, el isoteniscopio es aplicable a sólidos y a líquidos.
En el caso de los líquidos, la sustancia misma sirve de líquido de llenado para el manómetro auxiliar. Se pone en el isoteniscopio una cantidad de líquido suficiente para llenar el bulbo y el brazo corto de la sección manométrica. El isoteniscopio se une entonces al sistema de vacío; después de hacer el vacío en su interior, el isoteniscopio se llena de nitrógeno. La evacuación y la purga del sistema se repiten dos veces para eliminar el oxígeno residual. El isoteniscopio lleno se coloca en posición horizontal para que la muestra se extienda formando una fina capa en el bulbo de la muestra y en la sección manométrica (parte en U). La presión del sistema se reduce a 133 Pa y se calienta suavemente la muestra hasta que inicie la ebullición (eliminación de gases fijos disueltos). El isoteniscopio se cambia entonces de posición para que la muestra vuelva al bulbo y al brazo corto del manómetro, de forma que ambos queden totalmente llenos de líquido. La presión se mantiene al mismo nivel que para desgasificar; el extremo alargado del bulbo de la muestra se calienta con una pequeña llama hasta que el vapor originado por la muestra se expande suficientemente para desplazar parte de la muestra desde la porción superior del bulbo y del brazo del manómetro hacia la sección manométrica del isoteniscopio, creando así un espacio libre de nitrógeno y lleno de vapor.
El isoteniscopio se coloca entonces en un baño termostático y se ajusta la presión del nitrógeno hasta igualar a la presión de la muestra. El equilibrio de presiones viene indicado por la sección manométrica del isoteniscopio. En equilibrio, la presión de vapor del nitrógeno iguala a la presión de vapor de la sustancia.
En el caso de los sólidos, se utilizarán los líquidos manométricos citados en el punto 1.6.2.1, según el intervalo de temperatura y presión. El líquido manométrico desgasificado se introduce en un engrasamiento del brazo largo del isoteniscopio. A continuación se introduce en el bulbo el sólido problema y se desgasifica a temperatura elevada. Después se inclina el isoteniscopio de forma que el líquido manométrico se introduzca en el tubo de U. La medida de la presión de vapor en función de la temperatura se realiza con arreglo al punto 1.6.2.
1.6.4. Método de efusión: Balanza de presión de vapor
1.6.4.1. Aparato
En la referencia 1 se encontrará la descripción de varios modelos de aparatos. El aparato representado en la figura 4 ilustra los principios generales del presente método. Esta figura muestra los principales componentes del aparato, que está formado por un recipiente de vidrio o de acero inoxidable resistente al vacío elevado, un equipo para producir y medir el vacío y componentes incorporados para medir la presión de vapor en una balanza. En el aparato se incluyen los siguientes componentes incorporados:
|
— |
un horno de evaporación con reborde y alimentador giratorio. Es un recipiente cilíndrico, hecho, por ejemplo, de cobre o de una aleación resistente químicamente con buena conductividad térmica. También puede utilizarse un recipiente de vidrio con pared de cobre. El horno tiene un diámetro aproximado de 3 a 5 cm y una altura de 2 a 5 cm. Está provisto de 1 a 3 aberturas de diferentes tamaños para la corriente devapor. El calentamiento se efectúa o bien mediante una placa calefactora por debajo del horno o bien por una espiral calefactora alrededor del exterior del horno. Para evitar que se disipe calor a la placa de la base, el elemento calefactor se une a la placa de la base mediante un metal de baja conductividad térmica (níquel-plata o acero al cromo-níquel), por ejemplo, un tubo de níquel-plata unido a un alimentador giratorio si se utiliza un horno con varias aberturas. Esta disposición tiene la ventaja de permitir la introducción de una barra de cobre, lo que hace posible enfriar desde el exterior por medio de un baño refrigerante, |
|
— |
si la tapa del horno de cobre tiene tres aberturas de diferentes diámetros, situados a 90o uno de otro, pueden cubrirse varios intervalos de presión de vapor dentro del intervalo global de medida (aberturas con diámetros entre 0,30 y 4,50 mm, aproximadamente). Las aberturas grandes se utilizan para las presiones de vapor pequeñas y viceversa. Girando el horno puede ponerse la abertura deseada o una postura intermedia en la corriente de vapor (abertura del homo-escudo-platillo de balanza) y la corriente de moléculas se libera o se desvía a través de la abertura del horno hacia el platillo de la balanza. Para medir la temperatura de la sustancia se coloca en un lugar adecuado un termopar o un termómetro de resistencia, |
|
— |
por encima del escudo hay un platillo que forma parte de una microbalanza muy sensible (véase más abajo). El platillo de balanza tiene unos 30 mm de diámetro y puede estar hecho de aluminio dorado, |
|
— |
el platillo de balanza está rodeado por un recinto cilíndrico de refrigeración de bronce o cobre. Según el tipo de balanza, este recinto tiene aberturas para el astil de la balanza y una abertura con escudo para la corriente de moléculas, y debe garantizar la condensación completa del vapor en el platillo de balanza. La disipación de calor al exterior se realiza, por ejemplo, mediante una barra de cobre conectada al recinto de refrigeración. La barra pasa a través de la placa de la base y se aisla térmicamente de esta, por ejemplo con un tubo de acero al cromo-níquel. La barra se introduce en un frasco Dewar con nitrógeno líquido bajo la placa de la base o se hace circular nitrógeno líquido a través de la barra. De esta manera, la temperatura de la barra se mantiene a - 120 oC aproximadamente. El platillo de la balanza se enfría exclusivamente por radiación, lo que es válido para el intervalo de presiones estudiadas (el enfriamiento debe iniciarse alrededor de 1 hora antes de que empiece la medición), |
|
— |
la balanza se coloca por encima del recinto de refrigeración. Como ejemplos de balanzas adecuadas pueden citarse una microbalanza electrónica muy sensible de 2 brazos (8) o un instrumento de bobina móvil (véase la Test Guideline 104 de la OCDE, edición de 12.05.81), |
|
— |
la placa de base también lleva conexiones eléctricas para los termopares (o termómetros de resistencia) y resistencias calefactoras, |
|
— |
el vacío en el recipiente se produce por medio de una bomba de vacío parcial o una bomba de alto vacío (el vacío necesario es, aproximadamente, de 1 a 2 . 10-3 Pa, después de 2 h de bombeo). La presión se regula con un manómetro adecuado de ionización. |
1.6.4.2. Método de medida
El receptáculo se llena con la sustancia problema y se cierra la tapa. El escudo y el recinto de refrigeración se deslizan encima del horno. Se cierra el aparato y se conectan las bombas de vacío. La presión final antes de empezar a tomar medidas debe ser aproximadamente 10-4 Pa. El enfriamiento del recinto de refrigeración se iniciará a 10-2 Pa.
Una vez obtenido el vacío necesario, se inicia la serie de calibración a la temperatura mínima exigida. Se pone la abertura correspondiente de la tapa, la corriente de vapor pasa a través del escudo directamente a la parte superior y choca con el platillo enfriado de la balanza. Este platillo debe ser bastante grande para garantizar que choca contra él todo el chorro de vapor llegado a través del escudo. El momento de la corriente de vapor actúa como una fuerza contra el platillo de la balanza y las moléculas se condensan en su superficie enfriada.
El momento y la condensación simultánea producen una señal en el registrador. La evaluación de las señales proporciona dos datos:
|
1) |
en el aparato aquí descrito, la presión de vapor se determina directamente a partir del momento sobre el platillo de la balanza [para esto no es necesario conocer el peso molecular (2)]. Para evaluar las lecturas hay que tener en cuenta factores geométricos, como la abertura del horno y el ángulo de la corriente de moléculas; |
|
2) |
la masa del condensado puede medirse a la vez y, a partir de este dato, puede calcularse la velocidad de evaporación. La presión de vapor puede calcularse también a partir de la velocidad de evaporación y del peso molecular mediante la ecuación de Hertz (2): |

donde
|
G |
= |
velocidad de evaporación (kg s-1 m-2) |
|
M |
= |
masa molecular (g mol-1) |
|
T |
= |
temperatura (K) |
|
R |
= |
constante universal molar de los gases (J mol-1 K-1) |
|
p |
= |
presión de vapor (Pa) |
Cuando se consigue el vacío necesario, se inicia la serie de medidas a la temperatura más baja prevista.
En las medidas siguientes, se aumenta la temperatura a pequeños intervalos hasta el momento en que se consiga la temperatura más alta prevista. Entonces se enfría la muestra una vez más y puede registrarse una segunda curva de presión de vapor. Si esta segunda serie no confirma los resultados de la primera, es posible que la sustancia se descomponga en el intervalo de temperatura que se utiliza en la determinación.
1.6.5. Método de efusión: Pérdida de peso
1.6.5.1. Aparato
El aparato de efusión consta de las siguientes partes básicas:
|
— |
un recinto que puede termostatizarse y someterse a vacío, donde se sitúan las células de efusión, |
|
— |
una bomba de alto vacío (por ejemplo, bomba de difusión o bomba turbomolecular), con un manómetro de vacío, |
|
— |
un sifón, con nitrógeno líquido o hielo seco. |
Como ejemplo, se presenta en la figura 5 un recinto de aluminio, calentado eléctricamente y que puede someterse a vacío, con 4 células de efusión de acero inoxidable. La hoja de acero inoxidable de unos 0,3 mm de grosor tiene un orificio de efusión de 0,2 a 1,0 mm de diámetro y está unida a la célula de efusión por una tapa de rosca.
1.6.5.2. Método de medida
Las sustancias problema y de referencia se ponen en cada célula de efusión, el diafragma metálico con el orificio se sujeta con la tapa de rosca y se pesa cada célula con precisión de 0,1 mg. Se coloca la célula en el aparato termostatizado, que se somete a vado hasta alcanzar una presión por debajo de un décimo de la presión prevista. A intervalos determinados entre 5 y 30 horas se deja entrar aire en el aparato, y la pérdida de masa de la célula de efusión se determina repitiendo la pesada.
Con el fin de garantizar que los resultados no se alteran por la presencia de impurezas volátiles, se vuelve a pesar la célula a intervalos determinados para confirmar que la velocidad de evaporación es constante al menos durante dos intervalos de tiempo.
La presión de vapor p en la célula de efusión viene dada por:
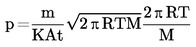
donde
|
p |
= |
presión de vapor (Pa) |
|
m |
= |
masa de sustancia que sale de la célula durante el tiempo t (kg) |
|
t |
= |
tiempo (s) |
|
A |
= |
área del orificio (m2) |
|
K |
= |
factor de corrección |
|
R |
= |
constante universal de los gases (J mol-1K-1) |
|
T |
= |
temperatura (K) |
|
M |
= |
masa molecular (kg mol-1) |
El factor de corrección K depende de la relación entre la longitud y el radio del orificio cilíndrico:
|
relación: |
0,1 |
0,2 |
0,6 |
1,0 |
2,0 |
|
K: |
0,952 |
0,909 |
0,771 |
0,672 |
0,514 |
La ecuación anterior puede escribirse:

donde  es la constante de la célula de efusión
es la constante de la célula de efusión
Esta constante de la célula de efusión E puede determinarse con sustancias de referencia (2,9) según la siguiente ecuación:
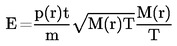
donde
|
p(r) |
= |
presión de vapor de la sustancia de referencia (Pa) |
|
M(r) |
= |
masa molecular de la sustancia de referencia (kg.mol-1) |
1.6.6. Método de saturación de gases
1.6.6.1. Aparato
El aparato utilizado en el presente método se compone típicamente de cierto número de elementos que se representan en la figura 6a y se describen a continuación (1).
Gas inerte:
El gas de arrastre no debe reaccionar químicamente con la sustancia problema. El nitrógeno sirve en la mayoría de los casos pero, a veces, pueden ser necesarios otros gases (10). El gas elegido debe estar seco (véase el número 4 de la figura 6a: sonda de humedad relativa).
Control de flujo gaseoso:
Un sistema adecuado de control de gases es indispensable para lograr un flujo constante y determinado a través de la columna de saturación.
Colectores de vapor:
Su elección depende de las características de la muestra, así como del método de análisis utilizado. El vapor debe recogerse cuantitativamente y de manera que permita el análisis posterior. Con determinadas sustancias, se utilizarán colectores con líquidos como el hexano o el etilenglicol. Con otras, se podrán utilizar absorbentes sólidos.
Como alternativa a la recogida de vapor y su análisis posterior, pueden utilizarse técnicas analíticas incorporadas en serie, como la cromatografía, para determinar la cantidad de material arrastrado por una cantidad conocida de gas de arrastre. Además, puede medirse la pérdida de masa de la muestra.
Intercambiador de calor:
Para determinaciones a diferentes temperaturas, quizá sea necesario incluir en el aparato un intercambiador de calor.
Columna de saturación
La sustancia problema se deposita a partir de una solución sobre un soporte inerte, que se introduce una vez recubierto en la columna de saturación. Las dimensiones de esta y la velocidad de salida deben elegirse de forma que garanticen la saturación completa del gas de arrastre. La columna debe estar termostatizada. En las determinaciones efectuadas a temperaturas superiores a la del ambiente, se debe calentar el espacio entre la columna y los colectores para impedir la condensación de la sustancia.
Con el fin de disminuir el arrastre de masa debido a la difusión, puede colocarse un capilar tras la columna de saturación (figura 6b).
1.6.6.2. Método de medida
Preparación de la columna de saturación:
Una vez disuelta en un disolvente muy volátil, parte de la sustancia se añade a un volumen adecuado de material de soporte. La cantidad de sustancia añadida debe ser suficiente para mantener la saturación durante todo el ensayo. El disolvente se evapora por completo al aire o en un rotavapor y se introduce el material bien homogeneizado en la columna. Una vez termostatizada la muestra, se hace pasar a través del aparato una corriente de nitrógeno seco.
Medida:
Se conectan los colectores o el detector incorporado en serie al tubo de salida de la columna y se registra el tiempo. Se comprueban la velocidad de flujo al comienzo de la experiencia y a intervalos regulares durante la misma, mediante un medidor de flujo de burbuja (o también, de forma continua, con uno de masa).
Debe medirse la presión a la salida de la columna de saturación:
|
a) |
intercalando un indicador de presión entre el saturador y los colectores ¡esto puede no ser adecuado por el aumento del espacio muerto y de la superficie de adsorción), o bien |
|
b) |
determinando los descensos de presión a través del sistema de recogida utilizado, en función de la velocidad de flujo en una prueba aparte (este método puede resultar poco satisfactorio en los colectores de líquido). |
El tiempo que se precise para recoger la cantidad de sustancia necesaria para los distintos métodos de análisis se determina durante ensayos preliminares o por estimación. Como alternativa a la recogida de sustancia problema para su análisis posterior, puede utilizarse alguna técnica analítica cuantitativa incorporada (por ejemplo, la cromatografía). Antes de calcular la presión de vapor a una temperatura dada, conviene efectuar pruebas preliminares para determinar la velocidad de flujo máxima a la que el gas de arrastre se satura completamente con vapor de la sustancia. Esto queda garantizado si el gas de arrastre pasa a través del saturador con la suficiente lentitud para que una velocidad más reducida no determine una presión de vapor calculada mayor.
La elección del método de análisis estará en función de la naturaleza de la sustancia que se vaya a ensayar (por ejemplo, cromatografía en fase gaseosa o gravimetría).
Se determina la cantidad de sustancia transportada por un volumen conocido de gas de arrastre.
1.6.6.3. Cálculo de la presión de vapor
La presión de vapor se calcula a partir de la densidad del vapor (W/V), por medio de la ecuación siguiente:
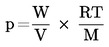
donde
|
p |
= |
presión de vapor (Pa) |
|
W |
= |
masa de sustancia problema evaporada (g) |
|
V |
= |
volumen de gas saturado (m3) |
|
R |
= |
constante universal molar de los gases (J mol-1 K-1) |
|
T |
= |
temperatura (K) |
|
M |
= |
masa molar de la sustancia problema (g mol-1) |
Los volúmenes medidos deben corregirse para tener en cuenta las diferencias de presión y temperatura entre el medidor de flujo y el saturador termostatizado. Si el medidor de flujo está situado después del colector de vapor, pueden ser necesarias ciertas correcciones para tener en cuenta la evaporación de productos contenidos en los colectores (1).
1.6.7. Rotor (8, 11, 13)
1.6.7.1. Aparato
La técnica de rotor puede aplicarse utilizando un viscosímetro de rotor como el que se muestra en la figura 8. En la figura 7 aparece un esquema de montaje experimental.
El aparato de medida consiste típicamente en una cabeza de medida de rotor, incluida en un recinto termostatizado (regulado con una precisión de 0,1 oC). El recipiente con la muestra se coloca en un recinto termostatizado (regulado con una precisión de 0,01 oC) y todas las demás partes del montaje se mantienen a una temperatura superior con el fin de evitar la condensación. Una bomba de alto vacío se conecta al sistema por medio de llaves de alto vacío.
La cabeza de medida de rotor consiste en una bola de acero (de 4 a 5 mm de diámetro) dentro de un tubo. La bola se suspende y estabiliza en un campo magnético, generalmente mediante la combinación de imanes permanentes y de bobinas de control.
Se hace girar la bola por campos giratorios producidos por las bobinas. Unas bobinas de recepción, que miden la magnetización continua, baja y lateral de la bola hacen que se pueda medir la velocidad de giro.
1.6.7.2. Método de medida
Cuando la bola alcanza una velocidad angular dada v(o) (generalmente, unas 400 revoluciones por segundo), se interrumpe la activación y se produce el frenado debido a la fricción con el gas.
El descenso de la velocidad angular se mide en función del tiempo. Como la fricción causada por la suspensión magnética es despreciable en comparación con la fricción por el gas, la presión p del gas viene dada por la siguiente fórmula:

donde
|
|
= |
velocidad media de las moléculas del gas |
|
r |
= |
radio de la bola |
|
p |
= |
densidad másica de la bola |
|
a |
= |
coeficiente de transferencia del momento tangencial (o = 1 para superficie esférica ideal de la bola) |
|
t |
= |
tiempo |
|
v(t) |
= |
velocidad angular al cabo del tiempo t |
|
v(o) |
= |
velocidad angular inicial |
Esta ecuación puede escribirse también:
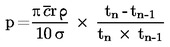
donde tn y tn-1 son los tiempos necesarios para que se dé un número N determinado de revoluciones. Estos intervalos de tiempo, tn y tn-1, son sucesivos y tn > tn-1.
La velocidad media E![]() de las moléculas de gas es:
de las moléculas de gas es:
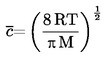
donde
|
T |
= |
temperatura |
|
R |
= |
constante universal molar de los gases |
|
M |
= |
masa molar |
2. RESULTADOS
Cualquiera que sea el método elegido, la presión de vapor debe determinarse, al menos, a dos niveles de temperatura. Son preferibles tres niveles, o más, entre 0 oC y 50 oC, para comprobar la linealidad de la curva de la presión de vapor.
3. INFORME
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
método utilizado, |
|
— |
especificación precisa de la sustancia (identidad e impurezas) y, en su caso, fase de purificación previa, |
|
— |
dos valores al menos de presión de vapor y temperatura, preferentemente entre 0 y 50 oC, |
|
— |
todos los datos originales, |
|
— |
una gráfica de log p frente a 1/T, |
|
— |
una estimación de la presión de vapor a 20 o 25 oC. |
En caso de transición (cambio de estado, descomposición), hay que incluir:
|
— |
descripción del cambio, |
|
— |
temperatura a la que se produce la presión atmosférica, |
|
— |
presión de vapor a 10 y a 20 oC por debajo de la temperatura de transición, así como a 10 y 20 o C por encima de dicha temperatura (excepto en el caso de transición del estado sólido al estado gaseoso). |
Deben señalarse todas las informaciones y observaciones útiles para la interpretación de los resultados, en particular lo referente a las impurezas y al estado físico de la sustancia.
4. BIBLIOGRAFÍA
|
(1) |
OCDE, París, 1981 Test Guideline 104, Decision of the Council C(81) 30 final. |
|
(2) |
Ambrose, B. Le Neindre, B.; B. Vodar, (Eds.): Experimental Thermodynamics, Butterworths, Londres, 1975, vol. II. |
|
(3) |
R. Weissberger ed.: Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed. Chapter IX, Interscience Publ., Nueva York, 1959, Vol. I, Pan I. |
|
(4) |
Knudsen, M. Ann. Phys. Lpz., 1909, vol. 29, 1979; vol, 34, 593. |
|
(5) |
NF T 20-048 AFNOR (Sept. 85). Chemical producís for industria) use — Determination of vapour pressure of solids and liquids within range from 10-1 to 105 Pa — Static method. |
|
(6) |
NF T 20-047 AFNOR (Sept. 85). Chemical products for industrial use — Determination of vapour pressure of solids and liquids within range from 10-3 to 1 Pa — Vapor pressure balance method. |
|
(7) |
ASTM D 2879-86, Standard test method for vapour pressure- temperature relationship and inicial decomposition temperature of liquids by isoteniscope. |
|
(8) |
G. Messer, P. Röhl, G. Grosse y W. Jitschin. J. Vac. Sci. Technol.(A), 1987, vol. 5, (4), 2440. |
|
(9) |
Ambrose, D.; Lawrenson, I..; Sprake, C.H.S. J. Chem. Thermodynamics 1975, vol. 7, 1173. |
|
(10) |
B.F. Rordorf. Thermochimica Acta, 1985, vol, 85, 435. |
|
(11) |
G. Comsa, J.K. Fremerey y B. Lindenau. J. Vac. Sci. Technol., 1980, vol. 17 (2), 642. |
|
(12) |
G. Reich. J. Vac. Sci. Technol., 1982, vol. 20 (4), 1148 |
|
(13) |
J.K. Fremerey. J. Vac. Sci. Technol.(A), 1985, vol. 3 (3), 1715, |
Apéndice 1
Método de estimación
INTRODUCCIÓN
Los valores calculados de presión de vapor pueden utilizarse:
|
— |
para decidir cuál de los métodos experimentales es el adecuado, |
|
— |
para dar un valor estimado o un valor límite en casos en que el método experimental no pueda aplicarse por causas técnicas (incluida la extrema debilidad de la presión de vapor), |
|
— |
para contribuir a señalar aquellos casos en que la omisión de la medida experimental se justifica por la probabilidad de que la presión de vapor sea < 10-5 Pa a temperatura ambiente. |
MÉTODO DE ESTIMACIÓN
La presión de vapor de los sólidos y líquidos puede estimarse con la correlación modificada de Watson (a). El único dato experimental necesario es el punto normal de ebullición. El método puede aplicarse en el intervalo de presiones de 105 Pa a 10 -5 Pa.
En el «Handbook of Chemical Property Estimation Methods» viene información detallada sobre el método (b).
CÁLCULO
Según (b), la presión de vapor se calcula de la forma siguiente:

donde
T= temperatura correspondiente
Tb= punto normal de ebullición
Pvp= presión de vapor a la temperatura T
AHVp= calor de vaporización
AZb= factor de compresibilidad (estimado en 0,97)
m= factor empírico que depende del estado físico a la temperatura correspondiente
Además,
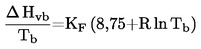
donde Kf es un factor empírico que tiene en cuenta la polaridad de la sustancia. La referencia (b) da los factores Kf de varios tipos de compuestos.
Es bastante frecuente tener datos en que figura un punto de ebullición a presión reducida. En estos casos, según (b), la presión de vapor se calcula con arreglo a la fórmula:

donde T1 es el punto de ebullición a la presión reducida P1.
INFORME
Cuando se siga el método de estimación, el informe incluirá una extensa documentation de los cálculos.
BIBLIOGRAFÍA
|
(a) |
K. M. Watson, Ind. Eng. Chem., 1943, vol. 35, 398. |
|
(b) |
W.J. Lyman, W.F. Reehl, D.H. Rosenblatt. Handbook of Chemical Property Estimation Methods, Ac Graw-Hill, 1982. |
Apéndice 2
Figura 1
Aparato para determinar la curva de presión de vapor, según el método dinámico

Figura 2a
Aparato para determinar la curva de presión de vapor según el método estático (con manómetro de tubo en U)

Figura 2b
Aparato para determinar la curva de presión de vapor según el método estático (con indicador de presión)
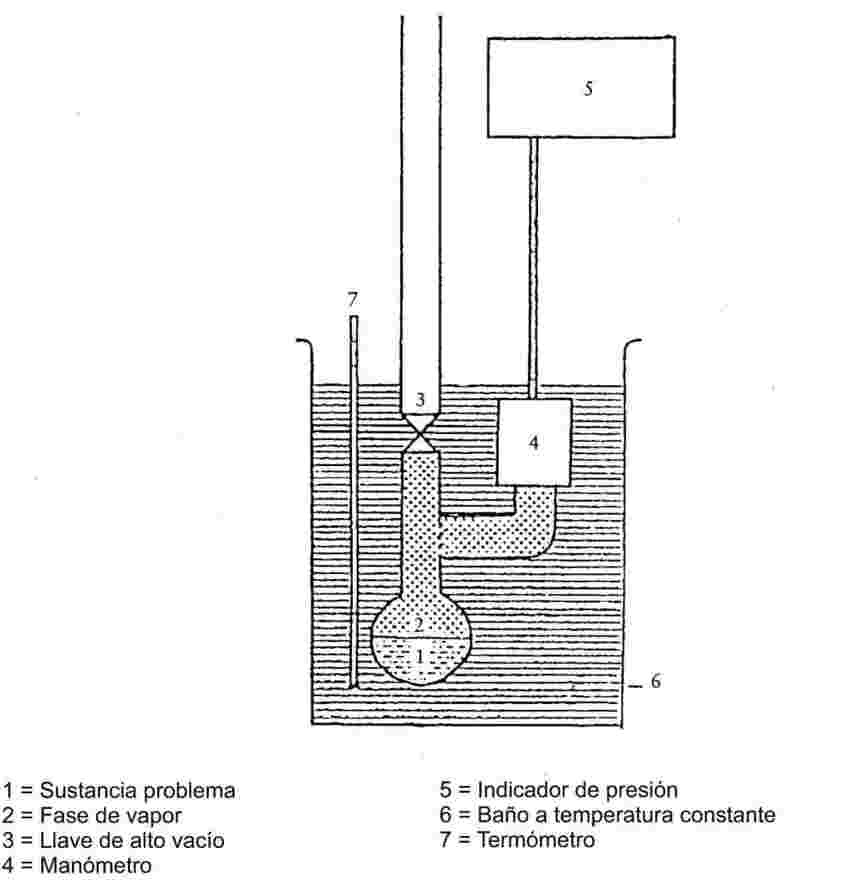
Figura 3
Isoteniscopio (véase la referencia 2)
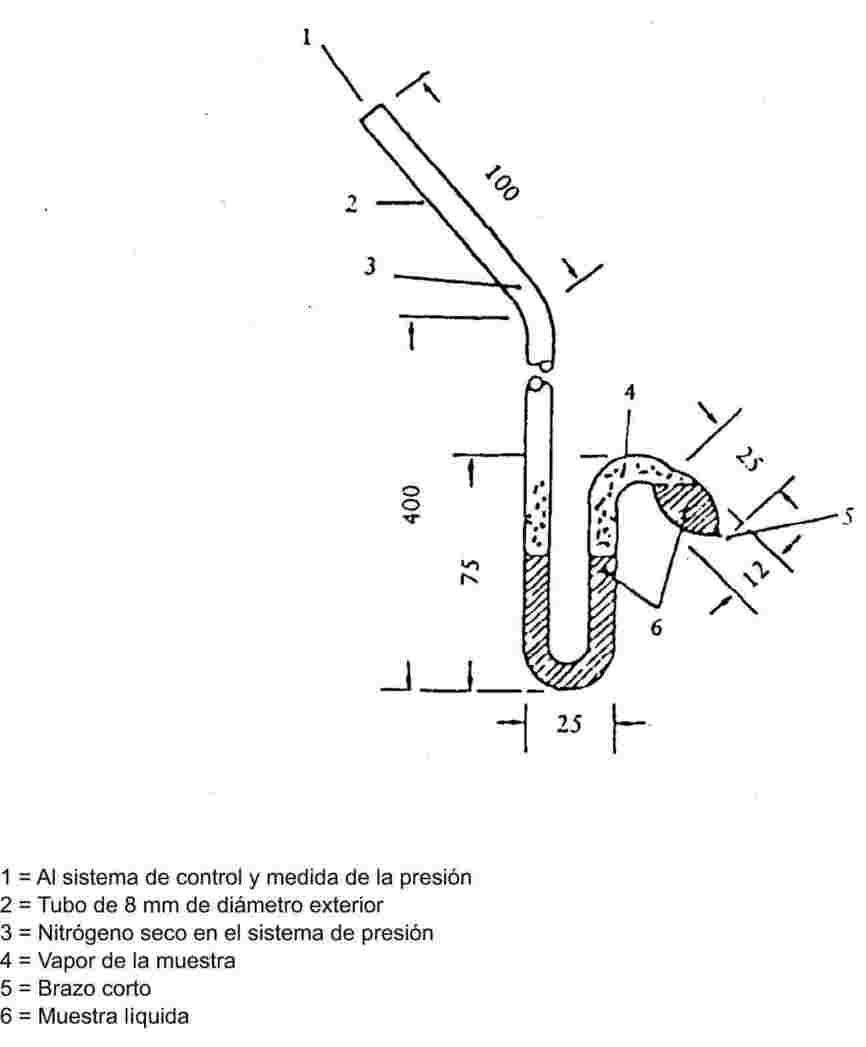
Figura 4
Aparato para determinar la curva de presión de vapor, según el método de la balanza de presión de vapor
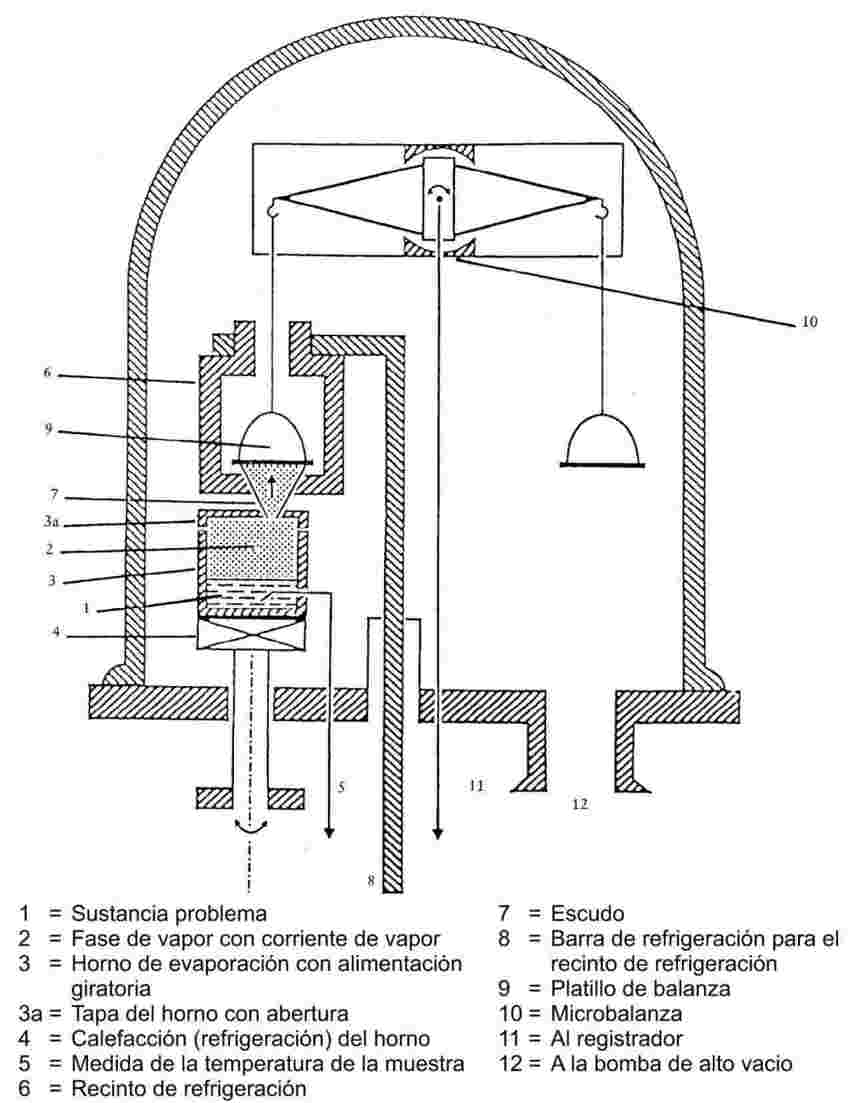
Figura 5
Ejemplo de aparato de evaporación a baja presión por el método de efusión, con una célula de efusión de 8 cm3 de volumen

Figura 6a
Modelo de aparato que permite determinar la presión de vapor, por el método de saturación de gases
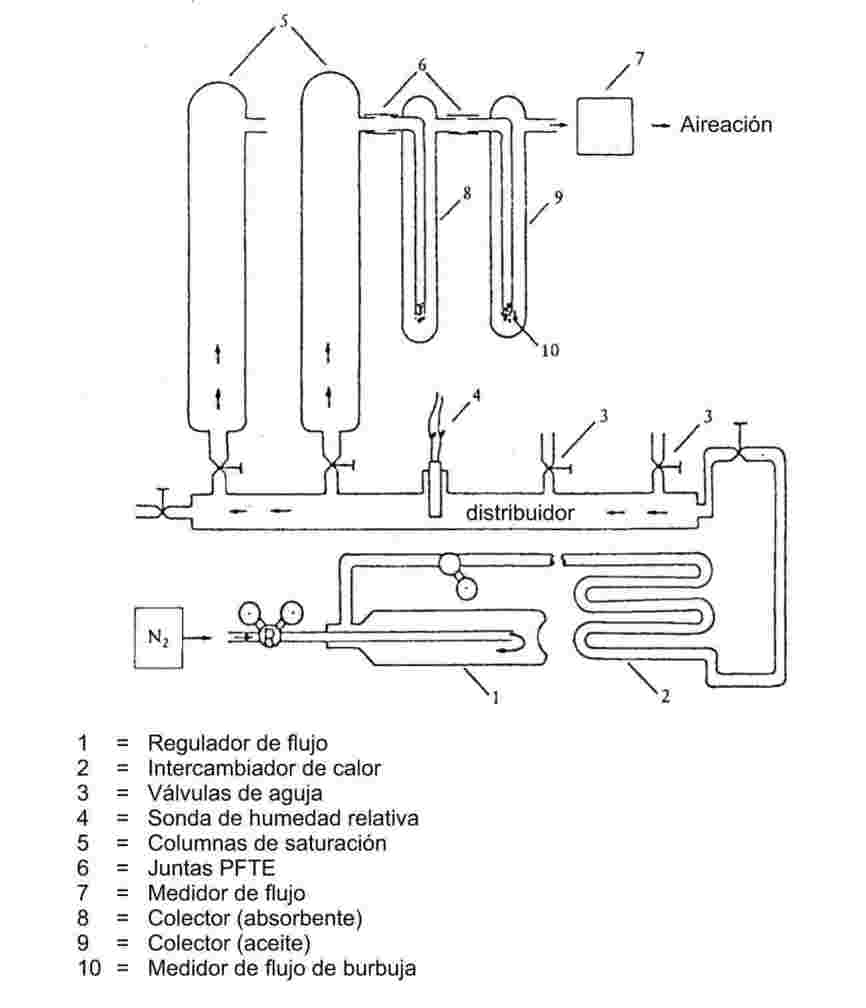
Figura 6b
Modelo de aparato para la determinación de la presión de vapor por el método de saturación de gases, con un capilar tras la cámara de saturación

Figura 7
Ejemplo de montaje experimental para el método del rotor
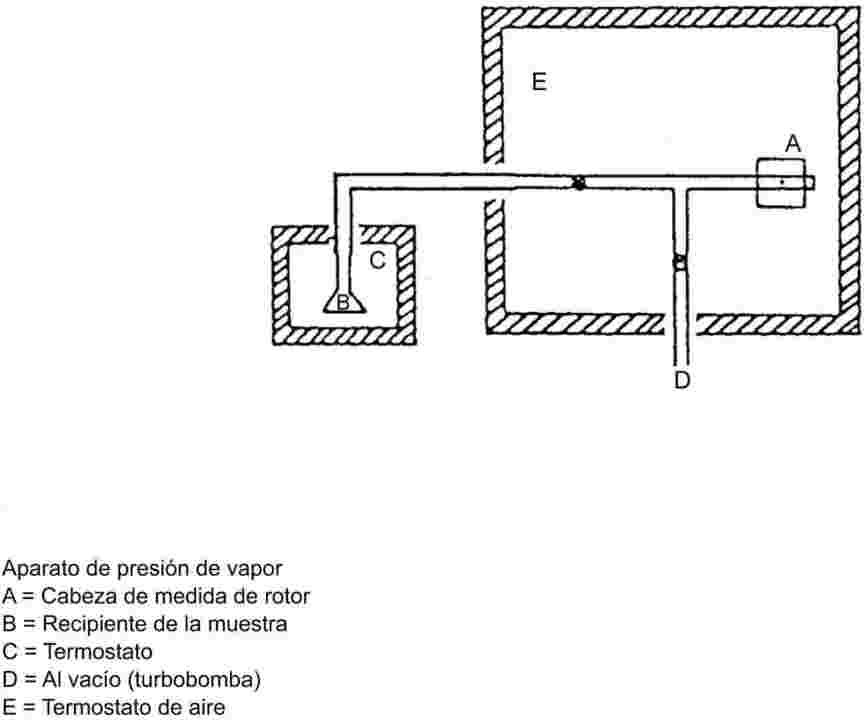
Figura 8
Ejemplo de cabeza de medida de rotor

A.5. TENSIÓN SUPERFICIAL
1. MÉTODO
Los métodos descritos se basan en las líneas directrices de la OCDE (1). Los principios fundamentales se dan en la referencia (2).
1.1. INTRODUCCIÓN
Los métodos descritos se aplican a la medida de la tensión superficial de las soluciones acuosas.
Antes de efectuar el ensayo, es conveniente disponer de información preliminar sobre la solubilidad en el agua, la estructura, las propiedades de hidrólisis y la concentración crítica para la formación de micelas de la sustancia.
Los siguientes métodos son aplicables a la mayor parte de las sustancias químicas, cualquiera que sea su grado de pureza.
Las medidas de la tensión superficial por el método del tensiómetro de anillo se limita a las soluciones acuosas con una viscosidad dinámica inferior a 200 mPa s, aproximadamente.
1.2. DEFINICIONES Y UNIDADES
La entalpia de superficie libre por unidad de superficie constituye la tensión superficial.
Esta última se expresa en:
N/m (en unidades SI) o
mN/m (en subunidades SI)
1 N/m = 103 dinas/cm
1 mN/m = 1 dina/cm en el sistema CGS (en desuso).
1.3. SUSTANCIAS DE REFERENCIA
No es necesario emplear sustancias de referencia cada vez que se estudie una sustancia nueva. Se deben utilizar esencialmente para comprobar la validez del método de vez en cuando y para poder comparar con los resultados obtenidos según otros métodos.
En las referencias (1) y (3) se señalan sustancias de referencia que cubren una amplia gama de tensiones superficiales.
1.4. PRINCIPIOS DE LOS MÉTODOS
Los métodos se basan en la medida de la fuerza máxima que es preciso ejercer verticalmente sobre una brida o anillo en contacto con la superficie del líquido estudiado, colocado en un recipiente adecuado, a fin de separarlo de dicha superficie, o sobre una placa, uno de cuyos bordes esté en contacto con la superficie, a fin de elevar la película formada.
Las sustancias que sean hidrosolubles al menos a la concentración de 1 mg/1 se someterán al ensayo en solución acuosa a una sola concentración.
1.5. CRITERIOS DE CALIDAD
La precisión de los métodos descritos es mayor de la que normalmente se requiere en la evaluación del riesgo para el medio ambiente.
1.6. DESCRIPCIÓN DE LOS MÉTODOS
Se prepara una solución de la sustancia en agua destilada. La concentración de esta solución debe ser el 90 % de la concentración de saturación de la sustancia en agua; si esta concentración es superior a 1 g/1, se utilizará para el ensayo la concentración de 1 g/1. Las sustancias cuya solubilidad en agua es inferior a 1 mg/1 no tienen que someterse al ensayo.
1.6.1. Método de la placa
Véase ISO 304 y NF T 73-060 (Tensioactivos: determinación de la tensión superficial por elevación de películas líquidas).
1.6.2. Método de la brida
Véase ISO 304 y NF T 73-060 (Tensioactivos: determinación de la tensión superficial por elevación de películas líquidas).
1.6.3. Método del anillo
Véase ISO 304y NF T 73-060 (Tensioactivos: determinación de la tensión superficial por elevación de películas líquidas).
1.6.4. Método de) anillo armonizado por la OCDE
1.6.4.1. Aparato
Para efectuar esta medida pueden emplearse tensiómetros comerciales. Constan de los elementos siguientes:
|
— |
un portamuestras móvil, |
|
— |
un dinamómetro, |
|
— |
un cuerpo de medida (anillo), |
|
— |
un recipiente de medida.1.6.4.1.1. |
1.6.4.1.1. Portamuestras móvil
El portamuestras móvil sirve de soporte al recipiente de medida termostatizado, que contiene el líquido problema. Va montado en la misma peana que el dinamómetro.
1.6.4.1.2. Dinamómetro
El dinamómetro (véase la figura) va colocado por encima del portamuestras. El error en la medida de la fuerza no debe pasar de ± 10-6 N, lo que equivale a un límite de error de ±0,1 mg en una medida de masa. En la mayoría de los casos, la escala de medidas de los tensiómetros vendidos en el mercado está calibrada en mN/m, de manera que se puede leer directamente la tensión superficial en mN/m con una precisión de 0,1 mN/m.
1.6.4.1.3. Cuerpo de medida (anillo)
El anillo suele estar formado por un hilo de platino-iridio de 0,4 mm de grosor, aproximadamente, y una circunferencia media de 60 mm. Dicho anillo está suspendido horizontalmente de una armadura que va unida por una varilla metálica al dinamómetro (véase la figura).
Figura
Cuerpo de medida
(todas las dimensiones se expresan en milímetros)
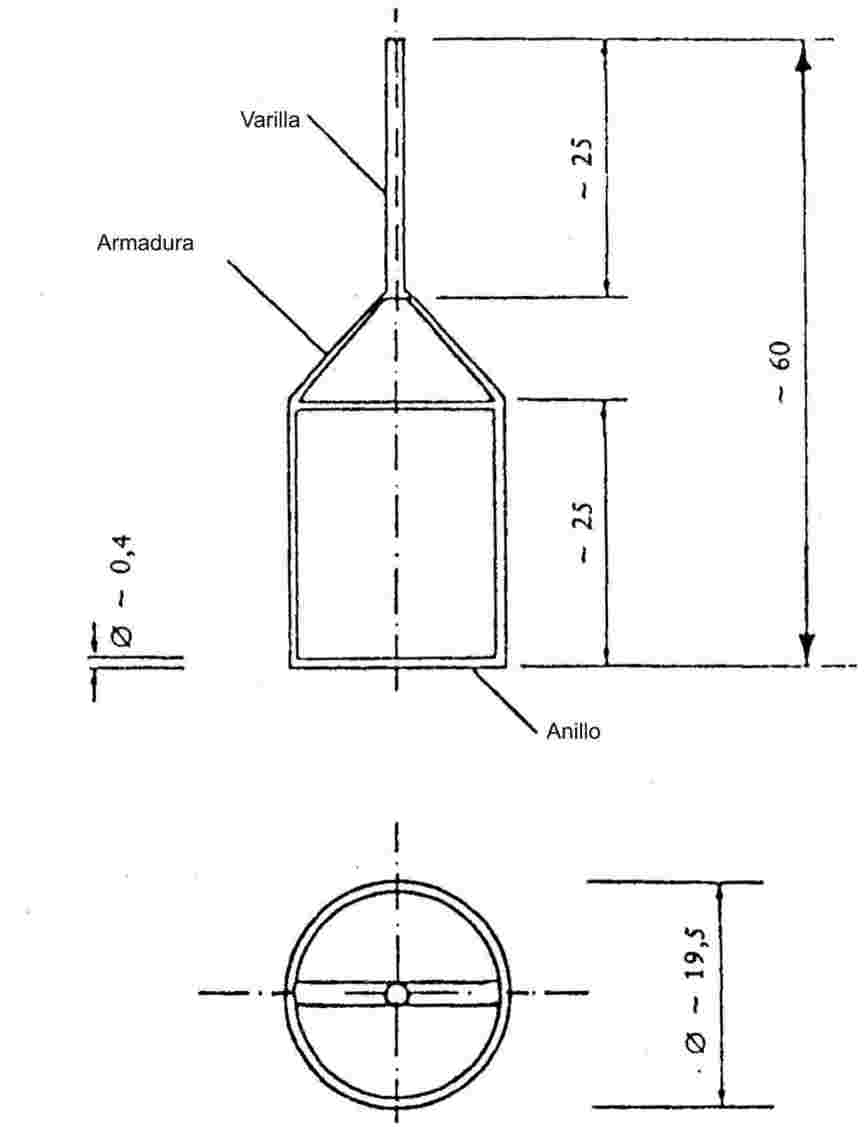
1.6.4.1.4. Recipiente de medida
El recipiente de medida que contiene la solución problema debe ser un recipiente de vidrio termostatizado. Debe estar diseñado de manera que, durante la determinación, la temperatura del líquido y de la fase gaseosa por encima de su superficie se mantenga constante y no haya evaporación. Los recipientes cilíndricos de vidrio de un diámetro interior de, al menos, 45 mm cumplen estos requisitos.
1.6.4.2. Preparación del aparato
1.6.4.2.1. Limpieza
Los recipientes de vidrio deben limpiarse con cuidado. Si es necesario se lavarán con mezcla sulfocrómíca caliente y, a continuación, con ácido fosfórico viscoso (83 a 98 % en peso de H3PO4); se enjuagarán abundantemente con agua corriente y luego con agua bidestilada hasta el momento en que se obtenga reacción neutra; finalmente, se secarán o bien se enjuagarán con parte del líquido que se va a medir.
El anillo se limpiará con abundante agua para eliminar todos los restos de sustancias solubles en agua, se sumergerá unos segundos en mezcla sulfocrómica, se enjuagará con agua bidestilada hasta que se obtenga reacción neutra y, finalmente, se secará rápidamente sobre una llama de metanol.
Nota:
Los restos de sustancias que no se disuelvan ni se destruyan con la mezcla sulfocrómica ni con el ácido fosfórico, como las siliconas, deberán ser eliminadas con un disolvente orgánico apropiado.
1.6.4.2.2. Calibrado del aparato
La validación del aparato consiste en comprobar el punto cero y en ajustarlo de tal manera que la indicación dada por el aparato permita una determinación fiable en mN/m.
Montaje:
Se nivelará el aparato, por ejemplo con un nivel de burbuja colocado sobre la peana y con tornillos de nivelación.
Ajuste del punto cero:
Después de montar el anillo en el aparato y antes de sumergirlo en el líquido, debe ajustarse a cero el indicador del tensiómetro y comprobarse el paralelismo del anillo con la superficie del líquido. A tal fin, puede utilizarse la superficie del líquido como espejo.
Calibrado:
El calibrado en sí del aparato puede hacerse con uno de los dos métodos siguientes:
|
a) |
con una masa: este procedimiento consiste en utilizar cumiapesos de masa conocida (entre 0,1 g y 1,0 g), que se colocan sobre el anillo. El factor de calibrado Φa, por el que habrá que multiplicar todas las lecturas del instrumento, se determina mediante la ecuación (1):
donde
|
|
b) |
con agua: este procedimiento utiliza agua pura cuya tensión superficial, por ejemplo a 23 oC, es igual a 72,3 mN/m. Es más rápido que el sistema anterior, pero siempre existe el riesgo de que ¡a tensión superficial del agua se vea modificada por restos de tensioactivos. El factor de calibradoΦb, por el que habrá que multiplicar todas las lecturas del aparato, se determina mediante la ecuación siguiente (2);
donde
|
1.6.4.3. Preparación de la muestra
Las soluciones acuosas de la sustancia se prepararán teniendo en cuenta las concentraciones exigidas. Es imprescindible que la disolución de la sustancia sea completa.
La solución así preparada debe mantenerse a una temperatura constante (±0,5 oC). Dado que la tensión superficial de una solución colocada en el recipiente de medida se modifica después de cierto tiempo, deben efectuarse varias medidas en momentos diferentes y hay que trazar una curva de la tensión superficial en función del tiempo. Cuando ya no haya modificaciones, se habrá alcanzado un estado de equilibrio.
El polvo o los vapores de otras sustancias falsean la medida. La operación debe realizarse bajo una campana de protección.
1.6.5. Condiciones del ensayo
Las medidas deben efectuarse a una temperatura de unos 20 oC, sin variaciones de temperatura superiores a ±0,5 oC.
1.6.6. Desarrollo del ensayo
Se vierte en el recipiente de medida, cuidadosamente limpio, la solución que se desea medir, procurando evitar la formación de burbujas y, a continuación, se coloca el recipiente de medida en el portamuestras, elevándolo hasta que el anillo quede sumergido bajo la superficie de la solución. Entonces se vuelve a bajar gradual y uniformemente (a una velocidad de unos 0,5 cm/min) el portamuestras para separar ei anillo de la superficie hasta llegar a la fuerza máxima. La capa de líquido adherida al anillo no debe desprenderse de este. Una vez acabada la medida, se sumerge de nuevo el anillo y se repiten las medidas hasta que se obtenga un valor de tensión superficial constante. En cada determinación se anotará el tiempo que haya transcurrido desde la transferencia de la solución al recipiente de medida. Las lecturas deben registrarse en el valor máximo de la fuerza necesaria para retirar el anillo de la superficie del líquido.
2. RESULTADOS
Para calcular la tensión superficial se multiplicará primero el valor leído en el aparato en mN/m por el factor de calibrado Φa o Φb(según el procedimiento de calibrado utilizado). Se obtendrá entonces un valor solo aproximadoque, a continuación, deberá corregirse.
Harkins y Jordán (4) han establecido de manera empírica factores de corrección para los valores de tensión superficial obtenidos por el método del anillo, que dependen de las dimensiones del anillo, de la densidad del líquido y de su tensión superficial.
Dada la laboriosidad del proceso de determinación del factor de corrección para cada medida a partir de las tablas de Harkins y Jordán, en las soluciones acuosas se admite la utilización de un procedimiento simplificado de lectura de la tensión superficial tomando directamente los valores corregidos de la tabla siguiente (se recurrirá a la interpolación en las lecturas que se sitúen entre dos valores de la tabla).
Tabla
Corrección de los valores medidos de la tensión superficial
Válido solamente para las soluciones acuosas p = 1 g/cm3
|
R |
= 9,55 mm (radio medio del anillo) |
|
r |
= 0,185 mm (radio del hilo que constituye el anillo) |
|
Valor experimental (mN/m) |
Valor corregido (mN/m) |
|
|
Calibrado con contrapeso [véase el punto 1.6.4.2.2.(a)] |
Calibrado con agua [véase el punto 1.6.4.2.2.(b)] |
|
|
20 |
16,9 |
18,1 |
|
22 |
18,7 |
20,1 |
|
24 |
20,6 |
22,1 |
|
26 |
22,4 |
24,1 |
|
28 |
24,3 |
26,1 |
|
30 |
26,2 |
28,1 |
|
32 |
28,1 |
30,1 |
|
34 |
29,9 |
32,1 |
|
36 |
31,8 |
34,1 |
|
38 |
33,7 |
36,1 |
|
40 |
35,6 |
38,2 |
|
.42 |
37,6 |
40,3 |
|
44 |
39,5 |
42,3 |
|
46 |
41,4 |
44,4 |
|
48 |
43,4 |
46,5 |
|
50 |
45,3 |
48,6 |
|
52 |
47,3 |
50,7 |
|
54 |
49,3 |
52,8 |
|
56 |
51,2 |
54,9 |
|
58 |
53,2 |
57,0 |
|
60 |
55,2 |
59,1 |
|
62 |
57,2 |
61,3 |
|
64 |
59,2 |
63,4 |
|
66 |
61,2 |
65,5 |
|
68 |
63,2 |
67,7 |
|
70 |
65,2 |
69,9 |
|
72 |
67,2 |
72,0 |
|
74 |
69,2 |
— |
|
76 |
71,2 |
— |
|
78 |
73,2 |
— |
Esta tabla se ha elaborado a partir de la corrección de Harkins Jordan, y es similar a la de la norma DIN (DIN 53914) para agua y soluciones acuosas (densidad p = 1 g/cm3); se utiliza con un anillo existente en el mercado cuyas dimensiones son R = 9,55 mm (radio medio del anillo) y r = 0,185 mm (radio del hilo que constituye el anillo). La tabla da los valores corregidos de las medidas de tensión superficial efectuadas tras un calibrado con contrapesos o con agua.
Otra posibilidad, sin calibrado previo, consiste en calcular la tensión superficial por medio de la fórmula siguiente:
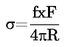
donde
|
F |
= |
fuerza medida en el dinamómetro en el punto de ruptura de la película |
|
R |
= |
radio del anillo |
|
f |
= |
factor de corrección (1). |
3. INFORME
3.1. INFORME DEI ENSAYO
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
método utilizado, |
|
— |
tipo de agua o de solución utilizada, |
|
— |
especificación precisa de la sustancia (identidad e impurezas), |
|
— |
resultados de las medidas: tensión superficial (leída) incluyendo a la vez las diferentes lecturas y su medida aritmética, así como el valor medio corregido (teniendo en cuenta el factor específico del equipo y la tabla de corrección), |
|
— |
concentración de la solución, |
|
— |
temperatura del ensayo, |
|
— |
tiempo de la solución empleada; en particular, el tiempo transcurrido entre la preparación de la solución y su medida, |
|
— |
evolución de la tensión superficial de la solución con el tiempo a partir del momento en que se haya transferido al recipiente de medida, |
|
— |
deben señalarse todas las informaciones y observaciones que sean útiles para la interpretación de los resultados, especialmente lo referente a las impurezas y al estado físico de la sustancia. |
3.2. INTERPRETACIÓN DE LOS RESULTADOS
Dado que el agua destilada tiene una tensión superficial de 72,75 mN/m a 20 oC, se considerarán sustancias tensioactivas aquellas que presenten una tensión superficial inferior a 60 mN/m en las condiciones de este método.
4. BIBLIOGRAFÍA
|
(1) |
OCDE, París, 1981, Test Guideline 115, Decision of the Council C(81) 30 final. |
|
(2) |
R. Weissberger ed., Technique of Organic Chemistry, Chapter XIV, Physical Methods of Organic Chemistry, 3rd ed. Interscience Publ., Nueva York, 1959, vol. I, Part I. |
|
(3) |
Pure Appl. Chem., 1976, Vol. 48, 511 |
|
(4) |
Harlcins, W.D., Jordan, H.F., J. Amer. Chem. Soc, 1930, Vol. 52, 1751. |
A.6. HIDROSOLUBILIDAD
1. MÉTODO
Los métodos descritos se basan en las líneas directrices de la OCDE (1).
1.1. INTRODUCCIÓN
Antes de proceder al ensayo, es conveniente disponer de información sobre la fórmula desarrollada, la presión de vapor, la constante de disociación y la hidrólisis (como función del pH) de la sustancia.
No existe un solo método que cubra toda la gama de solubilidades en el agua.
Los dos métodos de ensayo descritos a continuación abarcan la gama completa de solubilidades pero no son aplicables a sustancias volátiles:
|
— |
el primero, denominado en lo sucesivo «método de elución en columna», se aplica a las sustancias esencialmente puras con escasa solubilidad (< 10-2 g/l) y estables en el agua, |
|
— |
el segundo, denominado en lo sucesivo «método del frasco», se aplica a las sustancias esencialmente puras de solubilidad mayor (> 10-2 g/l) y estables en el agua. |
La hidrosolubilidad de la sustancia puede verse considerablemente afectada por la presencia de impurezas.
1.2. DEFINICIONES Y UNIDADES
La hidrosolubilidad de una sustancia es la concentración de saturación de masa de la sustancia en el agua a una temperatura determinada. Se expresa en unidades de masa por volumen de solución. La unidad SI es el kg/m3 (se puede utilizar también g/l).
1.3. SUSTANCIAS DE REFERENCIA
No es necesario emplear sustancias de referencia cada vez que se analice una sustancia nueva. Deben servir esencialmente para controlar de vez en cuando la validez del método y permitir la comparación con los resultados obtenidos según otros métodos.
1.4. PRINCIPIO DEL MÉTODO DE ENSAYO
La cantidad aproximada de muestra y el tiempo necesario para conseguir la concentración de saturación de masa deben determinarse mediante un ensayo preliminar sencillo.
1.4.1. Método de elución en columna
Este método se basa en la elución con agua de la sustancia problema en una microcolumna cargada con un material soporte inerte como, por ejemplo, bolas de cristal o arena, cubierto con un exceso de sustancia problema. La hidrosolubilidad se determina cuando la concentración en masa del eluato es constante, lo que se indica con una meseta de la gráfica en función del tiempo.
1.4.2. Método del frasco
En este método, la sustancia (los sólidos deben estar pulverizados) se disuelve en agua a una temperatura algo superior a la temperatura de ensayo. Cuando se alcanza la saturación, se enfría la mezcla, se mantiene a la temperatura de ensayo y se agita hasta alcanzar el equilibrio. Otra posibilidad es realizar la medida directamente a la temperatura de ensayo si, mediante un muestreo adecuado, se garantiza que se ha alcanzado el equilibrio de saturación. Luego, se determina con un método analítico apropiado la concentración en masa de la sustancia en la solución acuosa, que no debe contener ninguna partícula sin disolver.
1.5. CRITERIOS DE CALIDAD
1.5.1. Repetibilidad
En lo que respecta al método de elución en columna, se puede conseguir < 30 %; en cuanto al método del frasco, la repetibilidad deberá ser inferior al 15 %.
1.5.2. Sensibilidad
Depende del método de análisis, pero la concentración se puede determinar hasta 10-6 g/l.
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Condiciones del ensayo
El ensayo debe efectuarse, a ser posible, a 20 oC ±0,5 oC. Si se teme que la temperatura repercuta en la solubilidad (> 3 % por oC), se utilizarán también otras dos temperaturas, una superior y otra inferior al menos en 10 oC a la temperatura inicial elegida. En este caso, la temperatura debe ajustarse a ±0,1 oC. La temperatura elegida se mantendrá constante en todas las partes importantes del equipo.
1.6.2. Ensayo preliminar
En una probeta de 10 ml con tapón de vidrio, que contenga alrededor de 0,1 g de muestra (las sustancias sólidas deben estar reducidas a polvo), añadir volúmenes crecientes de agua destilada a temperatura ambiente, siguiendo la progresión indicada en el cuadro siguiente:
|
0,1 g soluble en «x» ml de agua |
0,1 |
0,5 |
1 |
2 |
10 |
100 |
> 100 |
|
solubilidad aproximada (g/l) |
> 1 000 |
1 000-200 |
200-100 |
100-50 |
50-10 |
10-1 |
< 1 |
Después de la adición de cada una de las cantidades de agua indicadas, agitar vigorosamente la mezcla durante 10 minutos y luego comprobar visualmente si contiene partículas de muestra no disueltas. Si después de añadir 10 mm de agua, la muestra o determinadas partes de esta no se han disuelto, hay que repetir el experimento en una probeta de 100 ml con mayores volúmenes de agua. Si la solubilidad es escasa, el tiempo necesario para disolver la sustancia puede ser considerablemente más largo (hay que dejar al menos 24 horas). La solubilidad aproximada se indica, en el cuadro, bajo el volumen de agua añadida en que se efectúa la disolución completa de la muestra. Si la sustancia se mantiene aparentemente insoluble, hay que dejar más de 24 horas (96 horas como máximo) o bien proceder a una nueva dilución a fin de determinar si hay que utilizar el método de elución en columna o el método de solubilidad en frasco.
1.6.3. Método de elución en columna
1.6.3.1. Material soporte, disolvente y eluyente
En el método de elución en columna, el material soporte debe ser inerte. Se pueden emplear bolas de cristal y arena. Para aplicar la sustancia problema al soporte, se utilizará un disolvente volátil adecuado de pureza de reactivo para análisis. Debe emplearse como eluyente agua bidestilada procedente de un aparato de vidrio o de cuarzo.
Nota:
No debe utilizarse agua procedente directamente de un intercambiador de iones orgánico.
1.6.3.2. Carga del soporte
Pesar unos 600 mg de material soporte y pasarlos a un matraz de fondo redondo de 50 ml.
Disolver una cantidad idónea y pesada de la sustancia problema en el disolvente elegido. Añadir al soporte una cantidad adecuada de esta solución. El disolvente debe evaporarse por completo, por ejemplo en un rotavapor; de lo contrario, no se daría la saturación en agua del soporte debido al efecto de partición en la superficie del soporte.
La carga del soporte puede plantear dificultades (resultados erróneos) si la sustancia problema se deposita en forma de aceite o en una fase cristalina diferente. La dificultad debe examinarse experimentalmente y recoger los datos correspondientes.
Dejar que se empape el soporte cargado de esta forma durante unas 2 horas en 5 ml de agua, aproximadamente, y luego pasar la suspensión a la microcolumna. También se podrá verter en esta última el soporte cargado seco, después de haber llenado previamente la microcolumna de agua y se equilibra después durante unas 2 horas.
Procedimiento:
La elución de la sustancia a partir del soporte puede efectuarse mediante dos sistemas diferentes:
|
— |
bomba de recirculación (véase la figura 1), |
|
— |
recipiente de nivelación (véase la figura 4). |
1.6.3.3. Método de elución en columna con bomba de recirculación
Aparato
En la figura 1 se muestra el esquema de un sistema que se utiliza normalmente. La figura 2 representa una microcolumna idónea, pero cualquier otra de dimensiones diferentes es aceptable, siempre que se mantengan los criterios de reproducibilidad y de sensibilidad. La columna debe tener un espacio de cabeza correspondiente al menos a 5 volúmenes de lecho de agua y una capacidad mínima de 5 muestras. No obstante, se puede reducir la dimensión si los 5 volúmenes de lecho iniciales, eliminados con las impurezas, se sustituyen por disolvente de relleno.
La columna debe estar unida a una bomba de recirculación que garantice un caudal aproximado de 25 ml/hora. Las conexiones de la bomba serán de politetrafluoretileno (PTFE) o vidrio. La columna y la bomba, cuando estén unidas, deben permitir la toma de muestras del efluente y el equilibrado a presión atmosférica del espacio de cabeza. El material de la columna se retendrá con un pequeño tapón de lana de vidrio (5 mm) que servirá también para separar las partículas por filtración. Como bomba de recirculación se podrá utilizar, por ejemplo, una bomba peristáltica o una bomba de membrana (hay que procurar que el material del tubo no sea causa de contaminación ni adsorción).
Procedimiento de medida
Se inicia la circulación a través de la columna. El caudal recomendado es de 25 ml/hora, aproximadamente (unos 10 volúmenes de lecho por hora para la columna arriba descrita). Los 5 primeros volúmenes de lecho (mínimo) se descartan a fin de eliminar las impurezas solubles en el agua. Luego se conecta la bomba de recirculación, haciendo funcionar el aparato hasta llegar al estado de equilibrio, definido por 5 muestras sucesivas cuyas concentraciones no difieran de forma aleatoria en más de ± 30 %. Dichas muestras deben estar separadas entre sí por un intervalo de tiempo correspondiente al paso de al menos 10 volúmenes de lecho del eluyente.
1.6.3.4. Método de elución en columna con recipiente de nivelación
Aparato (véanse las figuras 3 y 4)
Recipiente de nivelación: la conexión con este recipiente se hace mediante una junta de vidrio esmerilado, unida por tubos de politetrafluoretileno. El caudal recomendado es de unos 25 ml/hora. Se recogerán las sucesivas fracciones de eluato y se analizarán según el método elegido.
Procedimiento de medida:
Para determinar la hidrosolubilidad se utilizarán las fracciones procedentes del intervalo medio de elución cuyas concentraciones sean constantes (± 30 %) al menos en 5 fracciones consecutivas.
En ambos casos (con bomba de recirculación o con recipiente de nivelación) se repetirá la operación, reduciendo el caudal a la mitad. Si los resultados de las dos operaciones concuerdan, la prueba es satisfactoria; si se observa una solubilidad aparente más elevada con el caudal inferior, se continuará con la reducción del caudal a la mitad hasta que se obtenga la misma solubilidad en dos operaciones sucesivas.
En los dos casos (bomba de recirculación o recipiente de nivelación), se debe controlar la posible presencia de materia coloidal en las fracciones estudiando la aparición del efecto Tyndall (dispersión de la luz). La presencia de tales partículas falsea los resultados y el ensayo deberá repetirse mejorando la eficacia de la filtración de la columna.
Anotar el pH de cada muestra. Efectuar una segunda operación a la misma temperatura.
1.6.4. Método del frasco
1.6.4.1. Aparatos
Para este método se requiere el material siguiente:
|
— |
material de vidrio e instrumentos de laboratorio normales, |
|
— |
dispositivo adecuado para agitar las disoluciones a temperaturas constantes controladas, |
|
— |
centrífuga preferiblemente termostatizada y, si fuere necesario, con emulsiones, |
|
— |
equipo para la determinación analítica. |
1.6.4.2. Procedimiento de medida
Evaluar, a partir del ensayo preliminar, la cantidad de producto necesario para saturar el volumen de agua elegido. El volumen de agua necesario dependerá del método analítico y del intervalo de solubilidad. Poner una cantidad de material aproximadamente cinco veces superior a la cantidad determinada antes, en cada uno de tres recipientes de vidrio con tapón, también de vidrio (por ejemplo, tubos de centrífuga, matraces). Añadir a cada recipiente el volumen de agua elegido y taparlo herméticamente. Agitar los recipientes, debidamente cerrados, a 30 oC (utilizar un dispositivo agitador o mezclador que pueda actuar a temperatura constante como, por ejemplo, un agitador magnético en baño de agua controlado por termostato). Un día después, retirar uno de los recipientes y volver a equilibrar durante 24 horas a la temperatura del ensayo, agitando de vez en cuando. El contenido del recipiente se centrifuga luego a la temperatura del ensayo y se determina la concentración de la sustancia problema en la fase acuosa clara utilizando un método analítico adecuado. Los otros dos matraces se tratan de la misma manera, después de un primer equilibrado a 30 oC durante dos y tres días, respectivamente. Si al menos las concentraciones de los dos últimos recipientes concuerdan con la reproducibilidad requerida, el ensayo es satisfactorio. Si los resultados de los recipientes 1, 2 y 3 acusan una tendencia a la progresión, repetir de nuevo todo el ensayo utilizando tiempos de equilibrado más largos.
El procedimiento de medida puede realizarse también sin la preincubación a 30 oC. Para evaluar la velocidad de adquisición del equilibrio de saturación se irán tomando muestras hasta que el tiempo de agitación deje de influir en la concentración de la solución problema.
Debe anotarse el pH de cada muestra.
1.6.5. Análisis
Para efectuar estas determinaciones es preferible utilizar un método de análisis que sea específico de la sustancia, pues pequeñas cantidades de impurezas solubles pueden originar errores sensibles en la solubilidad medida. Pueden citarse como ejemplos los siguientes métodos: cromatografía en fase gaseosa o líquida, métodos volumétricos, métodos fotométricos, métodos voltamétricos.
2. RESULTADOS
2.1. MÉTODO DE ELUCIÓN EN COLUMNA
En cada operación debe calcularse el valor medio de al menos 5 muestras consecutivas tomadas durante el equilibrio de saturación, así como la desviación típica. Los resultados deben expresarse en unidades de masa por volumen de solución.
Las medias calculadas en dos pruebas con diferentes caudales se compararán para comprobar que su repetibilidad es inferior al 30 %.
2.2. MÉTODO DE FRASCO
Deben indicarse los resultados individuales de cada uno de los tres frascos; se hará la media de los resultados considerados constantes (repetibilidad inferior al 15 %) y se expresarán en unidades de masa por volumen de solución. Dicha operación podrá requerir la conversión de las unidades de masa en unidades de volumen, utilizando la densidad cuando la solubilidad sea muy elevada (> 100 g/l).
3. INFORME
3.1. MÉTODO DE ELUCIÓN EN COLUMNA
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
resultados del ensayo preliminar, |
|
— |
especificación precisa de la sustancia (identidad e impurezas), |
|
— |
concentraciones individuales, caudales y pH de cada muestra, |
|
— |
desviación típica y media de al menos 5 muestras tomadas durante el equilibrio de saturación de cada operación, |
|
— |
media de dos operaciones consecutivas aceptables, |
|
— |
temperatura del agua durante el proceso de saturación, |
|
— |
método de análisis utilizado, |
|
— |
naturaleza del material de soporte, |
|
— |
carga del material de soporte, |
|
— |
disolvente utilizado, |
|
— |
cualquier indicación de inestabilidad química de la sustancia durante el ensayo y método utilizado, |
|
— |
todas las observaciones útiles para la interpretación de los resultados, especialmente lo referente a las impurezas y al estado físico de la sustancia. |
3.2. MÉTODO DEL FRASCO
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
resultados del ensayo preliminar, |
|
— |
especificación precisa de la sustancia (identidad e impurezas), |
|
— |
resultados analíticos individuales y su media cuando se determine más de un valor para un mismo recipiente, |
|
— |
pH de cada muestra, |
|
— |
media de los valores de los diferentes recipientes cuando concuerden, |
|
— |
temperatura del ensayo, |
|
— |
método analítico utilizado, |
|
— |
cualquier indicación de inestabilidad química de la sustancia durante el ensayo y método utilizado, |
|
— |
todas las observaciones útiles para la interpretación de los resultados, especialmente lo referente a las impurezas y al estado físico de la sustancia. |
4. BIBLIOGRAFÍA
|
(1) |
OCDE, París, 1981, Test Guideline 105, Decision of the Council C(81) 30 final. |
|
(2) |
NF T 20-045 (AFNOR) (Sept. 85). Chemical products for industrial use — Determination of water solubility of solids and liquids with low solubility — Column elution method |
|
(3) |
NF T 20-046 (AFNOR) (Sept. 85). Chemical products for industrial use — Determination of water solubility of solids and liquids with high solubility — Flask method |
Apéndice
Figura 1
Método de elución en columna con bomba de recirculación

Figura 2
Microcolumna tipo
(dimensiones en milímetros)
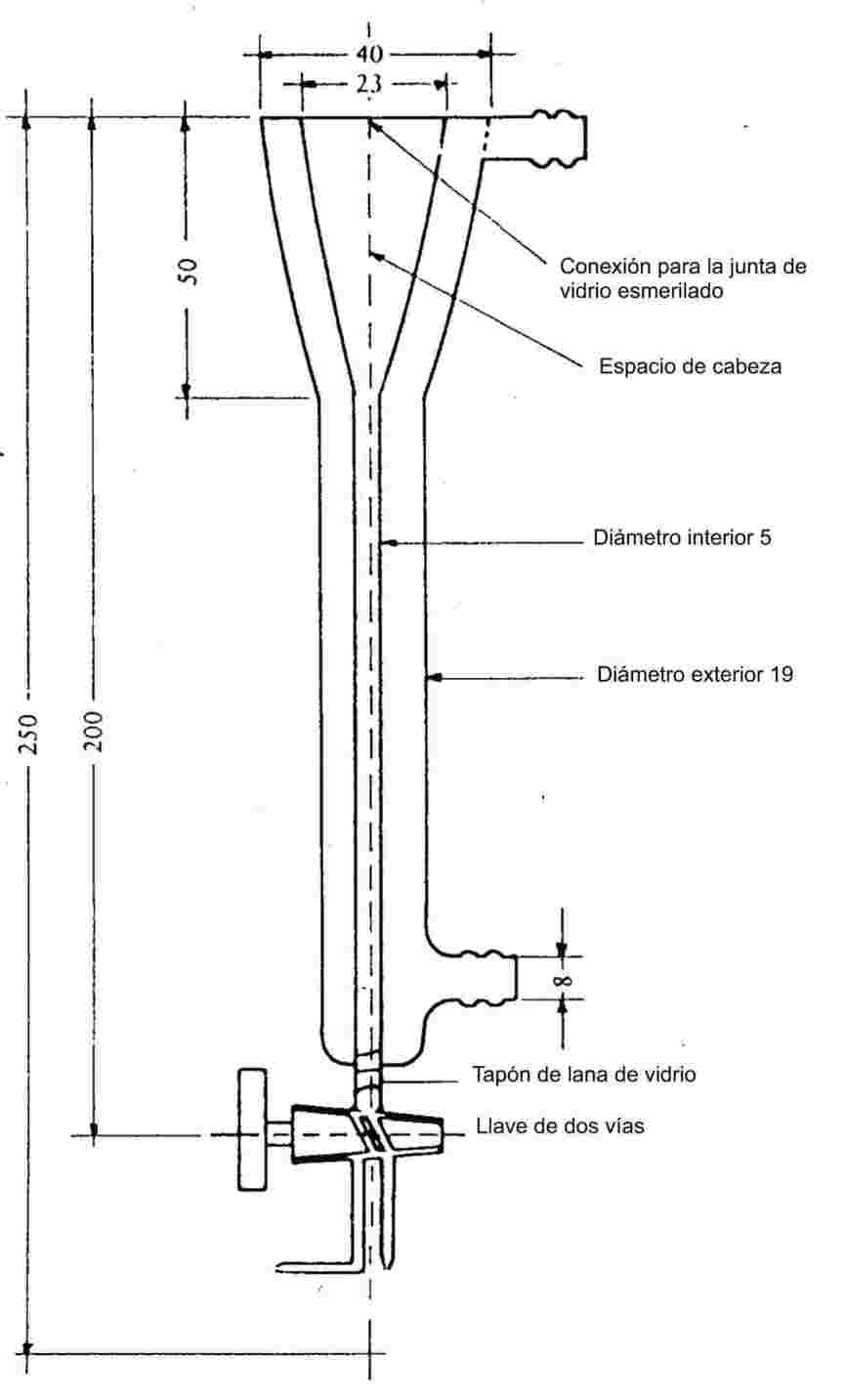
Figura 3
Microcolumna tipo
(dimensiones en milímetros)

Figura 4
Método de elución en columna con recipiente de nivelación
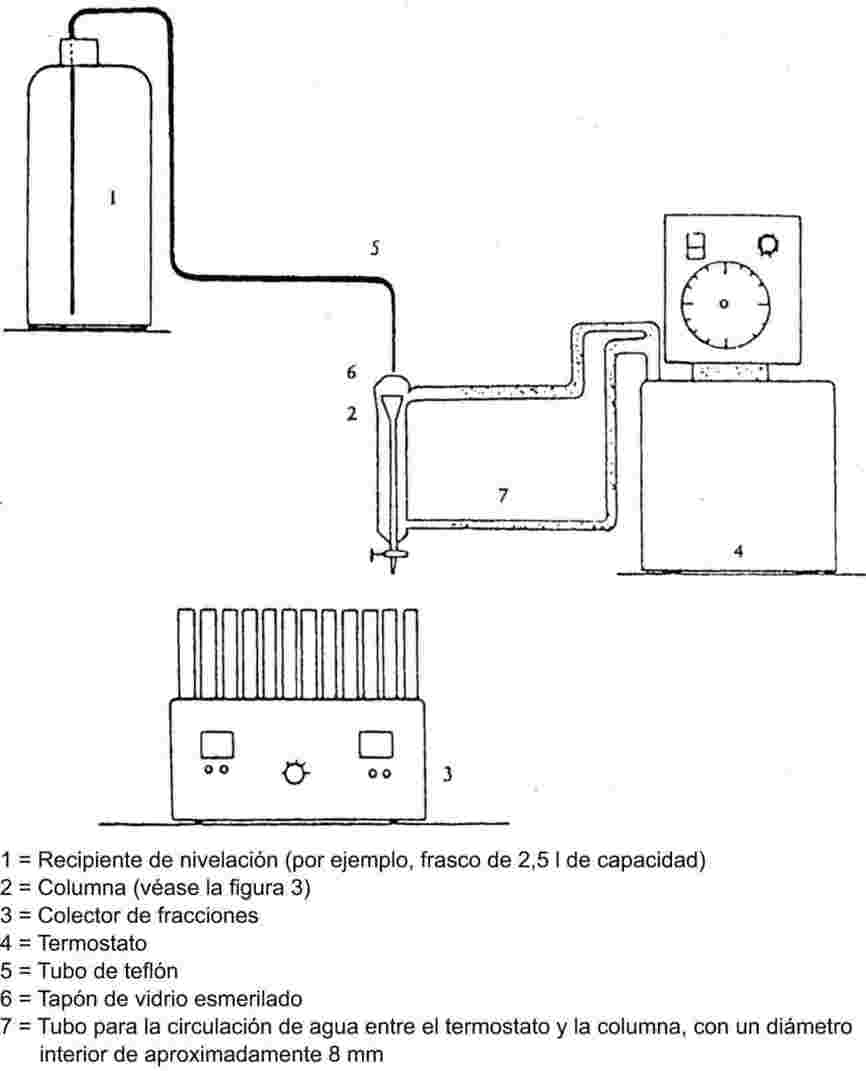
A.8. COEFICIENTE DE REPARTO
1. MÉTODO
El método de frasco de agitación se basa en las líneas directrices de la OCDE (1).
1.1. INTRODUCCIÓN
Es conveniente disponer de información preliminar sobre la fórmula desarrollada, la constante de disociación, la hidrosolubilidad, la hidrólisis, la solubilidad en n-octanol y la tensión superficial de la sustancia antes de proceder al ensayo.
Las medidas relativas a sustancias ionizables deben hacerse solo con la forma no ionizada (ácido libre o base libre) obtenida mediante el uso de una solución amortiguadora con un pH que esté al menos una unidad de pH por debajo (ácido libre) o por encima (base libre) del pK.
Este método de ensayo incluye dos procedimientos distintos: el método de frasco de agitación y la cromatografía líquida de alta resolución (CLAR). El primero puede aplicarse cuando el valor de log Pow (véanse más abajo las definiciones) está en el intervalo de - 2 a 4, y el segundo cuando está en el intervalo de 0 a 6. Antes de llevar a cabo ninguno de los procedimientos experimentales es necesario obtener una estimación previa del coeficiente de reparto.
El método de frasco de agitación es aplicable solo a sustancias esencialmente puras solubles en agua y n-octanol, y no es aplicable a productos tensoactivos (para los que debe darse su valor calculado o una estimación basada en las solubilidades en n-octanol y agua por separado).
El método de CLAR no puede aplicarse a ácidos o bases fuertes, complejos metálicos, materiales tensoactivos ni sustancias que reaccionen con el eluyente. Para estos materiales debe darse un valor calculado o una estimación basada en las solubilidades en n-octanol y agua por separado.
El método de CLAR es menos sensible a la presencia de impurezas en el compuesto problema que el método de frasco de agitación. Sin embargo, las impurezas pueden hacer en algunos casos que la interpretación de los resultados sea difícil porque la atribución de los picos resulte dudosa. En el caso de mezclas que den una banda sin resolución, hay que establecer los límites superior e inferior de log P.
1.2. DEFINICIONES Y UNIDADES
El coeficiente de reparto (P) se define como la relación de las concentraciones en equilibrio (ci) de una sustancia disuelta en un sistema bifásico consistente en dos disolventes considerablemente inmiscibles. En el caso del n-octanol y del agua:
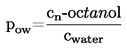
El coeficiente de reparto (P) es, pues, el cociente de dos concentraciones; se indica generalmente en forma de su logaritmo decimal (log P).
1.3. SUSTANCIAS DE REFERENCIA
Método de frasco de agitación
No es necesario utilizar sustancias de referencia cada vez que se investiga una nueva sustancia. Deben servir esencialmente para comprobar de vez en cuando la eficacia del método y para comparar con los resultados obtenidos según otros métodos.
Método de CLAR
Con el fin de relacionar los datos medidos según la CLAR de un compuesto con su P, hay que trazar una gráfica de calibración (log P — datos cromatográficos) con al menos 6 puntos de referencia. La selección de las sustancias de referencia adecuadas depende del criterio del analista. Siempre que sea posible, al menos una sustancia de referencia debe tener un Pow superior al de la sustancia problema, y otra un Pow inferior al de la sustancia problema. En el caso de valores de log P inferiores a 4, la calibración puede basarse en datos obtenidos por el método de frasco de agitación. En el caso de valores de log P superiores a 4, la calibración puede basarse en valores validados aparecidos en la bibliografía, siempre que estos valores estén de acuerdo con los valores calculados. Para mayor precisión, es preferible elegir compuestos de referencia que estén relacionados estructuralmente con la sustancia problema.
Se dispone de amplias listas de valores de log Pow de muchos grupos de productos químicos (2) (3). Si no se tienen datos sobre los coeficientes de reparto de compuestos relacionados estructuralmente, puede utilizarse entonces una calibración más general, establecida con otros compuestos de referencia.
En el apéndice 2 se recoge una relación de sustancias de referencia recomendadas con sus valores de Pow.
1.4. PRINCIPIO DEL MÉTODO
1.4.1. Método de frasco de agitación
Para determinar un coeficiente de reparto, es necesario conseguir un equilibrio entre los elementos constitutivos del sistema que se influencien mutuamente y determinar las concentraciones de las sustancias disueltas en las dos fases. Examinando la bibliografía existente sobre este tema, se encuentra que pueden utilizarse varias técnicas diferentes para resolver este problema, es decir, la mezcla completa de las dos fases, seguida de su separación, que permita determinar la concentración en equilibrio de la sustancia de ensayo.
1.4.2. Método de CLAR
La CLAR se realiza en columnas analíticas rellenas de una fase sólida comercializada con cadenas hidrocarbonadas largas (C8, C18, por ejemplo) unidas por enlaces químicos a la sílice. Los productos químicos inyectados en esta columna la recorrerán a diferente velocidad debido a los diferentes grados de reparto entre la fase móvil y la fase estacionaria hidrocarbonada. Las mezclas de productos químicos se eluyen por orden de hidrofobicidad, saliendo antes los productos hidrosolubles y después los liposolubles, según su coeficiente de reparto hidrocarburo-agua. Esto permite establecer una relación entre el tiempo de retención en una columna de este tipo (de fase inversa) y el coeficiente de reparto n-octanol/agua. El coeficiente de reparto se deduce del factor de capacidad k, dado por la expresión
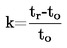
donde tR = tiempo de retención de la sustancia problema y to = tiempo medio que necesita una molécula de disolvente para recorrer la columna (tiempo muerto).
No es necesario utilizar métodos analíticos cuantitativos, sino solo determinar los tiempos de elución.
1.5. CRITERIOS DE CALIDAD
1.5.1. Repetibilidad
Método de frasco de agitación
Para obtener un coeficiente de reparto preciso, se harán determinaciones por duplicado en tres series de condiciones de ensayo diferentes, pudiendo variar tanto la cantidad de sustancia especificada como la relación de volumen de los disolventes. Los logaritmos de los valores determinados del coeficiente de reparto deberán situarse en un intervalo de ±0,3 unidades logarítmicas.
Método de CLAR
Para aumentar la confianza de la medida, deben hacerse las determinaciones por duplicado. Los valores de log P procedentes de las distintas medidas deben estar dentro de un intervalo de ±0,1 unidades logarítmicas.
1.5.2. Sensibilidad
Método de frasco de agitación
El intervalo de medida del método está determinado por el límite de detección del procedimiento analítico. Esto debe ser suficiente para evaluar los valores de log Pow en el intervalo de - 2 a 4 (a veces, bajo ciertas condiciones, este intervalo puede ampliarse hasta un log Pow de 5), cuando la concentración del soluto en cada fase no sobrepase los 0,01 moles por litro.
Método de CLAR
El método de CLAR permite la evaluación de coeficientes de reparto en el intervalo de log Pow entre 0 y 6.
En principio, el coeficiente de reparto de un compuesto puede estimarse en el margen de ± 1 unidad logarítmica del valor obtenido con el frasco de agitación. En la bibliografía se encuentran correlaciones típicas (4) (5) (6) (7) (8). Normalmente puede obtenerse mayor precisión cuando los gráficos de correlación se basan en compuestos de referencia relacionados estructuralmente (9).
1.5.3. Especificidad
Método de frasco de agitación
La ley de reparto de Nernst solo se aplica a temperatura, presión y pH constantes para las soluciones diluidas. Estrictamente se aplica a una sustancia pura dispersada entre dos disolventes puros. Los resultados pueden ser modificados por la aparición simultánea, en una o ambas fases, de varios solutos diferentes.
La disociación o la asociación de las moléculas disueltas se traduce en desviaciones respecto a la ley de reparto citada. Estas desviaciones se observan porque el coeficiente de reparto pasa a depender entonces de la concentración de la solución.
Debido a los múltiples equilibrios implicados, este método de ensayo no debe aplicarse sin corregir a los compuestos ionizables. Para estos compuestos debe considerarse la utilización de soluciones amortiguadoras en lugar de agua; el pH de la solución amortiguadora debe diferir del pKa de la sustancia al menos en una unidad de pH y hay que tener en cuenta la importancia de este pH para el medio ambiente.
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Estimación preliminar del coeficiente de reparto
El valor del coeficiente de reparto puede estimarse preferentemente mediante un método de cálculo (véase el apéndice 1), o bien, cuando sea adecuado, utilizando la relación de las solubilidades de la sustancia de ensayo en los disolventes puros (10).
1.6.2. Método de frasco de agitación
1.6.2.1. Preparación
n-Octanol: la determinación del coeficiente de reparto debe hacerse recurriendo a un reactivo de calidad analítica.
Agua: utilizar agua destilada o bidestilada procedente de un aparato de cristal o de cuarzo. En el caso de compuestos ionizables, puede ser necesario utilizar soluciones amortiguadoras en lugar de agua.
Nota:
Evitar el agua extraída directamente de un intercambiador de iones.
1.6.2.1.1. Presaturación de los disolventes
Antes de determinar un coeficiente de reparto, se saturan recíprocamente las fases del sistema de disolventes agitándolas a la temperatura de ensayo. Para realizar esto, es preciso agitar durante 24 horas, en un agitador mecánico, dos grandes frascos que contengan n-octanol puro de calidad analítica o agua, con una cantidad suficiente del otro disolvente y dejarlos reposar hasta que se separen las fases y se alcance el estado de saturación.
1.6.2.1.2. Preparación para el ensayo
El volumen del sistema bifásico debe llenar casi completamente el recipiente de ensayo para evitar cualquier pérdida de materia debida a la volatilización. La relación de volumen y las cantidades de sustancia que deben utilizarse se determinan de la siguiente manera:
|
— |
estimación preliminar del coeficiente de reparto (véase más arriba), |
|
— |
cantidad mínima de sustancia de ensayo requerida para el procedimiento analítico utilizado, |
|
— |
límite de concentración máxima en cada fase de 0,01 moles por litro. |
Se efectúan tres ensayos. En el primero se utiliza la relación calculada de volúmenes de n-octanol y agua, en el segundo se divide por dos esta relación, y en el tercero se multiplica por dos esta relación (por ejemplo, 1:1, 1:2, 2:1).
1.6.2.1.3. Sustancia de ensayo
Se prepara una solución de reserva en n-octanol presaturado con agua. La concentración de esta solución de reserva debe determinarse con precisión antes de utilizarla para determinar el coeficiente de reparto. Esta solución deberá guardarse en condiciones que garanticen su estabilidad.
1.6.2.2. Condiciones de ensayo
La temperatura de ensayo debe mantenerse constante (± 1 oC) y situarse en el intervalo de 20 oC a 25 oC.
1.6.2.3. Procedimiento de medida
1.6.2.3.1. Establecimiento del equilibrio de reparto
Para cada serie de condiciones de ensayo, preparar recipientes de ensayo por duplicado con las cantidades requeridas, cuidadosamente medidas, de los dos disolventes, así como la cantidad necesaria de solución de reserva.
Medir los volúmenes de las fases de n-octanol. Colocar en un agitador apropiado o agitar a mano los recipientes de ensayo. En caso de usar un tubo para centrifugar, un método que se recomienda consiste en hacer girar rápidamente 180 grados alrededor de su eje transversal el tubo, de forma que el aire que pudiera haber quedado retenido atraviese las dos fases. La experiencia muestra que 50 giros de este tipo suelen ser suficientes para establecer el equilibrio de reparto. Para mayor seguridad, se recomienda hacer 100 giros en 5 minutos.
1.6.2.3.2. Separación de las fases
Cuando sea necesario para separar las fases, centrifugar la mezcla. Efectuar esta operación con una centrífuga de laboratorio mantenida a temperatura ambiente o, si se utiliza una centrífuga sin control de temperatura, equilibrar los tubos de centrífuga a la temperatura de ensayo durante por lo menos una hora antes del análisis.
1.6.2.4. Análisis
Para determinar el coeficiente de reparto, es necesario analizar las concentraciones de la sustancia de ensayo en las dos fases. Para ello, se puede extraer una alícuota de cada una de las dos fases de cada tubo, para cada serie de condiciones de ensayo, y analizarlas según el procedimiento elegido. Debe calcularse la cantidad total de sustancia presente en las dos fases y compararla con la cantidad introducida inicialmente.
La muestra de la fase acuosa debe tomarse por un procedimiento que reduzca al mínimo el riesgo de incorporar trazas de n-octanol: para ello se puede utilizar una jeringuilla de cristal con aguja intercambiable; en primer lugar hay que llenar parcialmente de aire la jeringuilla y a continuación expulsarlo lentamente mientras se va introduciendo al mismo tiempo la aguja a través de la capa de n-octanol. Extraer un volumen adecuado de fase acuosa. Retirar rápidamente la jeringuilla de la solución y quitar la aguja. El contenido de la jeringuilla se podrá utilizar entonces como muestra acuosa. La concentración en las dos fases diferentes deberá determinarse, preferentemente, mediante un procedimiento específico de la sustancia. Algunos ejemplos de métodos analíticos que pueden ser adecuados son:
|
— |
métodos fotométricos, |
|
— |
cromatografía en fase gaseosa, |
|
— |
cromatografía líquida de alta resolución. |
1.6.3. Método de CLAR
1.6.3.1. Preparación
Aparatos
Se necesita un cromatógrafo de líquidos, dotado de una bomba de caudal constante y un instrumento de detección adecuado. Es recomendable el uso de una válvula de inyección con circuito de inyección. La presencia de grupos polares en la fase estacionaria puede afectar gravemente al funcionamiento de la columna de CLAR; por tanto, las fases estacionarias deben presentar la mínima proporción posible de grupos polares (11). Pueden utilizarse rellenos de fase inversa con micropartículas o columnas ya rellenas, disponibles comercialmente. Puede ponerse una columna de seguridad entre el sistema de inyección y la columna de análisis.
Fase móvil
Para preparar el disolvente de elución, del que se eliminarán los gases disueltos antes de utilizarlo, se empleará metanol para CLAR y agua para CLAR. Se empleará la elución isocrática. Se utilizarán mezclas de metanol/agua en que el contenido mínimo de agua será del 25 %. La mezcla típica de metanol-agua en la proporción de 3:1 (v/v) es satisfactoria para eluir compuestos de log P 6 en el plazo de una hora con un caudal de 1 ml/min. En el caso de compuestos de mayor log P puede ser necesario abreviar el tiempo de elución (y el de los compuestos de referencia) disminuyendo la polaridad de la fase móvil o la longitud de la columna.
Las sustancias muy poco solubles en n-octanol tienden a dar valores de log Pow anormalmente bajos con el método de CLAR; los picos de estos compuestos acompañan a veces al frente de disolvente. Esto se debe probablemente al hecho de que el proceso de reparto es demasiado lento como para alcanzar el equilibrio en el tiempo necesario normalmente en una separación por CLAR. Si se disminuye el caudal y/o se baja la proporción metanol/agua es posible obtener un valor fiable.
Las sustancias de ensayo y de referencia deben ser solubles en la fase móvil en concentraciones suficientes para permitir su detección. Solo podrán añadirse aditivos a la mezcla metanol-agua en casos excepcionales, ya que los aditivos cambian las propiedades de la columna. Si se trata de cromatogramas con aditivos, es obligatorio utilizar una columna independiente del mismo tipo. Si la mezcla metanol-agua no es adecuada, pueden utilizarse otras mezclas de disolventes orgánicos con agua, como etanol-agua o acetonitrilo-agua.
El pH del eluyente es fundamental para los compuestos ionizables. Debe situarse en el intervalo de pH de funcionamiento de la columna, que suele estar entre 2 y 8. Se recomienda el uso de soluciones amortiguadoras, Hay que evitar la precipitación de sales y el deterioro de la columna que se producen con algunas mezclas de fase orgánica con soluciones amortiguadoras. Las medidas de CLAR con fases estacionarias a base de sílice con el pH superior a 8 no son recomendables porque el uso de una fase móvil alcalina puede provocar una rápida alteración de las características de la columna.
Solutos
Los compuestos de referencia deben tener la mayor pureza posible. Si es posible, los compuestos que se vayan a utilizar en el ensayo o en la calibración se disolverán en la fase móvil.
Condiciones de ensayo
La temperatura durante las medidas no debe variar más de ± 2 K.
1.6.3.2. Medida
Cálculo del tiempo muerto t0
El tiempo muerto t0 puede determinarse utilizando una serie homóloga (por ejemplo, n-alquil-metil-cetonas) o bien compuestos orgánicos no retenidos (por ejemplo, tiourea o formamida). Para calcular el tiempo muerto t0 utilizando una serie homóloga, se inyectarán al menos 7 miembros de una serie homóloga y se determinarán sus respectivos tiempos de retención. Los tiempos de retención, medidos directamente, tr (nc + 1), se representan gráficamente en función de tr(nc) y se determinan la ordenada en el origen a y la pendiente b de la ecuación de regresión:
tr(nc + 1) = a + b tr(nc)
siendo nc = número de átomos de carbono. El tiempo muerto t0 viene dado por:
t0 = a/(1 - b)
Gráfico de calibración
El paso siguiente es construir una gráfica de correlación de log k frente a log P para los compuestos de referencia adecuados. En la práctica, se inyecta simultáneamente una serie de 5 a 10 compuestos patrón de referencia cuyo log P esté alrededor del intervalo supuesto, y se determinan los tiempos de retención, preferiblemente con un integrador-registrador unido al sistema de detección. Se calculan los respectivos logaritmos de los factores de capacidad, log K, y se representan en función del log P determinado según el método de frasco de agitación. La calibración se realiza a intervalos regulares, al menos una vez al día, de forma que puedan tenerse en cuenta los posibles cambios en las características de la columna.
Determinación del factor de capacidad de la sustancia de ensayo
Se inyecta la cantidad más pequeña posible de sustancia de ensayo en fase móvil. Se determina (por duplicado) el tiempo de retención para calcular el factor de capacidad k. A partir de la gráfica de correlación de los compuestos de referencia puede interpolarse el coeficiente de reparto de la sustancia de ensayo. Cuando los coeficientes de reparto son muy altos o muy bajos es necesario hacer una extrapolación y en estos casos hay que tener muy en cuenta los límites de confianza de la línea de regresión.
2. RESULTADOS
Método de frasco de agitación
La fiabilidad de los valores determinados de P puede verificarse procediendo a una comparación de las medias de las determinaciones efectuadas por duplicado frente a la media general.
3. INFORME
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
especificación precisa de la sustancia (identidad e impurezas), |
|
— |
cuando no puedan aplicarse los métodos (por ejemplo, con productos tensoactivos), habrá que dar un valor calculado o una estimación a partir de las solubilidades en n-octanol y agua por separado, |
|
— |
toda la información y las observaciones que sean útiles para la interpretación de los resultados, especialmente en lo relativo a las impurezas y al estado físico de la sustancia. |
Para el método de frasco de agitación:
|
— |
resultado de la estimación previa, en su caso, |
|
— |
temperatura de la determinación, |
|
— |
datos sobre los procedimientos analíticos utilizados para determinar las concentraciones, |
|
— |
tiempo y velocidad de la centrifugación, en su caso, |
|
— |
concentraciones medidas en ambas fases en cada determinación (es decir, hay que indicar un total de 12 concentraciones), |
|
— |
peso de la sustancia de ensayo, volumen de cada fase empleado en cada recipiente de ensayo y cantidad calculada total de sustancia de ensayo presente en cada fase tras el equilibrado, |
|
— |
valores calculados del coeficiente de reparto (P) y media de cada conjunto de condiciones de ensayo, así como la media de todas las determinaciones. Si hay alguna indicación de que el coeficiente de reparto puede variar con la concentración, habrá que señalarlo en el informe, |
|
— |
desviación típica de los distintos valores de P respecto a su media, |
|
— |
media del valor P de todas las determinaciones, expresada también en logaritmo decimal, |
|
— |
valor teórico calculado de Pow cuando este valor se haya determinado o cuando el valor medido sea superior a 104, |
|
— |
pH del agua utilizada y fase acuosa durante la prueba, |
|
— |
si se utilizan soluciones amortiguadoras, hay que justificar su uso en lugar del agua e indicar su composición, concentración y pH, así como el pH de la fase acuosa antes y después del ensayo. |
Para el método de CLAR:
|
— |
resultado de la estimación previa, en su caso, |
|
— |
sustancias de ensayo y de referencia, con sus purezas, |
|
— |
intervalo de temperatura de las determinaciones, |
|
— |
pH al que se hacen las determinaciones, |
|
— |
características de las columnas de análisis y de seguridad, fase móvil y sistema de detección, |
|
— |
datos de la retención y valores de log P de la bibliografía correspondiente a las sustancias de referencia utilizadas en la calibración, |
|
— |
datos de la recta de regresión ajustada (log K frente a log P), |
|
— |
datos de retención media y valor interpolado de log P del compuesto problema, |
|
— |
descripción del equipo y condiciones de funcionamiento, |
|
— |
perfiles de elución, |
|
— |
cantidades de sustancias problema y de referencia introducidas en la columna, |
|
— |
tiempo muerto y método para medirlo. |
4. BIBLIOGRAFÍA
|
(1) |
OCDE, París, 1981, Test Guideline 107, Decision of the Council C(81) 30 final. |
|
(2) |
C. Hansch y A.J. Leo, Substituent Constants for Correlaticn Analysis in Chemistry and Biology, John Wiley, Nueva York, 1979. |
|
(3) |
Log P and Parameter Database, A tool for the quantitative prediction of bioactivity (C. Hansch, chairman; A.J. Leo, dir.) — Available from Pomona College Medical Chemistry Proiect. 1982, Pomona College, Claremont, California 91711. |
|
(4) |
L. Renberg, G. Sundström y K. Sundh-Nygärd, Chemosphere, 1981, vol. 80, 683. |
|
(5) |
H. Ellgehausen, C. D'Hondt y R. Fuerer, Pestic. Sci., 1981, vol. 12, 212. |
|
(6) |
B. McDuffie, Chemosphere, 1981, vol. 10, 73. |
|
(7) |
W.E. Hammers et al., J. Chromatogr., 1982, vol. 247, 1. |
|
(8) |
J.E. Haky y A.M. Young, J. Liq. Chromat., 1984, vol. 7, 675. |
|
(9) |
S. Fujisawa y E. Masuhara, J. Biomed. Mat. Res., 1981, vol. 15, 787. |
|
(10) |
O. Jubermann, Verteilen und Extrahieren, in Methoden der Organischen Chemie (Houben Weyl), Allgemeine Laboratoriumpraxis (editado por E. Muller), Georg Thieme Verlag, Stuttgart, 1958, Band I/1, 223-339. |
|
(11) |
R.F. Rekker y H.M. de Kort, Euro. J Med. Chem., 1979, vol. 14, 479. |
|
(12) |
A. Leo, C. Hansch y D. Elkins, Partition coefficients and their uses. Chem. Rev., 1971, Vol. 71, 525. |
|
(13) |
R.F. Rekker, The Hydrophobic Fragmenta] Constant, Elsevicr, Amsterdam, 1977. |
|
(14) |
NF T 20-043 AFNOR (1985). Chemical products for industrial use — Determination of partition coefficient — Flask shaking method. |
|
(15) |
C.V. Eadsforth y P. Moser, Chemosphere, 1983, vol. 12, 1 459. |
|
(16) |
A. Leo, C. Hansch y D. Elkins, Chem. Rev, 1971, vol. 71, 525. |
|
(17) |
C. Hansch, A Leo, S.H. Unger, K.H. Kim, D. Nikaitani y E.J. Lien, J. Med. Chem., 1973, vol. 16, 1 207. |
|
(18) |
W.B. Neely, D.R. Branson y G.E. Blau, Environ. Sci. Technol., 1974, vol. 8, 1 113. |
|
(19) |
D.S. Brown y E.W. Flagg,]. Environ. Qual., 1981, vol. 10, 382. |
|
(20) |
J.K. Seydel y K.J. Schaper, Chemische Struktur und biologische Aktivitát von Wirkstoffen, Verlag Chemie, Weínheim, Nueva York, 1979. |
|
(21) |
R. Franke, Theoretical Drug Design Methods, Elsevier, Amsterdam, 1984. |
|
(22) |
Y.C. Martin, Quantitative Drug Design, Marcel Dekker, Nueva York, Basilea, 1978. |
|
(23) |
N.S. Nirrlees, S.J. Noulton, C.T. Murphy, P.J. Taylor; J. Med. Chem., 1976, vol. 19, 615. |
Apéndice 1
Métodos de cálculo y estimación
INTRODUCCIÓN
En Handbook of Chemical Property Estimation Methods (a) se ofrece una introducción general de los métodos de cálculo, junto con datos y ejemplos.
Los valores calculados de Pow pueden utilizarse:
|
— |
para decidir cuál de los métodos experimentales es adecuado (intervalo del método de frasco de agitación: log Pow entre — 2 y 4; intervalo del método de CLAR: log Pow entre 0 y 6), |
|
— |
para seleccionar las condiciones de ensayo adecuadas (por ejemplo, sustancias de referencia para procedimientos de CLAR, relación de volúmenes n-octanol/agua para el método de frasco de agitación), |
|
— |
como control interno del laboratorio para detectar posibles errores experimentales, |
|
— |
para dar una estimación de Pow en casos en que los métodos experimentales no puedan aplicarse por causas técnicas. |
MÉTODO DE ESTIMACIÓN
Estimación previa del coeficiente de reparto
El valor del coeficiente de reparto puede estimarse a partir de las solubilidades de la sustancia problema en los disolventes puros:

MÉTODOS DE CÁLCULO
Principio de los métodos de cálculo
Todos los métodos de cálculo se basan en la fragmentación formal de la molécula en subestructuras adecuadas de las que se conocen incrementos fiables de log Pow. El log Pow de toda la molécula se calcula entonces como la suma de los valores de los fragmentos correspondientes más unos términos de corrección por las interacciones intramoleculares.
Existen listas de constantes de fragmentos y términos de corrección (b) (c) (d) (e). Algunas se actualizan periódicamente (b).
Criterios cualitativos
En general, la habilidad del método de cálculo va disminuyendo al aumentar la complejidad del compuesto estudiado. En el caso de moléculas simples de bajo peso molecular y uno o dos grupos funcionales, puede esperarse una desviación de 0,1 a 0,3 unidades logarítmicas de Pow entre los resultados de los diferentes métodos de fragmentación y el valor medido. Si se trata de moléculas más complejas, el margen de error puede ser mayor, dependiendo de la fiabilidad y disponibilidad de constantes de fragmentos, así como de la capacidad de detectar interacciones intramoleculares (por ejemplo, enlaces de hidrógeno) y del uso adecuado de los términos de corrección (problema menos arduo con los programas de ordenador CLOGP-3) (b). En el caso de compuestos ionizados, es importante considerar correctamente la carga o el grado de ionización.
Procedimientos de cálculo
Método de π de Hansch
La constante del sustituyente hidrofóbico original, π, introducida por Fujita et al. (f) se define como:
πx = log Pow (PhX) - log Pow (PhH)
donde Pow (PhX) es el coeficiente de reparto de un derivado aromático y Pow (PhH) el del compuesto original.
(P.y. πCl = log Pow (C6H5Cl) - log Pow (C6H6) = 2,84 - 2,13 = 0,71).
Según esta definición, el método de π es aplicable principalmente a la sustitución aromática. Se han tabulado los valores de π de gran número de sustituyentes (b) (c) (d) y se utilizan para calcular el log Pow de moléculas o subestructuras aromáticas.
Método de Rekker
Según Rekker (g), el valor de log Pow se calcula de la manera siguiente:
donde fi representa las constantes de los diferentes fragmentos moleculares y ai la frecuencia de su aparición en la molécula estudiada. Los términos de corrección pueden expresarse como integral múltiple de una sola constante Cm (llamada «constante mágica»). Las constantes de fragmentos fi y Cm se han obtenido a partir de una lista de 1 054 valores experimentales de Pow (825 compuestos) mediante análisis de regresión múltiple (c) (h). La determinación de los términos de interacción se lleva a cabo según normas establecidas recogidas en a bibliografía (e) (h) (i).
Método de Hansch-Leo
Según Hansch y Leo (c), el valor de log Pow se calcula de la forma siguiente:

donde fi representa las constantes de los diferentes fragmentos moleculares; Fj, los términos de corrección, y ai y bj las frecuencias correspondientes de presencia. A partir de valores experimentales de Pow, se establecieron por el método de tanteo una lista de valores de fragmentos atómicos y de grupos y una lista de términos de corrección Fj (llamados «factores»). Los términos de corrección se han ordenado en varias clases diferentes (a) (c). Es bastante complicado y exige mucho tiempo el tener en cuenta todas las normas y los términos de corrección. Se han ideado programas informáticos (b).
Método combinado
El cálculo de log Pow de moléculas complejas puede mejorarse considerablemente si se divide la molécula en grandes subestructuras de las que se conozcan valores fiables de log Pow a partir de listas (b) (c) o por mediciones propias. Estos fragmentos (por ejemplo, heterociclos, antraquinona, azobenceno) pueden combinarse con los valores π de Hansch o con las constantes de fragmentos de Rekker o Leo.
Observaciones
|
i) |
Los métodos de cálculo solo pueden aplicarse a compuestos parcial o completamente ionizados, si es posible tener en cuenta los necesarios factores de corrección. |
|
ii) |
Si puede suponerse la presencia de enlaces de hidrógeno intramoleculares, hay que añadir los correspondientes términos de corrección (aproximadamente, de +0,6 a +1,0 unidades logarítmicas de Pow) (a). La presencia de tales enlaces puede suponerse a partir de modelos estéricos o datos espectroscópicos de la molécula. |
|
iii) |
Si son posibles varias formas tautoméricas, debe usarse como base para el cálculo la forma más probable. |
|
iv) |
Hay que seguir cuidadosamente las revisiones de las listas de constantes de fragmentos. |
Informe
Cuando se utilicen métodos de cálculo/estimación, el informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
descripción de la sustancia (mezcla, impurezas, etc.), |
|
— |
indicación de cualquier posible enlace de hidrógeno intramolecular, disociación, carga o cualquier otro efecto poco común (por ejemplo, tautomería), |
|
— |
descripción del método de cálculo, |
|
— |
indicación o suministro de la base de datos, |
|
— |
peculiaridades de la selección de los fragmentos, |
|
— |
documentación exhaustiva de los cálculos. |
BIBLIOGRAFÍA
|
(a) |
W.J. Lyman, W.F. Reehl y D.H. Rosenblatt (ed.), Handbook of Chemical Property Estimation Methods, McGraw-Hill, Nueva York, 1983. |
|
(b) |
Pomona College, Medical Chemistry Project, Claremont, California 91711, USA, Log P Database and Med. Chem. Software (Program CLOGP-3). |
|
(c) |
C. Hansch, A.J. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology, John Wiley, Nueva York, 1979. |
|
(d) |
A. Leo, C. Hansch, D. Elkins, Chem. Rev., 1971, vol. 71, 525. |
|
(e) |
R.F. Rekker, H.M. de Kort, Eur. J. Med. Chem. — Chim. Ther., 1979, vol. 14, 479. |
|
(f) |
T. Fujita, J. Iwasa y C. Hansch, J. Amer. Chem. Soc, 1964, vol. 86, 5175. |
|
(g) |
R.F. Rekker, The Hydrophobic Fragmental Constant, Pharmacochemistry Library, vol. 1 Elsevier, Nueva York, 1977. |
|
(h) |
C.V. Eadsforth, P. Moser, Chemosphere, 1983, vol. 12, 1459. |
|
(i) |
R.A. Scherrer, ACS — American Chemical Society, Washington D. C, 1984, Symposium Series 255, p. 225. |
Apéndice 2
Sustancias de referencia recomendadas para el método de CLAR
|
№ |
Sustancia de referencia |
log Pow |
pKa |
|
1 |
2-Butanona |
0,3 |
|
|
7 |
4-Acetilpiridina |
0,5 |
|
|
3 |
Anilina |
0,9 |
|
|
4 |
Acetanilida |
1,0 |
|
|
5 |
Alcohol bencílico |
1,1 |
|
|
6 |
p-Metoxifenol |
1,3 |
pKa = 10,26 |
|
7 |
Ácido fenoxiacético |
1,4 |
pKa = 3,12 |
|
8 |
Fenol |
1,5 |
pKa = 9,92 |
|
9 |
2,4-Dinitrofenol |
1,5 |
pKa = 3,96 |
|
10 |
Benzonitrilo |
1,6 |
|
|
11 |
Fenilacetonitrilo |
1,6 |
|
|
12 |
Alcohol 4-metilbencílico |
1,6 |
|
|
13 |
Acetofenona |
1,7 |
|
|
14 |
2-Nitrofenol |
1,8 |
pKa = 7,17 |
|
15 |
Ácido 3-nitrobenzoico |
1,8 |
pKa = 3,47 |
|
16 |
4-Cloranilina |
1,8 |
pKa = 4,15 |
|
17 |
Nitrobenceno |
1,9 |
|
|
18 |
Alcohol cinámico |
1,9 |
|
|
19 |
Ácido benzoico |
1,9 |
pKa = 4,19 |
|
20 |
p-Cresol |
1,9 |
pKa = 10,17 |
|
21 |
Ácido cinámico |
2,1 |
pKa = 3,89 cis 4,44 trans |
|
22 |
Anisol |
2,1 |
|
|
23 |
Metilbenzoato |
2,1 |
|
|
24 |
Benceno |
2,1 |
|
|
25 |
Ácido 3-metilbenzoico |
2,4 |
pKa = 4,27 |
|
26 |
4-Clorofenol |
2,4 |
pKa = 9,1 |
|
27 |
Tricloroetileno |
2,4 |
|
|
28 |
Atrazina |
2,6 |
|
|
29 |
Etilbenzoato |
2,6 |
|
|
30 |
2,6-Diclorobenzonitrilo |
2,6 |
|
|
31 |
Ácido 3-clorobenzoico |
2,7 |
pKa = 3,82 |
|
32 |
Tolueno |
2,7 |
|
|
33 |
1-Naftol |
2,7 |
pKa = 9,34 |
|
34 |
2,3-Dicloroanilina |
2,8 |
|
|
35 |
Clorobenceno |
2,8 |
|
|
36 |
Alil-feniléter |
2,9 |
|
|
37 |
Bromobenceno |
3,0 |
|
|
38 |
Etilbenceno |
3,2 |
|
|
39 |
Benzofenona |
3,2 |
|
|
40 |
4-Fenilfenol |
3,2 |
pKa = 9,54 |
|
41 |
Timol |
3,3 |
|
|
42 |
1,4-Diclorobenceno |
3,4 |
|
|
43 |
Difenilamina |
3,4 |
pKa = 0,79 |
|
44 |
Nafraleno |
3,6 |
|
|
45 |
Fenilbenzoato |
3,6 |
|
|
46 |
Isopropilbenceno |
3,7 |
|
|
47 |
2,4,6-Triclorofenol |
3,7 |
pKa = 6 |
|
48 |
Bifenilo |
4,0 |
|
|
49 |
Bencilbenzoato |
4,0 |
|
|
50 |
2,4-Dinitro-6 sec. butilfenol |
4,1 |
|
|
51 |
1,2,4-Triclorobenceno |
4,2 |
|
|
52 |
Ácido dodecanoico |
4,2 |
|
|
53 |
Difeniléter |
4,2 |
|
|
54 |
n-Butilbenceno |
4,5 |
|
|
55 |
Fenantreno |
4,5 |
|
|
56 |
Fluoranteno |
4,7 |
|
|
57 |
Dibencilo |
4,8 |
|
|
58 |
2,6-Difenilpiridina |
4,9 |
|
|
59 |
Trifenilamina |
5,7 |
|
|
60 |
DDT |
6,2 |
|
|
Otras sustancias de referencia de bajo log Pow |
|||
|
1 |
Ácido nicotínico |
-0,07 |
|
A.9. PUNTO DE INFLAMACIÓN
1. MÉTODO
1.1. INTRODUCCIÓN
Es conveniente disponer de datos preliminares sobre la inflamabilidad de la sustancia antes de proceder al ensayo. Este procedimiento es aplicable a las sustancias líquidas cuyos vapores pueden ser inflamados por fuentes de ignición. Los métodos de ensayo descritos en el presente documento solo son válidos para los intervalos de punto de inflamación especificados en cada uno de los distintos métodos.
A la hora de seleccionar el método hay que tener en cuenta las posibles reacciones químicas entre la sustancia y el soporte de la muestra.
1.2. DEFINICIONES Y UNIDADES
El punto de inflamación es la temperatura mínima, corregida a una presión de 101,325 kPa, a la cual un líquido desprende vapores, en las condiciones definidas en el método de ensayo, en una cantidad tal que se produzca una mezcla vapor/aire inflamable en el recipiente del ensayo.
Unidad: oC
t = T - 273,15
(t en oC y T en K)
1.3. SUSTANCIAS DE REFERENCIA
No es necesario utilizar sustancias de referencia cada vez que se examina una nueva sustancia. Su principal función es la de servir para comprobar las características del método de vez en cuando y comparar con los resultados obtenidos según otros métodos.
1.4. PRINCIPIO DEL MÉTODO
Se coloca la sustancia en un recipiente de ensayo y se calienta o se enfría hasta la temperatura de ensayo según el procedimiento descrito en cada método concreto. Se hacen pruebas de ignición para observar si la muestra se inflama o no a la temperatura de ensayo.
1.5. CRITERIOS DE CALIDAD
1.5.1. Repetibilidad
La repetibilidad depende del intervalo del punto de inflamación y del método de ensayo utilizado; máximo 2 oC.
1.5.2. Sensibilidad
La sensibilidad depende del método de ensayo utilizado.
1.5.3. Especificidad
La especificidad de ciertos métodos de ensayo se limita a algunos intervalos de punto de inflamación y depende de las características de la sustancia (por ejemplo, alta viscosidad).
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Preparación
Se coloca una muestra de la sustancia problema en un aparato de ensayo, de conformidad con los puntos 1.6.3.1 y/o 1.6.3.2.
Por seguridad, se recomienda para sustancias energéticas o tóxicas seguir un método que exija una muestra pequeña, alrededor de 2 cm3.
1.6.2. Condiciones del ensayo
Se debe instalar el aparato lejos de corrientes de aire, siempre que esto no suponga ningún problema de seguridad.
1.6.3. Desarrollo del ensayo
1.6.3.1. Método de equilibrio
Véanse las normas ISO 1516, ISO 3680, ISO 1523 e ISO 3679.
1.6.3.2. Métodos de no equilibrio
Aparato Abel:
Véanse las normas BS 2000, parte 170, NF M07-011 y NF T66-009.
Aparato Abel-Pensky:
Véanse las normas EN 57, DIN 51755 primera parte (para temperaturas de 5 a 65 oC), DIN 51755 segunda parte (para temperaturas inferiores a 5 oC) y NF M07-036.
Aparato Tag:
Véase la norma ASTM D-56.
Aparato Pensky-Martens:
Véanse las normas ISO 2719, EN 11, DIN 51758, ASTM D 93, BS 2000-34 y NF M07-019.
Observaciones:
Cuando el punto de inflamación determinado por un método de no equilibrio del punto 1.6.3.2 tiene los siguientes valores: 0 ± 2 oC, 21 ± 2 oC o 55 ± 2 oC, es conveniente confirmarlo mediante un método de equilibrio utilizando el mismo aparato.
Únicamente se pueden utilizar para la notificación los métodos que puedan dar la temperatura del punto de inflamación.
Para determinar el punto de inflamación de líquidos viscosos (pinturas, gomas, etc.) que contengan disolventes, solo se pueden utilizar los aparatos y métodos de ensayo que permitan determinar el punto de inflamación de los líquidos viscosos.
Véanse las normas ISO 3679, ISO 3680, ISO 1523 y DIN 53213 primera parte.
|
2. |
RESULTADOS |
3. INFORME
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
especificación precisa de la sustancia (identificación e impurezas), |
|
— |
descripción del método utilizado, así como cualquier desviación, |
|
— |
los resultados y cualquier otra información u observación que pueda ser útil para la interpretación de los resultados. |
4. BIBLIOGRAFÍA
Ninguna.
A.10. INFLAMABILIDAD (SÓLIDOS)
1. MÉTODO
1.1. INTRODUCCIÓN
Es conveniente disponer de datos preliminares sobre las posibles propiedades explosivas de la sustancia antes de proceder al ensayo.
El presente método solo es aplicable a las sustancias en polvo, granulosas o pastosas.
Para no englobar todas las sustancias que pueden inflamarse, sino únicamente aquellas que se queman muy rápidamente o cuya forma de combustión es, de una forma u otra, particularmente peligrosa, solo se considerarán como fácilmente inflamables las sustancias cuya velocidad de combustión sobrepase un cierto límite.
Puede ser especialmente peligroso que la incandescencia se propague a través de un polvo metálico debido a las dificultades para apagar un fuego. Los polvos metálicos se considerarán fácilmente inflamables si permiten la difusión de la incandescencia en toda su masa en un tiempo especificado.
1.2. DEFINICIONES Y UNIDADES
Tiempo de combustión expresado en segundos.
1.3. SUSTANCIAS DE REFERENCIA
No se especifican.
1.4. PRINCIPIO DEL MÉTODO
La sustancia se dispone formando una mecha o cinta continua de unos 250 mm de longitud y se hace un ensayo exploratorio previo para determinar si, al aplicar una llama de gas, se produce la propagación de la combustión con llama o sin ella. Si se produce la propagación a lo largo de 200 mm de la mecha dentro de un tiempo dado, hay que realizar un ensayo completo para determinar la velocidad de combustión.
1.5. CRITERIOS DE CALIDAD
No se indican.
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Ensayo exploratorio previo
Se pone la sustancia formando una mezcla o cinta continua de unos 250 mm de longitud, 20 mm de anchura y 10 mm de altura, sobre una placa incombustible, no porosa y de baja conductividad térmica. Una llama fuerte procedente de un mechero de gas (diámetro mínimo: 5 mm) se aplica a un extremo de la mecha hasta que el polvo empieza a arder o durante un máximo de 2 minutos (5 minutos si se trata de polvos de metales o de aleaciones metálicas). Hay que observar si la combustión se propaga a lo largo de 200 mm de la mecha durante un período de prueba de 4 minutos (o 40 minutos si se trata de polvos metálicos). Si la sustancia no se enciende ni propaga la combustión ardiendo con llama o sin ella a lo largo de 200 mm de la mecha en el plazo de 4 minutos (o 40 minutos), entonces la sustancia no debe considerarse fácilmente inflamable y no es necesario seguir con las pruebas. Si la sustancia propaga la combustión a lo largo de 200 mm de longitud de la mecha en menos de 4 minutos (o menos de 40 minutos si se trata de polvos metálicos), hay que aplicar el procedimiento descrito a continuación (puntos 1.6.2 y siguientes).
1.6.2. Ensayo de la velocidad de combustión
1.6.2.1. Preparación
En el caso de sustancias en polvo o granulosas se vierte la sustancia sin comprimir dentro de un molde de 250 mm de longitud con una sección transversal triangular de 10 mm de altura interior y 20 mm de anchura. A ambos lados del molde, en sentido longitudinal, se colocan dos placas metálicas a modo de soportes laterales, que deben sobrepasar en 2 mm el borde superior de la sección triangular transversal del molde (véase la figura). A continuación se deja caer tres veces el molde desde una altura de 2 cm, sobre una superficie dura. Si es necesario, se completa el molde de nuevo. A continuación, se retiran las placas laterales y se enrasa. Se coloca sobre el molde una placa no combustible, no porosa y de baja conductividad térmica, se le da la vuelta y se desmolda.
Las sustancias pastosas se extienden sobre una superficie no combustible, no porosa y de baja conductividad térmica, formando un cordón de 250 mm de longitud y alrededor de 1 cm2 de sección.
1.6.2.2. Condiciones del ensayo
En el caso de sustancias sensibles a la humedad, hay que efectuar el ensayo lo más rápidamente posible después de retirar la sustancia del recipiente.
1.6.2.3. Desarrollo del ensayo
Colocar el dispositivo en el tiro de una campana de gases.
La velocidad del aire debe ser suficiente para evitar que los humos pasen al laboratorio y no debe variar mientras dure el ensayo. Alrededor del dispositivo se pondrá una pantalla.
Se utiliza una llama fuerte procedente de un mechero de gas (diámetro mínimo: 5 mm) para encender un extremo de la sustancia. Cuando la mecha ha ardido a lo largo de 80 mm, se procede a medir la velocidad de combustión a lo largo de los siguientes 100 mm.
El ensayo se repite seis veces, usando cada vez una placa fría y limpia, salvo que se obtenga antes un resultado positivo.
2. RESULTADOS
Para proceder a la evaluación es preciso conocer el tiempo de combustión del ensayo exploratorio previo (1.6.1) y el menor tiempo de combustión de un máximo de seis ensayos (1.6.2.3).
3. INFORME
3.1. INFORME DEL ENSAYO
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
especificación precisa de la sustancia (identificación e impurezas), |
|
— |
descripción de la sustancia de ensayo y su estado físico, incluida la tasa de humedad, |
|
— |
resultados del ensayo exploratorio previo y del ensayo de velocidad de combustión, en su caso, |
|
— |
cualquier observación complementaria que pueda ser útil para la interpretación de los resultados. |
3.2. INTERPRETACIÓN DE LOS RESULTADOS
Las sustancias pulverulentas, granulosas o pastosas se deberán considerar fácilmente inflamables cuando el tiempo de combustión de cualquier ensayo efectuado según el procedimiento descrito en el punto 1.6.2 sea inferior a 45 segundos. Deberá considerarse que los polvos metálicos o de aleaciones metálicas son fácilmente inflamables cuando puedan encenderse y la llama o la zona de reacción se extienda a toda la muestra en 10 minutos o menos.
4. BIBLIOGRAFÍA
NF T 20-042 (SEPT 85). Chemical products for industrial use. Determination of the flammability of solids.
Apéndice
Figura
Molde y accesorios necesarios para la formación de las mechas
(todas las dimensiones expresadas en milímetros)

A.11. INFLAMABILIDAD (GASES)
1. MÉTODO
1.1. INTRODUCCIÓN
El presente método permite determinar si los gases mezclados con el aire a temperatura (alrededor de 20 oC) y presión ambiente son inflamables y, en caso positivo, en qué intervalo de concentraciones. Se exponen a una chispa eléctrica mezclas que contengan concentraciones crecientes de gas problema y se observa si se produce la inflamación.
1.2. DEFINICIONES Y UNIDADES
El intervalo de inflamabilidad es el intervalo de concentración entre los límites de explosión superior e inferior. Los límites de explosión superior e inferior son los límites de concentración del gas inflamable en mezcla con el aire a los que el fuego no se propaga.
1.3. SUSTANCIA DE REFERENCIA
No se especifica.
1.4. PRINCIPIO DEL MÉTODO
Se aumenta gradualmente la concentración del gas en el aire y, en cada etapa, se expone la mezcla a una chispa eléctrica.
1.5. CRITERIOS DE CALIDAD
No se indican.
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Equipo
El recipiente de ensayo es un cilindro de cristal de un diámetro interior de al menos 50 mm y una altura mínima de 300 mm, que se pone verticalmente. Los electrodos de ignición distan de 3 a 5 mm uno del otro y están situados a 60 mm del fondo del cilindro. El cilindro está equipado con una válvula para reducir la presión. El aparato debe estar protegido por un blindaje para limitar los daños de una posible explosión.
La fuente de ignición es una chispa inductiva constante de 0,5 segundos de duración, producida por un transformador de alta tensión con una tensión de salida de 10 a 15 kV (la potencia máxima es de 300 W). En la referencia (2) se describe un ejemplo de equipo adecuado.
1.6.2. Condiciones del ensayo
El ensayo debe efectuarse a temperatura ambiente (alrededor de 20 oC).
1.6.3. Desarrollo del ensayo
Mediante bombas dosificadoras se llena el cilindro de cristal con una mezcla de aire y gas de concentración conocida. Se hace saltar una chispa en esta mezcla y se observa si se desprende una llama de la fuente de ignición y se propaga independientemente. Se irá aumentando la concentración de gas en un 1 % de volumen cada vez, hasta que se produzca la inflamación descrita anteriormente.
Si la estructura química del gas indica que debe ser ininflamable y puede calcularse la composición de la mezcla estequiométrica con el aire, solo será necesario someter a ensayo, en etapas del 1 %, mezclas que estén en el intervalo entre el 10 % por debajo y el 10 % por arriba de la composición estequiométrica.
2. RESULTADOS
La propagación de la llama constituye el único dato válido para la determinación de esta propiedad.
3. INFORME
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
especificación precisa de la sustancia (identificación e impurezas), |
|
— |
descripción del aparato utilizado, mencionando las dimensiones, |
|
— |
temperatura en el momento del ensayo, |
|
— |
concentraciones de ensayo, así como los resultados obtenidos, |
|
— |
resultado del ensayo: gas no inflamable o fácilmente inflamable, |
|
— |
si se ha llegado a la conclusión de que el gas no es inflamable, se debe indicar el intervalo de concentraciones que se ha sometido a ensayo en etapas del 1 %, |
|
— |
cualquier información u observación que pueda ser útil para la interpretación de los resultados. |
4. BIBLIOGRAFÍA
|
(1) |
NF T 20-041 (SEPT 85). Chemical products for industrial use. Determination of the flammability of gases. |
|
(2) |
W. Berthold, D. Conrad, T. Grewer, H. Grosse-Wortmann, T. Redeker y H. Schacke. «Entwicklung einer Standard-Apparatur zur Messung von Explosionsgrenzen». Chem.-Ing.-Tech., 1984, vol. 56, 126-127. |
A.12. INFLAMABILIDAD (EN CONTACTO CON EL AGUA)
1. MÉTODO
1.1. INTRODUCCIÓN
Este método de ensayo puede utilizarse para determinar si la reacción de una sustancia con el agua o el aire húmedo ocasiona el desprendimiento de una cantidad peligrosa de un gas o de varios gases, que puedan ser fácilmente inflamables.
Puede aplicarse tanto a sustancias sólidas como líquidas, pero no a las sustancias que se inflaman espontáneamente en contacto con el aire.
1.2. DEFINICIONES Y UNIDADES
Sustancias fácilmente inflamables: preparados que, en contacto con el agua o el aire húmedo, desprenden una cantidad peligrosa de gases fácilmente inflamables, a una velocidad mínima de 1 l/kg por hora.
1.3. PRINCIPIO DEL MÉTODO
El ensayo incluye varias fases sucesivas que se describen a continuación; si la inflamación se produce en cualquiera de estas fases no es necesario proseguir el ensayo. Si se sabe que la sustancia no reacciona violentamente con el agua, se pasa directamente a la fase 4 (1.3.4).
1.3.1. Fase 1
Se coloca la sustancia problema en una cubeta que contenga agua destilada a 20 oC y se observa si el gas desprendido se inflama o no.
1.3.2. Fase 2
Se coloca la sustancia de ensayo en un papel de filtro que flote sobre un recipiente lleno de agua destilada a 20 oC y se observa si el gas que se desprende se inflama o no. El papel de filtro solo sirve para mantener la sustancia en un lugar, lo cual aumenta las probabilidades de inflamación.
1.3.3. Fase 3
Se forma con la sustancia de ensayo una pila de 2 cm de altura y 3 cm de diámetro, aproximadamente. Se añaden algunas gotas de agua a la pila y se observa si el gas que se desprende se inflama o no.
1.3.4. Fase 4
Se mezcla la sustancia de ensayo con agua destilada a 20 oC y se mide la velocidad de producción de gas durante 7 horas, a intervalos de una hora. Si al cabo de 7 horas la velocidad de producción es variable o va aumentando, debe prolongarse el tiempo de medida hasta un máximo de 5 días. Si, en un momento dado, la velocidad de producción supera 1 l/kg por hora, el ensayo puede darse por acabado.
1.4. SUSTANCIA DE REFERENCIA
No se especifica.
1.5. CRITERIOS DE CALIDAD
No se indican.
1.6. DESCRIPCIÓN DE LOS MÉTODOS
1.6.1. Fase 1
1.6.1.1. Condiciones del ensayo
El ensayo se realiza a temperatura ambiente (alrededor de 20 oC).
1.6.1.2. Desarrollo del ensayo
Se coloca una pequeña cantidad (aproximadamente unos 2 mm de diámetro) de la sustancia problema en una cubeta con agua destilada. Observar: i) si hay desprendimiento de gas, y ii) si el gas se inflama. Si se inflama, se considerará que la sustancia es peligrosa y se dará por finalizado el ensayo.
1.6.2. Fase 2
1.6.2.1. Equipo
Papel de filtro flotando sobre una superficie de agua destilada en un recipiente adecuado, como, por ejemplo, una cubeta de evaporación de 100 mm de diámetro.
1.6.2.2. Condiciones del ensayo
El ensayo se realiza a temperatura ambiente (alrededor de 20 oC).
1.6.2.3. Desarrollo del ensayo
Se coloca una pequeña cantidad de la sustancia problema (aproximadamente 2 mm de diámetro) sobre el centro del papel de filtro. Observar: i) si hay desprendimiento de gas, y ii) si el gas se inflama. Si se inflama, se considerará que la sustancia es peligrosa y se dará por finalizado el ensayo.
1.6.3. Fase 3
1.6.3.1. Condiciones del ensayo
El ensayo se realiza a temperatura ambiente (alrededor de 20 oC).
1.6.3.2. Desarrollo del ensayo
Se forma con la sustancia de ensayo una pila de 2 cm de altura y 3 cm de diámetro, aproximadamente, con un pequeño cráter en la cumbre. Se añaden algunas gotas de agua en el hueco y se observa: i) si hay desprendimiento de gas, y ii) si el gas se inflama. Si se inflama, se considerará que la sustancia es peligrosa y se dará por finalizado el ensayo.
1.6.4. Fase 4
1.6.4.1. Equipo
El equipo se monta según se muestra en la figura.
1.6.4.2. Condiciones del ensayo
Hay que asegurarse de que el recipiente que contiene la sustancia problema está libre de partículas pulverulentas (tamaño de partícula < 500 μm). Si estas representan más del 1 % en peso del total, o si la muestra es quebradiza, hay que reducir a polvo la sustancia antes de proceder al ensayo, para tener en cuenta la reducción del tamaño de las partículas durante su almacenamiento y manipulación; en caso contrario, la sustancia se utiliza en su forma original. Debe realizarse el ensayo a temperatura ambiente (alrededor de 20 oC) y a presión atmosférica.
1.6.4.3. Desarrollo del ensayo
Se ponen entre 10 y 20 ml de agua en el embudo cuentagotas del equipo y 10 g de sustancia en el matraz cónico. El volumen de gas producido puede medirse con cualquier método adecuado. Se abre la tapa del embudo cuentagotas para que penetre el agua en el matraz cónico y se pone en marcha un cronómetro. Se mide la producción de gas cada hora a lo largo de un período de 7 horas. Si durante este período la producción de gas es irregular o si al final del período la velocidad de producción de gas está en aumento, hay que continuar con las medidas hasta un máximo de 5 días. Si en cualquier momento de la medida la velocidad de producción de gas pasa de 1 l/kg por hora, puede interrumpirse el ensayo. Este ensayo debe realizarse por triplicado.
Si no se conoce la identidad química del gas, debe analizarse este. Si el gas contiene componentes fácilmente inflamables y si, además, se ignora si el conjunto de la mezcla es fácilmente inflamable, hay que preparar y someter a ensayo una mezcla de la misma composición, de conformidad con el método A.11.
2. RESULTADOS
La sustancia se considera peligrosa si:
|
— |
se produce ignición espontánea en cualquier fase del desarrollo del ensayo, o |
|
— |
se produce gas inflamable a una velocidad superior a 1 l/kg de sustancia por hora. |
3. INFORME
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
especificación precisa de la sustancia (identificación e impurezas), |
|
— |
datos sobre cualquier preparación inicial de la sustancia problema, |
|
— |
resultados de los ensayos (fases 1, 2, 3 y 4), |
|
— |
identidad química del gas desprendido, |
|
— |
la velocidad de formación del gas si se realiza la fase 4 (1.6.4), |
|
— |
cualquier observación complementaria que sea útil para la interpretación de los resultados. |
4. BIBLIOGRAFÍA
|
(1) |
Recommendations on the Transport of Dangerous Goods, test and criteria, 1990, Naciones Unidas, Nueva York. |
|
(2) |
NF T 20-040 (SEPT 85). Chemical products for industrial use. Determination of the flammability of gases formed by the hydrolysis of solid and liquid products. |
Apéndice
Figura
Equipo

A.13. PROPIEDADES PIROFÓRICAS DE SÓLIDOS Y LÍQUIDOS
1. MÉTODO
1.1. INTRODUCCIÓN
El procedimiento del ensayo es aplicable a las sustancias sólidas y liquidas que pueden inflamarse espontáneamente poco tiempo después de haber entrado en contacto con el aire a temperatura ambiente (alrededor de 20 oC).
Este método de ensayo no puede aplicarse a las sustancias que tienen que exponerse al aire durante varias horas o días a temperatura ambiente o a temperaturas elevadas antes de inflamarse.
1.2. DEFINICIONES Y UNIDADES
Se considera que una sustancia tiene propiedades pirofóricas si, en las condiciones descritas en el punto 1.6, se inflama o se carboniza.
También puede ser necesario estudiar la autoinflamabilidad de líquidos según el método A.15. Temperatura de autoinflamación (líquidos y gases).
1.3. SUSTANCIAS DE REFERENCIA
No se especifican.
1.4. PRINCIPIO DEL MÉTODO
Se añade la sustancia, sea sólida o líquida, a un soporte inerte y se pone en contacto con el aire a temperatura ambiente durante un período de 5 minutos. Si las sustancias líquidas no se inflaman, entonces se absorben en papel de filtro y se exponen al aire a temperatura ambiente (alrededor de 20 oC) durante 5 minutos. Si un sólido o líquido se inflama, o un líquido inflama o carboniza a un papel de filtro, entonces se considera que la sustancia es pirofórica.
1.5. CRITERIOS DE CALIDAD
Repetibilidad: debido a la importancia en relación con la seguridad, un solo resultado positivo será suficiente para considerar que la sustancia es pirofórica.
1.6. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
1.6.1. Equipo
Llenar una cubeta de porcelana de unos 10 cm de diámetro con una capa de tierra de infusorios, de unos 5 mm de espesor, a temperatura ambiente (alrededor de 20 oC).
Observaciones:
La tierra de infusorios, o cualquier otra sustancia inerte equivalente que se obtenga fácilmente, se tomará como representativa de suelo sobre el que pudiera esparcirse la sustancia en caso de accidente.
El papel de filtro seco es necesario para el ensayo de los líquidos que no se inflaman en contacto con el aire cuando están unidos a un soporte inerte.
1.6.2. Realización del ensayo
a) Sólidos pulverulentos
Se vierten 1 o 2 cm3 de la sustancia problema, desde una altura de alrededor de 1 m, sobre una superficie no combustible y se observa si la sustancia se inflama durante la caída o durante los primeros 5 minutos después de depositarse.
El ensayo se realiza seis veces, salvo que se produzca ignición.
b) Líquidos
Se vierten 5 cm3 del líquido problema en la cubeta de porcelana preparada y se observa si la sustancia se inflama en un tiempo de 5 minutos.
Si no se observa inflamación durante estos seis ensayos, hay que realizar los siguientes ensayos:
Con una jeringuilla se ponen 0,5 ml de la sustancia en un papel de filtro dentado y se observa si se produce inflamación o carbonización del papel de filtro en el plazo de 5 minutos desde la adición del líquido. El ensayo se realiza tres veces, salvo que se produzca inflamación o carbonización.
2. RESULTADOS
2.1. TRATAMIENTO DE LOS RESULTADOS
El ensayo puede interrumpirse tan pronto como se obtenga un resultado positivo en cualquiera de las pruebas.
2.2. EVALUACIÓN
Una sustancia se considera pirofórica si se inflama en el plazo de 5 minutos cuando se añade a un soporte inerte y se expone al aire, o bien cuando la sustancia líquida carboniza o inflama un papel de filtro en el plazo de 5 minutos desde que se pone en contacto con este y se expone al aire.
3. INFORME
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
especificación precisa de la sustancia (identificación e impurezas), |
|
— |
resultado del ensayo, |
|
— |
cualquier observación complementaria que sea útil para la interpretación de los resultados. |
4. BIBLIOGRAFÍA
|
(1) |
NF T 20-039 (SEPT 85). Chemical products for industrial use. Determination of the spontaneous flammability of solids and liquids. |
|
(2) |
Recommendations on the Transport of Dangerous Goods, Test and criteria, 1990, Naciones Unidas, Nueva York. |
A.14. PROPIEDADES EXPLOSIVAS
1. MÉTODO
1.1. INTRODUCCIÓN
Se trata de un método de ensayo que permite determinar si una sustancia sólida o pastosa presenta o no peligro de explosión cuando se expone al efecto de una llama (sensibilidad térmica) o a un choque o fricción (sensibilidad a estímulos mecánicos), y si una sustancia líquida presenta peligro de explosión cuando se expone al efecto de una llama o un choque.
El método comprende tres partes:
|
a) |
un ensayo de sensibilidad térmica (1); |
|
b) |
un ensayo de sensibilidad mecánica respecto al choque (1); |
|
c) |
un ensayo de sensibilidad mecánica respecto a la fricción (1). |
El método proporciona datos que permiten evaluar la probabilidad de inicio de una explosión por medio de algunos estímulos corrientes. No tiene por objeto determinar si una sustancia puede o no hacer explosión en cualesquiera condiciones.
El método sirve para, determinar si una sustancia presenta peligro de explosión (sensibilidad térmica y mecánica) en las condiciones específicas definidas por la Directiva. En el ensayo se utiliza un cierto número de equipos ampliamente utilizados internacionalmente (1) y que, por regla general, dan resultados convincentes. Hay que reconocer que el método no es definitivo. Pueden utilizarse equipos distintos a los especificados siempre que estén reconocidos internacionalmente y se pueda establecer una buena correlación entre los resultados obtenidos con el equipo alternativo y los del equipo especificado.
No es necesario realizar los ensayos si los datos termodinámicos disponibles (calor de formación, calor de descomposición, etc.) o la ausencia de ciertos grupos reactivos (2) en la fórmula desarrollada permiten establecer de forma razonablemente inequívoca que la sustancia no puede descomponerse rápidamente con formación de gases o liberación de calor (dicho de otro modo, si la materia no presenta ningún riesgo de explosión). No es necesario realizar un ensayo de sensibilidad a la fricción con las sustancias líquidas.
1.2. DEFINICIONES Y UNIDADES
Explosivos:
Sustancias que puedan hacer explosión bajo el efecto de una llama, o que sean sensibles al choque o a la fricción en el equipo especificado (o que sean más sensibles mecánicamente que el 1,3-dinitrobenceno en un equipo alternativo).
1.3. SUSTANCIAS DE REFERENCIA
1,3-dinitrobenceno, producto cristalino técnico pasado por tamiz de 0,5 mm, para los métodos de fricción y choque.
Perhidro-l,3,5-trinitro-l,3,5-triazina (RDX, hexogen, ciclonita — CAS 121-82-4), recristalizado a partir de ciclohexanona acuosa, tamizado por vía húmeda a través de un tamiz de 250 μm y retenido en un tamiz de 150 μm y secado a 103 ± 2 oC durante 4 horas para la segunda serie de ensayos de fricción y choque.
1.4. PRINCIPIO DEL MÉTODO
Se debe realizar un ensayo preliminar para determinar las condiciones de seguridad que deben presidir la ejecución de los tres ensayos de sensibilidad.
1.4.1. Pruebas de seguridad en la manipulación (3)
Por razones de seguridad, antes de pasar a los ensayos principales, se someten muestras muy reducidas (unos 10 mg) de sustancia a un calentamiento sin confinamiento con llama de gas, a choque con cualquier tipo de instrumento adecuado y a fricción utilizando un mazo y un yunque, o cualquier otro tipo de instrumento que sirva para producir fricción. El objetivo es determinar si la sustancia es tan sensible y explosiva que los ensayos de sensibilidad prescritos, especialmente el de sensibilidad térmica, deban realizarse con precauciones especiales a fin de evitar cualquier daño corporal al operario.
1.4.2. Sensibilidad térmica
Este método consiste en calentar la sustancia en un tubo de acero cerrado por placas horadadas con agujeros de diferentes diámetros, para determinar si la sustancia puede hacer explosión en condiciones de calor intenso y un confinamiento determinado.
1.4.3. Sensibilidad mecánica (choque)
El método consiste en someter la sustancia al choque producido por una masa especificada que se deja caer desde una altura también especificada.
1.4.4. Sensibilidad mecánica (fricción)
Este método consiste en someter la sustancia sólida o pastosa a una fricción entre superficies tipo, en condiciones específicas de carga y de movimiento relativo.
1.5. CRITERIOS DE CALIDAD
No se indican.
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Sensibilidad térmica (efecto de la llama)
1.6.1.1. Equipo
El equipo consiste en un tubo de acero no reutilizable con un sistema de cierre reutilizable (figura 1), instalado en un instrumento de calefacción y protección. Cada tubo se obtiene por embutido de una lámina de acero (véase el apéndice) y tiene 24 mm de diámetro interior, 75 mm de longitud y 0,5 mm de espesor de pared. Los tubos tienen un reborde en el extremo abierto para que se puedan cerrar con el dispositivo de la placa horadada. Este dispositivo consiste en una placa horadada resistente a la presión, con un orificio central, unido firmemente al tubo mediante una junta aterrajada en dos partes (tuerca y abrazadera de rosca); esta junta está hecha de acero al cromo-manganeso (véase el apéndice) exento de chispas hasta 800 oC. Las placas horadadas tienen 6 mm de espesor, están hechas de acero resistente al calor (véase el apéndice) y pueden escogerse con varios diámetros de abertura.
1.6.1.2. Condiciones del ensayo
Normalmente se somete a ensayo la sustancia tal como llega al laboratorio, aunque en algunos casos (por ejemplo, si está prensada, fundida o condensada de alguna otra forma) puede ser necesario estudiar la sustancia después de triturarla.
Si se trata de un sólido, la masa de material que debe utilizarse en cada ensayo se determina mediante un proceso seco de dos fases. Se llena un tubo tarado con 9 cm3 de sustancia y esta se apisona aplicando una fuerza de 80 N a toda la sección transversal del tubo. La forma de llenar el tubo puede ser distinta por razones de seguridad o en caso de que la forma física de la muestra pueda cambiar por la compresión; por ejemplo, si la sustancia es muy sensible a la fricción, no puede apisonarse. Si el material es compresible, se añade más cantidad y se vuelve a apisonar hasta que la altura del relleno queda a 55 mm de la boca. Se determina la masa total utilizada para llenar el tubo hasta el nivel de 55 mm y se hacen dos adiciones más, cada una apisonada con una fuerza de 80 N. Entonces, según sea necesario, se añade más material y se apisona o bien se extrae para dejar la altura del relleno a 15 mm de la boca. Se lleva a cabo un segundo proceso seco, empezando con una cantidad igual a un tercio de la masa total utilizada en el primer proceso seco, y se apisona esta cantidad. Se añaden otros dos incrementos de esta cantidad con apisonamiento de 80 N y se ajusta el nivel de la sustancia en el tubo a 15 mm de la boca por adición o extracción de material, según sea necesario. La cantidad de sólido determinada en el segundo proceso seco es la que se utilizará en cada ensayo; el llenado se hace en tres cantidades iguales, comprimida cada una a 9 cm3 mediante la fuerza que sea necesaria. (Puede ser más fácil utilizar anillos separadores.)
Los líquidos y geles se cargan en el tubo hasta una altura de 60 mm teniendo especial cuidado con los geles para evitar la formación de vacíos. La abrazadera de rosca se desliza sobre el tubo desde el fondo, se inserta la placa horadada correspondiente y se aprieta la tuerca después de poner un lubricante a base de disulfuro de molibdeno. Es muy importante comprobar que no queda nada de sustancia aprisionada entre el reborde y la placa o en las roscas.
La fuente de calor es propano procedente de una botella industrial, provisto de un regulador de presión (60 a 70 mbar), que pasa a través de un contador y mediante un distribuidor se reparte en cuatro mecheros de forma equilibrada (lo que se comprueba por observación visual de las llamas de los mecheros). Los mecheros se colocan alrededor del recinto de ensayo según se indica en la figura 1. Los cuatro mecheros suponen un consumo total de unos 3,2 litros de propano por minuto. Pueden utilizarse otros mecheros y gases combustibles pero la velocidad de calentamiento debe ser la especificada en la figura 3. Independientemente del equipo utilizado, es necesario comprobar periódicamente la velocidad de calentamiento mediante tubos llenos de ftalato de dibutilo, según se indica en la figura 3.
1.6.1.3. Desarrollo de los ensayos
Cada ensayo se realiza hasta que el tubo esté fragmentado o el tubo se haya calentado durante 5 minutos. Si el ensayo produce la fragmentación del tubo en tres o más piezas, que en algunos casos pueden estar unidas entre sí por bandas estrechas de metal, según ilustra la figura 2, entonces se considera que el ensayo produce explosión. Un ensayo en que se produzcan menos fragmentos o no haya fragmentación se considera que no produce explosión.
Se hace primero una serie de tres ensayos con una placa de 6,0 mm de diámetro de orificio y, si no se obtienen explosiones, se hace una segunda serie de tres ensayos con una placa de 2,0 mm de diámetro de orificio. Desde el momento en que se produzca una explosión en cualquiera de las dos series de ensayos no es necesario seguir realizando ensayos.
1.6.1.4. Evaluación
El resultado del ensayo se considera positivo si se produce una explosión en cualquiera de las dos series de ensayos.
1.6.2. Sensibilidad mecánica (choque)
1.6.2.1. Equipo (figura 4)
Las partes fundamentales de un equipo clásico de martinete son un bloque de acero fundido con base, yunque, columna, guías, pesos que caen, mecanismo de liberación y un soporte para las muestras. El yunque de acero (100 mm de diámetro por 70 mm de altura) se atornilla a la parte superior de un bloque de acero (230 mm de longitud por 250 mm de anchura por 200 mm de altura) con una base de fundición (450 mm de longitud por 450 mm de anchura por 60 mm de altura). En un soporte atornillado a la parte posterior del bloque de acero se fija la columna, hecha de tubo de acero estirado sin costuras. El aparato se fija con cuatro tornos a un bloque macizo de hormigón (de 60 × 60 × 60 cm) de forma que los raíles de guía sean absolutamente verticales y el peso caiga en caída libre. Pueden utilizarse pesos de 5 y 10 kg, de acero macizo. La cabeza de choque de cada peso está hecha de acero templado HRC 60 a 63 y tiene un diámetro mínimo de 25 mm.
La muestra problema se pone en un dispositivo de choque, formado por dos cilindros coaxiales de acero macizo, uno encima del otro, en un anillo de guía de acero en forma de cilindro hueco. Los cilindros de acero macizo deben tener 10 (-0,003, -0,005) mm de diámetro y 10 mm de altura, sus superficies deben estar pulidas, los bordes redondeados (0,5 mm de radio de curvatura) y su dureza debe ser de HRC 58 a 65. El cilindro hueco debe tener 16 mm de diámetro exterior, un orificio pulido de 10 (+0,005, +0,010) mm y 13 mm de altura. El dispositivo de choque se monta sobre un yunque intermedio (26 mm de diámetro y 26 mm de altura) hecho de acero y centrado por un anillo con perforaciones que dejen escapar los humos.
1.6.2.2. Condiciones de ensayo
El volumen de muestra debe ser de 40 mm3, o bien un volumen adecuado para otro tipo de aparato alternativo. Las sustancias sólidas deben someterse a ensayo en estado seco y prepararse de la forma siguiente:
|
a) |
las sustancias en polvo se pasan por tamiz de 0,5 mm; para las pruebas se usa la fracción que haya atravesado el tamiz; |
|
b) |
las sustancias prensadas, fundidas o condensadas de alguna otra forma se trituran en fragmentos pequeños y se tamizan; la fracción de diámetro comprendido entre 0,5 y 1 mm es la que se utiliza para los ensayos y debe ser representativa de la sustancia original. |
Las sustancias normalmente suministradas como pastas se someterán a ensayos en el estado más seco posible o en algún caso, suprimiendo al máximo posible la cantidad de diluyente. Las sustancias líquidas se estudiarán dejando una separación de 1 mm entre el cilindro de acero superior y el inferior.
1.6.2.3. Desarrollo de los ensayos
Se ejecuta una serie de seis ensayos dejando caer la masa de 10 kg desde la altura de 0,40 m (40 J). Si se obtiene alguna explosión en esta serie de 40 J, hay que realizar otra serie de seis ensayos, dejando caer la masa de 5 kg desde la altura de 0,15 m (7,5 J). En otros equipos, se compara la muestra con la sustancia de referencia elegida según un procedimiento establecido (por ejemplo, técnica de subida y bajada, etc.).
1.6.2.4. Evaluación
El resultado del ensayo se considera positivo si se produce una explosión (una inflamación y/o una detonación es equivalente a una explosión) al menos una vez en cualquiera de los ensayos con el equipo de choque especificado o si la muestra es más sensible que el 1,3-dinitrobenceno o el RDX en un ensayo de choque alternativo.
1.6.3. Sensibilidad mecánica (fricción)
1.6.3.1. Equipo (figura 5)
El equipo para el ensayo de fricción consiste en una base de acero fundido sobre la que se monta el dispositivo de fricción. Este consiste en una espiga fija de porcelana y un plato de porcelana móvil. El plato de porcelana se fija a una corredera que se desplaza sobre dos rieles. La corredera se conecta a un motor eléctrico por medio de una barra de conexión, una leva y un engranaje de transmisión adecuado, de forma que el plato de porcelana se desplace una sola vez hacia atrás y hacia adelante por debajo de la espiga de porcelana a lo largo de 10 mm. La espiga de porcelana puede cargarse, por ejemplo, con 120 o 360 N.
Los platos planos de porcelana están hechos de porcelana técnica blanca (rugosidad entre 9 y 32 μm) y sus dimensiones son 25 mm de longitud, 25 mm de anchura y 5 mm de altura. La espiga cilíndrica de porcelana también es de porcelana técnica blanca, mide 15 mm de longitud y 10 mm de diámetro y sus superficies extremas son esféricas y rugosas, con un radio de curvatura de 10 mm.
1.6.3.2. Condiciones de los ensayos
El volumen de la muestra debe ser de 10 mm3, o bien un volumen adecuado para otro tipo de aparato alternativo.
Las sustancias sólidas se someten a ensayo en estado seco y se preparan de la forma siguiente:
|
a) |
las sustancias en polvo se pasan por tamiz de 0,5 mm; para las pruebas se utiliza toda la fracción que haya atravesado el tamiz; |
|
b) |
las sustancias prensadas, fundidas o condensadas de alguna otra forma se trituran en fragmentos pequeños y se tamizan; para los ensayos se utiliza la fracción de diámetro inferior a 0,5 mm. |
Las sustancias normalmente suministradas como pastas se someterán a ensayos en el estado más seco posible. Si la sustancia no puede ser preparada en estado seco, la pasta (suprimiendo al máximo posible la cantidad de diluyente) es sometida a ensayo en forma de película de 0,5 mm de espesor, 2 mm de anchura y 10 mm de longitud, preparada con una plantilla.
1.6.3.3. Desarrollo de los ensayos
Se pone sobre la muestra la espiga de porcelana y se aplica la carga. Al realizar el ensayo, las marcas esponjosas del plato de porcelana deben ser transversales a la dirección del movimiento. Hay que vigilar para que la espiga se apoye en la muestra, que haya bastante sustancia problema bajo la espiga y que el plato se mueva correctamente bajo la espiga. En el caso de sustancias pastosas, para aplicar la sustancia al plato se utiliza un dosificador de 0,5 mm de espesor con una ranura de 2 × 10 mm. El plato de porcelana tiene que desplazarse 10 mm hacia adelante y hacia atrás bajo la espiga en el plazo de 0,44 segundos. Cada parte de la superficie del plato y de la espiga debe utilizarse solo una vez; los dos extremos de cada espiga sirven para dos ensayos y cada una de las dos superficies de un plato sirve para tres ensayos.
Se ejecuta una serie de seis pruebas con una carga de 360 N. Si se obtiene algún resultado positivo durante estas seis pruebas, hay que realizar otra serie de seis pruebas con una carga de 120 N. En otros equipos, se compara la muestra con la sustancia de referencia elegida mediante un procedimiento establecido (por ejemplo, técnica de subida y bajada, etc.).
1.6.3.4. Evaluación
El resultado del ensayo se considera positivo si se produce una explosión (una crepitación y/o una detonación o una inflamación equivale a una explosión) al menos una vez en cualquiera de las pruebas con el equipo especificado de fricción o si se satisfacen los criterios equivalentes de otro ensayo alternativo de fricción.
2. RESULTADOS
En principio, se considera que una sustancia presenta peligro de explosión en el sentido de la Directiva si se obtiene un resultado positivo en el ensayo de sensibilidad térmica, al choque o a la fricción.
3. INFORME
3.1. INFORME DEL ENSAYO
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
identidad, composición, pureza, grado higrométrico, etc., de la sustancia que se somete a ensayo, |
|
— |
forma física de la muestra y si se ha triturado, fragmentado o tamizado, |
|
— |
observaciones durante los ensayos de sensibilidad térmica (por ejemplo, masa de la muestra, número de fragmentos, etc.), |
|
— |
observaciones durante los ensayos de sensibilidad mecánica (por ejemplo, formación de cantidades considerables de humo o descomposición completa sin detonación, llamas, chispas, crepitación, etc.), |
|
— |
resultado de cada tipo de ensayo, |
|
— |
si se ha utilizado otro aparato alternativo, hay que indicar la justificación científica de dicha utilización, así como las pruebas de correlación entre los resultados obtenidos con el aparato especificado y los obtenidos con aparatos equivalentes, |
|
— |
cualquier observación que se considere útil, por ejemplo, referencia a ensayos realizados con productos similares, que puedan ser importantes para la interpretación correcta de los resultados, |
|
— |
cualquier otra observación importante para la interpretación de los resultados. |
3.2. INTERPRETACIÓN Y EVALUACIÓN DE LOS RESULTADOS
El informe debe indicar los resultados que se consideren erróneos, anormales o no representativos. Cuando se rechace un resultado, se facilitará una explicación y se indicarán los resultados del ensayo sustitutivo o complementario. Si un resultado anormal no pudiera explicarse, se deberá aceptar el valor obtenido y se utilizará para clasificar la sustancia en consecuencia.
4. BIBLIOGRAFÍA
|
(1) |
Recommendations on the Transport of Dangerous Goods: Tests and criteria, 1990, Naciones Unidas, Nueva York. |
|
(2) |
Bretherick, L., Handbook of Reactive Chemical Hazards, 4th edition, Butterworths, Londres, ISBN 0-750-60103-5, 1990. |
|
(3) |
Koenen, H., Ide, K.H. y Swart, K.H., Explosivstoffe, 1961, vol. 3, 6-13 y 30-42. |
|
(4) |
NF T 20-038 (Sept. 85). Chemical products for industrial use — Determination of explosion risk. |
Apéndice
Ejemplo de especificación de material para el ensayo de sensibilidad térmica (véase DIN 1623)
|
(1) |
Tubo: especificación de material no 1.0336.505 g |
|
(2) |
Placa horadada: especificación de material no 1.4873 |
|
(3) |
Tuerca y abrazadera de rosca: especificación de material no 1.3817 |
Figura 1
Equipo para el ensayo de sensibilidad térmica
(dimensiones en milímetros)
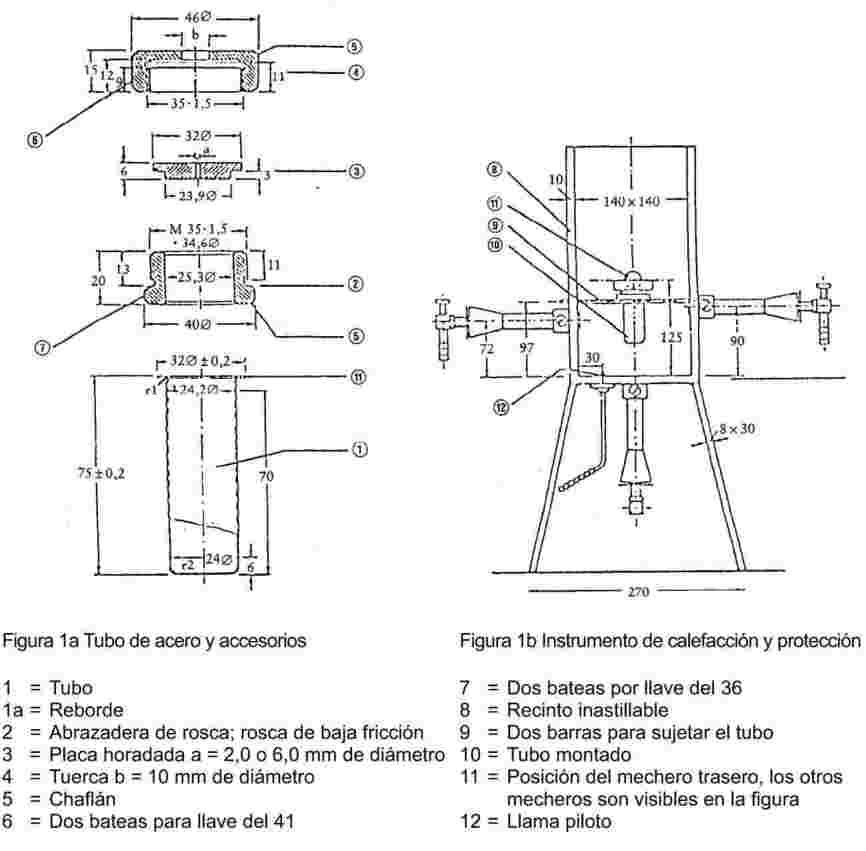
Figura 2
Ensayo de sensibilidad térmica
Ejemplos de fragmentación
Figura 3
Calibración de la velocidad de calentamiento en el ensayo de sensibilidad térmica
Curva temperatura/tiempo obtenida al calentar ftalato de dibutilo (27 cm3) en un tubo cerrado (placa con orificio de 1,5 mm) utilizando un caudal de propano de 3,2 l/min. La temperatura se mide con un termopar de 1 mm de diámetro de alumel/cromel revestido de acero inoxidable, situado en el centro a 43 mm por debajo del borde del tubo. La velocidad de calentamiento entre 135 oC y 285 oC debe estar entre 185 y 215 K/min.
Figura 4
Equipo del ensayo de choque
(dimensiones en milímetros)

Figura 4
Continuación

Figura 5
Equipo de sensibilidad a la fricción

A.15. TEMPERATURA DE AUTOINFLAMACIÓN (LÍQUIDOS Y GASES)
1. MÉTODO
1.1. INTRODUCCIÓN
No deben someterse a este ensayo las sustancias explosivas ni las que empiecen a arder espontáneamente en contacto con el aire a temperatura ambiente. El procedimiento de ensayo es aplicable a gases, líquidos y vapores que, en presencia de aire, puedan inflamarse por el contacto con una superficie caliente.
La temperatura de autoinflamación puede reducirse considerablemente por la presencia de impurezas catalíticas, por el material de la superficie o por un mayor volumen del recipiente de ensayo.
1.2. DEFINICIONES Y UNIDADES
El grado de autoinflamabilidad se expresa en términos de temperatura de autoinflamación. La temperatura de autoinflamación es la temperatura más baja a la que se inflama la sustancia problema, en presencia del aire y en las condiciones definidas por el método de ensayo.
1.3. SUSTANCIAS DE REFERENCIA
Las sustancias de referencia se citan en las normas (véase el punto 1.6.3). Sirven fundamentalmente para comprobar las características del método de vez en cuando y para poder comparar con los resultados obtenidos según otros métodos.
1.4. PRINCIPIO DEL MÉTODO
El método determina la temperatura mínima de la superficie interna de un recinto que provoca la inflamación de un gas, vapor o líquido inyectado en el recinto.
1.5. CRITERIOS DE CALIDAD
La repetibilidad varía según el intervalo de temperatura de autoinflamación y el método de ensayo utilizado.
La sensibilidad y la especificidad dependen del método de ensayo utilizado.
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Equipo
El equipo se describe en el método que se indica en el punto 1.6.3.
1.6.2. Condiciones del ensayo
Se somete a ensayo una muestra de la sustancia problema según el método indicado en el punto 1.6.3.
1.6.3. Desarrollo del ensayo
Véanse las normas CEI-79-4, DIN 51794, ASTM-E 659-78, BS 4056 y NF T 20-037.
2. RESULTADOS
Registrar la temperatura de ensayo, la presión atmosférica, la cantidad de muestra utilizada y el intervalo de tiempo que transcurre hasta que se produce la inflamación.
3. INFORME
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
especificación precisa de la sustancia (identificación e impurezas), |
|
— |
cantidad de muestra utilizada, presión atmosférica, |
|
— |
equipo utilizado, |
|
— |
resultados de las medidas (temperaturas de ensayo, resultados relativos a la inflamación, intervalos de tiempo correspondientes), |
|
— |
cualquier observación complementaria que pueda ser útil para la interpretación de los resultados. |
4. BIBLIOGRAFÍA
Ninguna.
A.16. TEMPERATURA RELATIVA DE AUTOINFLAMACIÓN DE SÓLIDOS
1. MÉTODO
1.1. INTRODUCCIÓN
Las sustancias explosivas y las sustancias que se inflaman espontáneamente en contacto con el aire a temperatura ambiente no deben someterse a este ensayo.
El objetivo del ensayo es proporcionar datos preliminares sobre la autoinflamabilidad de las sustancias sólidas a altas temperaturas.
Si el calor producido, bien por reacción de la sustancia con el oxígeno o bien por descomposición exotérmica, no se disipa con la suficiente rapidez en el ambiente, el autocalentamiento ocasiona la autoinflamación. La autoinflamación se produce, en consecuencia, cuando la velocidad de producción de calor sobrepasa la velocidad de disipación.
El procedimiento es útil como ensayo de selección preliminar para las sustancias sólidas. Teniendo en cuenta la naturaleza compleja de la inflamación y de la combustión de los sólidos, la temperatura de autoinflamación determinada según este método solo debe servir para hacer comparaciones.
1.2. DEFINICIONES Y UNIDADES
La temperatura de autoinflamación, tal como se determina por este método, es la temperatura ambiente mínima, expresada en grados centígrados ( oC), a la que se inflama cierto volumen de una sustancia en condiciones definidas.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
Se coloca un volumen definido de la sustancia problema en un horno a temperatura ambiente; se registra la curva de temperatura en el centro de la muestra frente al tiempo, elevando la temperatura del horno hasta 400 oC o hasta el punto de fusión, si este es menor, a razón de 0,5 oC por minuto. La temperatura del horno a la que la temperatura de la muestra alcanza los 400 oC por autocalentamiento es la que, a fines del presente ensayo, se denomina temperatura de autoinflamación.
1.5. CRITERIOS DE CALIDAD
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Equipo
1.6.1.1. Horno
Horno de laboratorio con temperatura programable (volumen: unos 2 litros) equipado con circulación natural de aire y una válvula de explosión. Se debe evitar que los gases de descomposición entren en contacto con las resistencias eléctricas para evitar cualquier peligro de explosión.
1.6.1.2. Cubo de tela de alambre
Siguiendo el modelo de la figura 1, se corta un pedazo de tela metálica de acero inoxidable con abertura de 0,045 mm. Se dobla la tela metálica y se sujeta con alambre para formar un cubo abierto por la parte superior.
1.6.1.3. Termopares
Termopares apropiados.
1.6.1.4. Registro
Cualquier registro de dos canales, calibrado en el intervalo de 0 a 600 oC o a una tensión correspondiente.
1.6.2. Condiciones del ensayo
El ensayo se efectúa con las sustancias tal y como se reciben.
1.6.3. Desarrollo del ensayo
Se llena el cubo con la sustancia problema, se comprime con cuidado y se añade más sustancia hasta llenar por completo el cubo. Se suspende este en el centro del horno a temperatura ambiente. Se coloca un termopar en el centro del cubo y otro entre el cubo y la pared del horno para registrar la temperatura de este último.
Las temperaturas del horno y de la muestra se registran continuamente elevando la temperatura del horno hasta los 400 oC o hasta el punto de fusión del sólido, si este valor es menor, a razón de 0,5 oC por minuto.
Cuando la sustancia se inflame, el termopar colocado en la muestra indicará una subida muy fuerte de la temperatura por encima de la temperatura del horno.
2. RESULTADOS
La temperatura del horno a la que la temperatura de la muestra alcanza los 400 oC por autocalentamiento es significativa para la evaluación (véase la figura 2).
3. INFORME
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
descripción de la sustancia problema, |
|
— |
resultados de las medidas, incluida la curva de temperatura-tiempo, |
|
— |
todas las observaciones complementarias que sean útiles para la interpretación de los resultados. |
4. BIBLIOGRAFÍA
NF T 20-036 (SEPT 85). Chemical products for industrial use. Determination of the relative temperatura of the spontaneous flammability of solids.
Figura 1
Modelo de cubo de ensayo de 20 mm
Figura 2
Curva tipo temperatura/tiempo

A.17. PROPIEDADES COMBURANTES (SÓLIDOS)
1. MÉTODO
1.1. INTRODUCCIÓN
Es conveniente disponer de información previa sobre las posibles propiedades explosivas de la sustancia antes de proceder al ensayo.
Este ensayo no es aplicable a líquidos, gases, sustancias explosivas o fácilmente inflamables ni a los peróxidos orgánicos.
Este ensayo es innecesario cuando el examen de la estructura química ponga de manifiesto que la sustancia no puede dar reacción exotérmica con un combustible.
Para saber si en este ensayo hay que tomar precauciones especiales, debe efectuarse una prueba preliminar.
1.2. DEFINICIONES Y UNIDADES
Tiempo de combustión: tiempo de reacción, expresado en segundos, necesario para que la zona de reacción se propague a través de la pila, según el procedimiento descrito en el punto 1.6.
Velocidad de combustión: expresada en milímetros por segundo.
Velocidad máxima de combustión: el valor más elevado entre las velocidades de combustión obtenidas con mezclas que contengan desde un 10 a un 90 % en peso de oxidante.
1.3. SUSTANCIAS DE REFERENCIA
Como sustancia de referencia se utiliza nitrato de bario (de grado analítico) tanto para el ensayo como para el ensayo preliminar.
La mezcla de referencia es la mezcla compuesta por nitrato de bario y celulosa en polvo, preparada según lo indicado en el punto 1.6, que tiene la velocidad máxima de combustión (se trata, generalmente, de una muestra con un 60 % en peso de nitrato de bario).
1.4. PRINCIPIO DEL MÉTODO
Por razones de seguridad se procede a un ensayo preliminar. Este ensayo será, por sí solo, suficiente si durante el mismo se observa claramente que la sustancia problema tiene propiedades comburantes. En caso contrario, la sustancia deberá someterse al ensayo completo.
Para efectuar este ensayo completo se mezclan en proporciones variables la sustancia problema y un combustible definido. Con cada una de las mezclas se forma una pila y esta se enciende por un extremo. La velocidad máxima de combustión observada se compara con la velocidad máxima de combustión de la mezcla de referencia.
1.5. CRITERIOS DE CALIDAD
Cualquier método de trituración y de mezcla que deba emplearse será válido siempre que la diferencia entre la velocidad máxima de combustión en los seis ensayos y la media aritmética no sobrepase el 10 %.
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Preparación
1.6.1.1. Sustancia problema
Con el fin de obtener una granulometría inferior a 0,125 mm, se somete la muestra al siguiente procedimiento: se tamiza, se tritura la parte que quede y se tamiza de nuevo, repitiendo la operación hasta que toda la muestra haya pasado por el tamiz.
Para triturar y tamizar puede utilizarse cualquier método, siempre que se cumplan los criterios de calidad requeridos.
Antes de hacer la mezcla se desecará la muestra a 105 oC hasta obtener peso constante. Si la temperatura de descomposición de la sustancia es inferior a 105 oC, se secará a una temperatura inferior adecuada.
1.6.1.2. Combustible
El combustible es celulosa en polvo, del tipo utilizado en cromatografía en capa fina y en columna. Se considera adecuada una celulosa en la que la longitud de más del 85 % de las fibras esté comprendida entre 0,020 y 0,075 mm. El polvo de celulosa se tamiza con una malla de 0,125 mm. Hay que usar el mismo lote de celulosa en todo el ensayo.
Antes de preparar la mezcla, hay que secar la celulosa a 105 oC hasta obtener el peso constante.
Si en el ensayo preliminar se utiliza serrín, se elegirá un serrín de madera blanda, que se pasará por un tamiz de 1,6 mm de malla, se mezclará bien y se secará a 105 oC durante 4 horas en capas de no más de 25 mm de espesor. Una vez enfriado, se debe guardar en un envase hermético, llenándolo lo más posible. Es preferible utilizar el serrín dentro de las 24 horas siguientes al secado.
1.6.1.3. Fuente de ignición
Debe utilizarse la llama fuerte de un mechero de gas (diámetro mínimo: 5 mm). Si se utiliza otra fuente de ignición (por ejemplo, si se hace el ensayo en atmósfera inerte) hay que dar su descripción y justificación.
1.6.2. Desarrollo del ensayo
Nota:
Las mezclas de comburante con celulosa o serrín deben considerarse potencialmente explosivas y tratarse con las debidas precauciones.
1.6.2.1. Ensayo preliminar
La sustancia secada se mezcla bien con celulosa o serrín seco, en una proporción en peso de 2 partes de sustancia problema por 1 parte de celulosa o serrín. Con la mezcla se forma una pila cónica de 3,5 cm de diámetro de base y 2,5 cm de altura, llenando sin comprimir un molde cónico (por ejemplo, un embudo de laboratorio de cristal con el vástago tapado).
La pila se coloca sobre una superficie fría, incombustible, no porosa y de baja conductividad térmica. El ensayo debe realizarse bajo una campana extractora (véase el punto 1.6.2.2).
La fuente de ignición se pone en contacto con el cono. Se observan y se anotan la intensidad y la duración de la reacción resultante.
Si la reacción es intensa, se considerará que la sustancia es comburante.
Cuando existan dudas respecto al resultado, será necesario efectuar el ensayo completo que se describe a continuación.
1.6.2.2. Ensayo completo
Se preparan mezclas de comburante y de celulosa que contengan de un 10 a un 90 % en peso de comburante, en incrementos del 10 %. Para los casos límite, hay que utilizar mezclas intermedias de comburante y celulosa para determinar la velocidad máxima de combustión con más precisión.
Se forma la pila con ayuda de un molde metálico de sección triangular de 250 mm de longitud, 10 mm de altura interior y 20 mm de anchura interior. A ambos lados de este molde se colocan en sentido longitudinal dos placas metálicas puestas como topes laterales que sobrepasen en 2 mm el borde superior de la sección triangular (véase la figura). Este montaje se llena, sin apretar, con un ligero exceso de mezcla. Después de haber dejado caer una vez el molde desde una altura de 2 cm sobre una superficie dura, se elimina la materia sobrante por medio de una lámina sostenida oblicuamente. Se quitan entonces los topes laterales y se aplana la superficie del polvo con ayuda de un rodillo. Se pone entonces sobre el molde una placa incombustible, no porosa y de baja conductividad térmica, se da la vuelta al conjunto y se quita el molde.
Se pone la pila en el tiro de una campana de gases.
Durante el ensayo, la velocidad de aspiración debe ser constante y suficiente para evitar que los humos se expandan por el laboratorio. Alrededor del aparato se coloca un cortaviento.
Dadas las propiedades higroscópicas de la celulosa y de algunas sustancias problema, el ensayo debe efectuarse lo más rápidamente posible.
Se enciende un extremo de la pila aplicándole la llama.
Se mide el tiempo de reacción sobre 200 mm después de que la zona de reacción haya recorrido una distancia inicial de 30 mm.
El ensayo se realiza con la sustancia de referencia y, por lo menos una vez, con cada una de las mezclas de sustancia problema con celulosa.
Si se observa una velocidad máxima de combustión significativamente mayor que la de la mezcla de referencia, puede detenerse el ensayo; si no es así, se repetirá de nuevo cinco veces con cada una de las tres mezclas que hayan dado las velocidades de combustión más elevadas.
Si se sospecha que el resultado es un falso positivo, hay que repetir el ensayo con una sustancia inerte que presente un tamaño de partícula similar, como el kieselguhr, en lugar de la celulosa. Otra posibilidad es someter a ensayo en atmósfera inerte (< 2 % v/v de oxígeno) la mezcla sustancia problema/celulosa que haya dado la mayor velocidad de combustión.
2. RESULTADOS
Por razones de seguridad, la velocidad máxima de combustión, en lugar de la media, será considerada como la propiedad comburante característica de la sustancia examinada.
En la evaluación se tendrá en cuenta la velocidad de combustión más elevada medida en una serie de seis ensayos con una mezcla dada.
Se representa gráficamente la velocidad de combustión más elevada de cada mezcla en función del contenido en comburante. La velocidad máxima de combustión se obtiene a partir de la gráfica.
Las seis velocidades de combustión medidas en una serie con la mezcla que haya dado la velocidad máxima de combustión, no deben diferir en más de 10 % de la media aritmética. En caso contrario, se deberán mejorar los métodos de trituración y mezcla.
Se compara la velocidad máxima de combustión obtenida con la velocidad máxima de combustión de la mezcla de referencia (véase el punto 1.3).
Si se hacen ensayos en atmósfera inerte, se comparará la velocidad máxima de reacción con la de la mezcla de referencia en atmósfera inerte.
3. INFORME
3.1. INFORME DEL ENSAYO
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
identidad, composición, pureza, humedad, etc., de la sustancia problema, |
|
— |
cualquier tratamiento a que se haya sometido la muestra (por ejemplo, triturado o secado), |
|
— |
fuente de ignición utilizada en los ensayos, |
|
— |
resultados de las medidas, |
|
— |
tipo de reacción (por ejemplo: combustión rápida superficial, combustión en toda la masa, cualquier observación que se refiera a los productos de combustión, etc.), |
|
— |
cualquier otra observación que resulte de utilidad para la interpretación de los resultados, incluida una descripción de la intensidad (llama, chispa, humo, combustión lenta sin llama, etc.) y la duración aproximada de la reacción observada durante el ensayo preliminar de seguridad/selección para la sustancia problema y la sustancia de referencia, |
|
— |
resultado de los ensayos con una sustancia inerte, en su caso, |
|
— |
resultado de los ensayos en atmósfera inerte, en su caso. |
3.2. INTERPRETACIÓN DE LOS RESULTADOS
Se considerará que una sustancia es comburante si:
|
a) |
hay una reacción intensa en el ensayo preliminar; |
|
b) |
durante el ensayo completo la velocidad máxima de combustión de las mezclas problema es superior o igual a la de la mezcla de referencia formada por celulosa y nitrato de bario. |
A fin de evitar los falsos positivos, a la hora de interpretar los resultados también habrá que tener en cuenta los valores obtenidos al estudiar la sustancia problema mezclada con material inerte o en atmósfera inerte.
4. BIBLIOGRAFÍA
NF T 20-035 (SEPT 85). Chemical products for industrial use. Determination of the oxidizing properties of solids.
Apéndice
Figura
Molde y accesorios necesarios para formar las pilas
(rodas las dimensiones expresadas en milímetros)

A.18. PESO MOLECULAR MEDIO EN NÚMERO Y DISTRIBUCIÓN DE LOS PESOS MOLECULARES DE LOS POLÍMEROS
1. MÉTODO
Este método de cromatografía de permeación sobre gel (CPG) es copia de las directrices TG 118 de la OCDE (1996). Los principios fundamentales y demás información técnica se dan en la referencia 1.
1.1. INTRODUCCIÓN
La gran diversidad de las propiedades de los polímeros impide describir un único método que fije precisamente las condiciones de separación y de evaluación que cubran todas las eventualidades y particularidades que se producen en la separación de los polímeros. En particular, es frecuente que los polímeros complejos no puedan someterse a la CPG. En este caso, puede determinarse el peso molecular diferentemente (véase el anexo). Deberán entonces indicarse todos los datos y la justificación del método utilizado.
El método descrito aquí se basa en la norma DIN 55672 (1). Se encontrará en esta norma DIN información precisa sobre la manera de llevar a cabo las pruebas y de evaluar los datos. Cuando sea indispensable introducir modificaciones de las condiciones experimentales, estos cambios deberán justificarse. Se podrán utilizar otras normas con las debidas referencias. El método descrito utiliza para la calibración muestras de poliestireno de polidispersidad conocida y puede ser necesario modificarlo para adaptarlo a algunos polímeros como, por ejemplo, los polímeros solubles en el agua o ramificados de cadena larga.
1.2. DEFINICIONES Y UNIDADES
El peso molecular medio en número Mn y el peso molecular medio en peso Mw se determinan con las siguientes ecuaciones:
|
|
|
donde
Hi = el nivel de la señal del detector a partir de la línea de base para el volumen de retención Vi
Mi = el peso molecular de la fracción polimérica de volumen de retención Vi
n = el número de puntos de medición.
La amplitud de la distribución de los pesos moleculares, que caracteriza la dispersidad del sistema, viene dada por la relación Mw/Mn.
1.3. SUSTANCIAS DE REFERENCIA
Como la CPG es un método relativo, es necesario efectuar una calibración. Para eso, se utilizan normalmente patrones de poliestireno de distribución estrecha y estructura lineal cuyos pesos moleculares medios Mn y Mw se conocen, así como su distribución de pesos moleculares. La curva de calibración solo puede servir para determinar el peso molecular de la muestra desconocida si las condiciones de separación de la muestra y de los patrones se han seleccionado de manera idéntica.
Una relación determinada entre el peso molecular y el volumen de elución solo es válida en las condiciones específicas de una experiencia particular. Estas condiciones son, sobre todo, la temperatura, el disolvente (o la mezcla de disolventes), las condiciones de la cromatografía y la columna o sistema de columnas de separación.
Los pesos moleculares de la muestra, determinados de esta manera, son valores relativos y se describen como «pesos moleculares equivalentes de poliestireno». En otros términos, según las diferencias estructurales y químicas entre la muestra y los patrones, los pesos moleculares pueden más o menos desviarse de los valores absolutos. Si se recurre a otros patrones como, por ejemplo, de políetilenglicol, de polióxido de etileno, de polimetacrilato de metilo, de poliácido acrilico, se explicará esta elección.
I.4. PRINCIPIOS DEL MÉTODO DE ENSAYO
La distribución de pesos moleculares de la muestra y los pesos moleculares medios (Mn, Mw) pueden determinarse por CPG. La CPG es una cromatografía líquida particular en la cual la muestra se separa según los volúmenes hidrodinámicos de sus componentes (2).
La separación se efectúa mientras que la muestra pasa por una columna rellena de un material poroso, normalmente un gel orgánico. Las moléculas pequeñas consiguen penetrar en los poros, mientras que las gruesas quedan excluidas. El recorrido de las moléculas grandes es, pues, más corto y se eluyen antes. Las moléculas de tamaño medio penetran en algunos poros y se eluyen posteriormente. Las moléculas más pequeñas, con un radio hidrodinámico medio más pequeño que los poros del gel, pueden penetrar en todos ellos y se eluyen al final.
En una situación ideal, la separación depende completamente del tamaño de las distintas moléculas, pero en la práctica es difícil evitar la interferencia de al menos algunos efectos de absorción. La situación empeora en caso de relleno irregular de la columna o de volúmenes muertos (2).
Se procede a la detección basándose, por ejemplo, en el índice de refracción o en la absorción UV para llegar a una curva de distribución simple. Sin embargo, para precisar sobre la curva valores efectivos de los pesos moleculares, es necesario calibrar la columna haciendo pasar polímeros de peso molecular conocido y, a ser posible, de estructura globalmente idéntica, por ejemplo, diversos patrones de poliestireno. Resulta de manera característica una curva de Gauss, a veces distorsionada por una pequeña cola hacia el lado de los pesos moleculares bajos; el eje vertical indica la cantidad, en peso, de las especies de distintos pesos moleculares eluídas, y el eje horizontal el logaritmo del peso molecular.
1.5. CRITERIOS DE CALIDAD
La repetibilidad (desviación típica relativa: RSD) del volumen de elución deberá sobrepasar el 0,3 %. La repetibilidad requerida del análisis estará garantizada por corrección mediante patrón interno si se evalúa un cromatograma en función del tiempo y no corresponde al criterio previamente mencionado (1). Las polidispersidades dependen de los pesos moleculares de los patrones. En el caso de los patrones de poliestireno, los valores característicos son:
|
Mp < 2 000 |
Mw/Mn < 1,20 |
|
2 000 < Mp < 106 |
Mw/Mn < 1,05 |
|
Mp > 106 |
Mw//Mn < 1,20 |
(Mp es el peso molecular del patrón en el máximo del pico)
1.6. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
1.6.1. Preparación de las soluciones patrón de poliestireno
Se disuelven los patrones de poliestireno mezclándolos cuidadosamente con el eluyente elegido. Se tendrán en cuenta las recomendaciones del fabricante para la preparación de las soluciones.
Las concentraciones de los patrones elegidos dependen de distintos factores como, por ejemplo, el volumen de inyección, la viscosidad de la solución y la sensibilidad del detector analítico. El volumen máximo de inyección debe adaptarse a la longitud de la columna, con el fin de evitar las sobrecargas. Generalmente, los volúmenes inyectados para las separaciones analíticas por CPG en una columna de 30 cm × ~ 7,8 mm se sitúan entre 40 y 100 ul. Es posible utilizar volúmenes mayores, pero no deben sobrepasar los 250 ul. La relación óptima entre el volumen de inyección y la concentración debe determinarse antes de la calibración efectiva de la columna.
1.6.2. Preparación de la solución de muestra
En principio, las mismas exigencias se aplican a la preparación de las soluciones de muestras. La muestra se disuelve en un disolvente conveniente como, por ejemplo, tetrahidrofurano (THF), agitando con cuidado. No debe nunca disolverse en baño de ultrasonidos. En caso necesario, la solución de muestra se purifica con un filtro de membrana cuyo tamaño de poro se sitúa entre 0,2 y 2 μm.
Debe señalarse en el informe final la presencia de partículas no disueltas, puesto que puede tratarse de sustancias de elevado peso molecular. Se utilizará un método conveniente para determinar el porcentaje en peso de partículas no disueltas. Las disoluciones se utilizarán en el plazo de 24 horas.
1.6.3. Equipo
|
— |
Depósito de disolvente. |
|
— |
Desgasificador (en su caso). |
|
— |
Bomba. |
|
— |
Amortiguador de pulsaciones (en su caso). |
|
— |
Sistema de inyección. |
|
— |
Columnas de cromatografía. |
|
— |
Detector. |
|
— |
Caudalímetro (en su caso). |
|
— |
Conjunto de registro-tratamiento de los datos. |
|
— |
Receptáculo de residuos. |
Es necesario garantizar que el sistema de CPG es inerte respecto a los disolventes utilizados (por ejemplo, utilizando capilares de acero para el disolvente THF).
1.6.4. Sistema de inyección y de distribución del disolvente
Un volumen definido de la solución de muestra se pasa a la columna con ayuda de un muestreador automático o bien manualmente, en una zona estrictamente definida. La liberación o la depresión manual demasiado rápida del émbolo de la jeringuilla pueden causar modificaciones en la distribución de pesos moleculares observada. El sistema de distribución del disolvente deberá, en la medida de lo posible, estar libre de pulsaciones, y lo ideal es que incluya un amortiguador de pulsaciones. El caudal es del orden de 1 ml/min.
1.6.5. Columna
Según la muestra, el polímero se caracterizará con ayuda de una columna simple o de varias columnas conectadas en secuencia. Está disponible en el comercio diverso material para columnas porosas, de propiedades definidas (por ejemplo, tamaño de poro, límites de exclusión). La elección del gel de separación o de la longitud de la columna depende tanto de las propiedades de la muestra (volúmenes hidrodinámicos, distribución de los pesos moleculares) como de las condiciones específicas de la separación, como el disolvente, la temperatura y el caudal (1) (2) (3).
1.6.6. Platos teóricos
La columna (o la combinación de columnas) utilizada para la separación debe caracterizarse por el número de platos teóricos. En el caso de tomar el THF como disolvente de elución, es necesario cargar una solución de etilbenceno u otro soluto homopolar adecuado en una columna de longitud conocida. El número de platos teóricos viene dado por la siguiente ecuación:
|
|
o |
|
donde
|
N |
= |
número de platos teóricos |
|
Ve |
= |
volumen de elución en el máximo del pico |
|
W |
= |
anchura del pico en la línea de base |
|
W1/2 |
= |
lanchura del pico a la mitad de la altura. |
1.6.7. Capacidad de separación
Además del número de platos teóricos, cantidad que determina la anchura de la banda, la capacidad de separación desempeña también un papel y está determinada por la pendiente de la curva de calibración. La capacidad de separación de una columna viene dada por la siguiente relación:

donde
|
Ve, Mx |
= |
volumen de elución de un poliestireno de peso molecular Mx |
|
Ve(I0Mx) |
= |
volumen de elución de un poliestireno de peso molecular diez veces superior. |
La resolución del sistema se define generalmente deí siguiente modo:

donde
|
Vel' Ve1 |
= |
volúmenes de elución de los dos patrones de poliestireno en el máximo del pico |
|
W1, W2 |
= |
anchuras de los picos en la línea de base |
|
M1, M2 |
= |
pesos moleculares en el máximo del pico (deberían diferir en un factor de 10) |
El valor R del sistema de columna debería sobrepasar 1.7 (4),
1.6.8. Disolvente
Todos los disolventes deben ser de gran pureza (para el THF, se exigirá una pureza del 99,5 %). El depósito de disolvente (en caso necesario, en una atmósfera de gas inerte) debe ser suficientemente grande para la calibración de la columna y varios análisis de muestras. El disolvente debe desgasificarse antes de su transporte a la columna por la bomba.
1.6.9. Ajuste de la temperatura
La temperatura de los componentes internos esenciales (circuito de inyección, columnas, detector y conducciones) será constante y en coherencia con la elección del disolvente.
1.6.10. Detector
El detector tiene por objeto registrar cuantitativamente la concentración de muestra eluida de la columna. Para evitar el ensanchamiento inútil de los picos, el volumen de la célula del detector debe ser lo más bajo posible. No debería exceder de 10 μl excepto en caso de detectores de dispersión de luz y de viscosidad. La detección se hace generalmente por refractometría diferencial. Sin embargo, si alguna propiedad de la muestra o del disolvente de elución lo impone, se puede recurrir a otros tipos de detectores, como por ejemplo, detectores de UV/visible, IR, viscosidad, etc.
2. RESULTADOS E INFORMES
2.1. RESULTADOS
Hay que remitirse a la norma DIN (1) respecto al detalle de los criterios de evaluación así como para los imperativos relativos a la recogida y tratamiento de los datos.
De cada muestra, deben realizarse dos experiencias independientes, que será necesaria analizar separadamente.
Mn, Mw, Mw/Mn y Mp deben establecerse en cada medición. Es necesario indicar explícitamente que los valores medidos son valores relativos equivalentes a los pesos moleculares de los patrones utilizados.
Después de la determinación de los volúmenes de retención o del tiempo de retención (eventualmente corregidos con ayuda de un patrón interno), los valores log Mp (Mp es el máximo del pico del patrón de calibración) se representan gráficamente en función de una de esas cantidades. Serán necesarios al menos dos puntos de calibración por década de pesos moleculares y al menos cinco puntos de medida para la curva entera, que debe cubrir el peso molecular estimado de la muestra. El peso molecular más bajo de la curva de calibración se define por-el n-hexilbenceno u otro soluto homopolar adecuado. La media en número y la media en peso de los pesos moleculares se determinan generalmente por tratamiento informático de los datos, sobre la base de las fórmulas del punto 1.2. En caso de digitalización manual, se puede consultar el documento ASTM D 3536-91 (3).
La curva de distribución debe darse en forma de cuadro o de gráfico (frecuencia diferencial o porcentajes de las sumas frente al log M). Para la representación gráfica, una década de pesos moleculares debería normalmente ocupar unos 4 cm de anchura y el máximo del pico debería encontrarse a unos 8 cm de altura. En el caso de curvas de distribución integral, la diferencia de ordenadas entre 0 y 100 % debería ser de cerca de 10 cm.
2.2. INFORME DEL ENSAYO
El informe del ensayo debe contener la siguiente información:
2.2.1. Sustancia de ensayo
|
— |
Información existente sobre la sustancia de ensayo (identidad, aditivos, impurezas). |
|
— |
Descripción del tratamiento de la muestra, observaciones, problemas. |
2.2.2. Equipo
|
— |
Depósito de eluyente, gas inerte, desgasificación del eluyente, composición del eluyente, impurezas. |
|
— |
Bomba, amortiguador de pulsaciones, sistema de inyección. |
|
— |
Columnas de separación (fabricante, toda la información sobre las características de las columnas como tamaño del poro, naturaleza del material de separación, etc., número, longitud y orden de las columnas utilizadas). |
|
— |
Número de platos teóricos de la columna (o combinación), capacidad de separación (resolución del sistema). |
|
— |
Información sobre la simetría de los picos. |
|
— |
Temperatura de la columna, naturaleza del ajuste de la temperatura. |
|
— |
Detector (principio de medida, volumen de la célula). |
|
— |
Caudalímetro si se utiliza (fabricante, principio de medida). |
|
— |
Sistema de registro y tratamiento de los datos (material y programas informáticos). |
2.2.3. Calibración del sistema
|
— |
Descripción precisa del método empleado para construir la curva de calibración. |
|
— |
Información sobre los criterios de calidad de este método (por ejemplo, coeficiente de correlación, suma de cuadrados de error, etc.). |
|
— |
Explicaciones sobre todas las extrapolaciones, hipótesis y aproximaciones hechas durante el procedimiento experimental y la evaluación y tratamiento de los datos. |
|
— |
Todas las medidas utilizadas para construir la curva de calibración deben precisarse en un cuadro que incluya la información siguiente de cada punto de calibración:
|
2.2.4. Evaluación
|
— |
Evaluación en función del tiempo: métodos utilizados para garantizar la reproducibilidad requerida (método de corrección, patrón interno, etc.). |
|
— |
Información sobre si la evaluación se ha realizado basándose en el volumen de elución o en el tiempo de retención. |
|
— |
Información sobre los límites de la evaluación si no se analiza completamente un pico. |
|
— |
Descripción de los métodos de suavizado, si se utilizan. |
|
— |
Procedimientos de preparación y de tratamiento preliminar de la muestra. |
|
— |
Posible presencia de partículas no disueltas. |
|
— |
Volumen de inyección (μl) y concentración de inyección (mg/ml). |
|
— |
Observaciones sobre los efectos que llevan a divergencias en relación con el perfil ideal de la CPG. |
|
— |
Descripción detallada de todas las modificaciones de los procedimientos de ensayo. |
|
— |
Precisiones sobre las gamas de errores. |
|
— |
Toda la información y observaciones pertinentes para la interpretación de los resultados. |
3. REFERENCIAS
|
(1) |
DIN 55672 (1995). Gelpermeationschromatographie (GPC) mit Tetrabydrofuran (THE) als Elutionsmittel, Teil 1. |
|
(2) |
Yau, W.W., Kirkland, J.J., and Bly, D.D. eds, (1979). Modern Size Exclusión Liquid Cbromato- grapby, J. Wiley and sons. |
|
(3) |
ASTM D 3536-91, (1991). Standard Test Method for Molecular Weight Averages and Molecular Weight Distribution by Liquid Exclusion Chromatography (Gel Permeation Cbromato- graphy-GPC). American Society for Testing and Materials, Philadelphia, Pennsylvania. |
|
(4) |
ASTM D 5296-92, (1992). Standard Test Method for Molecular Weight Averages and Molecular Weight Distribution of Polystirene by High Performance Size-Exclusion Chromatography. American Society for Testing and Materials, Philadelphia, Pennsylvania. |
Anexo
Ejemplos de otros métodos de determinación del peso molecular promedio en número (M) de los polímeros
La cromatografía por permeación sobre gel (CPG) es el método de elección para determinar Mn, en particular cuando se dispone de un conjunto de patrones que tienen una estructura comparable a la del polímero. Sin embargo, cuando surgen dificultades prácticas en la utilización de la CPG o si se espera por adelantado que la sustancia no cumpla algún criterio Mn reglamentario (y que exija confirmación) existen otros métodos, por ejemplo:
1. Utilización de las propiedades coligativas
|
1.1. |
Ebulloscopia/crioscopia: ponen en juego la medida de la subida del punto de ebullición (ebulloscopia) o del descenso del punto de congelación (crioscopia) de un disolvente, cuando se añade el polímero. El método se basa en el hecho de que el efecto del polímero disuelto sobre el punto de ebullición congelación del líquido depende del peso molecular del polímero (1) (2). Aplicabilidad, Mn < 20 000. |
|
1.2. |
Descenso de la presión de vapor: pone en juego la medida de la presión de vapor de un líquido de referencia determinado antes y después de la adición de cantidades conocidas de polímero (1) (2). Aplicabilidad, M < 20 000 (teóricamente, sin embargo, en la práctica, su valor es limitado). |
|
1.3 |
Osmometría de membrana: se basa en el principio de la osmosis, es decir, la tendencia natural de las moléculas de disolvente a atravesar una membrana semipermeable desde una solución diluida hacia una solución concentrada para llegar al equilibrio. En el ensayo, la solución diluida tiene una concentración cero, mientras que la solución concentrada contiene el polímero. El efecto del paso del disolvente a través de la membrana causa un diferencial de presión que depende de la concentración y del peso molecular del polímero (1) (3) (4). Aplicabilidad, Mn < 20 000 y 200 000. |
|
1.4 |
Osmometría en fase de vapor: compara la velocidad de evaporación de un aerosol de disolvente puro con al menos tres aerosoles que contiene el polímero a distintas concentraciones (l) (5) (6). Aplicabilidad, Mn < 20 000. |
2. Análisis de grupos terminales
Para utilizar este método, es necesario conocer al mismo tiempo la estructura global del polímero v la naturaleza de los grupos terminales de las cadenas (que debe ser distinguible del esqueleto principal por, por ejemplo, RMN o valoración/derivación). La determinación de la concentración molecular de los grupos terminales presentes en el polímero permite determinar un valor del peso molecular (7) (8)
Aplicabilidad, Mn hasta 50 000 (con fiabilidad decreciente).
3. referencias
|
(1) |
Billmeyer, F.W. Jr, (1984). Textbook of Polymer Science, 3rd Edn, John Viley, Nueva York. |
|
(2) |
Glover, CA., (1975). «Absolute Colligative Property Methods». Chapter 4 In: Polymer Molecular Weights, Part I, P.E. Slade, Jr. ed, Marcel Dekker, Nueva York. |
|
(3) |
ASTM D 3750-79, (1979). Standard Practicc for Determination of Number-Average Molecular Wcight of Polymers by Membrane Osnwmetry. American Society for Testing and Materials, Philadelphia, Pennsylvania. |
|
(4) |
Coll, H. (1989), «Membrane Osmometry. In: Determination of Molecular Weight, A.R. Cooper ed, I. W'iley and Sons. pp. 25-52». |
|
(5) |
ASTM 3592-77, (19"). Standard Recommended Practice for Determination of Molecular Weight by Vapour Pressure, American Society íor Testing and Materials, Philadelphia, Pennsylvania. |
|
(6) |
Morris, C.E.M., (1989), «Vapour Pressure Osmometry-. In: Determination of Molecular Weight, A.R. Cooper ed., John Wiley and Sons». |
|
(7) |
Schröder, E., Müller, G., y Arndt, K-F., (1989). Polymer Characterisation, Carl Hanser Verlag, Munich. |
|
(8) |
Garmon, R.G., (1975). «End-Group Determinations», Chapter 3 In: Polymer Molecular Weights, Part I, P.E. Slade, Jr. ed. Marcel Dekker, Nueva York. |
|
(9) |
Amiya, S, et al. (1990). Pure and Applied Chemistry, 62, 2139-2146. |
A.19. CONTENIDO DE SUSTANCIAS DE BAJO PESO MOLECULAR EN LOS POLÍMEROS
1. MÉTODO
Este método de cromatografía de permeación sobre el gel (CPG) es copia de las directrices TG 119 de la OCDE (1996). Los principios fundamentales y demás información técnica se dan en la referencia 1.
1.1. INTRODUCCIÓN
La gran diversidad de las propiedades de los polímeros impide describir un único método que fije precisamente las condiciones de separación y de evaluación que cubran todas las eventualidades y particularidades que se producen en la separación de los polímeros. En particular, es frecuente que los polímeros complejos no puedan someterse a la CPG. En este caso, puede determinarse el peso molecular diferentemente (véase el anexo). Deberán entonces indicarse todos los datos y la justificación del método utilizado.
El método descrito aquí se basa en la norma DIN 55672 (1). Se encontrará en esta norma DIN información precisa sobre la manera de llevar a cabo las pruebas y de evaluar los datos. Cuando sea indispensable introducir modificaciones de las condiciones experimentales, estos cambios deberán justificarse. Se podrán utilizar otras normas con las debidas referencias. El método descrito utiliza para la calibración muestras de poliestireno de polidispersidad conocida y puede ser necesario modificarlo para adaptarlo a algunos polímeros como, por ejemplo, los polímeros solubles en el agua o ramificados de cadena larga.
1.2. DEFINICIONES Y UNIDADES
Un peso molecular bajo se define arbitrariamente como un peso molecular por debajo de 1 000 dalton.
El peso molecular medio en número Mti y el peso molecular medio en peso Mw se determinan con las siguientes ecuaciones:
|
|
|
donde
|
Hi |
= |
nivel de la señal del detector a partir de la línea de base para el volumen de retención Vi |
|
Mi |
= |
peso molecular de la fracción polimérica de volumen de retención Vi y n es el número de puntos de medición. |
La amplitud de la distribución de los pesos moleculares, que caracteriza la dispersidad del sistema, viene dada por la relación Mw/Mn.
1.3. SUSTANCIAS DE REFERENCIA
Como la CPG es un método relativo, es necesario efectuar una calibración. Para eso, se utilizan normalmente patrones de poliestireno de distribución estrecha y estructura lineal cuyos pesos moleculares medios Mn y Mw se conocen, así como su distribución de pesos moleculares. La curva de calibración solo puede servir para determinar el peso molecular de la muestra desconocida si las condiciones de separación de la muestra y de los patrones se han seleccionado de manera idéntica.
Una relación determinada entre el peso molecular y el volumen de elución solo es válida en las condiciones específicas de una experiencia particular. Estas condiciones son, sobre todo, la temperatura, el disolvente (o la mezcla de disolventes), las condiciones de la cromatografía y la columna o sistema de columnas de separación,
Los pesos moleculares de la muestra, determinados de esta manera, son valores relativos y se describen como «pesos moleculares equivalentes de poliestireno». En otros términos, según las diferencias estructurales y químicas entre la muestra y los patrones, los pesos moleculares pueden más o menos desviarse de los valores absolutos. Si se recurre a otros patrones como, por ejemplo, de polietilenglícol, de polióxido de etileno, de polimetacrilato de metilo, de poliácido acrílico, se explicará esta elección.
1.4. PRINCIPIOS DEL MÉTODO DE ENSAYO
La distribución del peso molecular de la muestra y los pesos moleculares medios (Mn y Mw) pueden determinarse por CPG. La CPG es una cromatografía líquida particular en la cual la muestra se separa según los volúmenes hidrodinámicos de sus componentes (2).
La separación se efectúa mientras que la muestra pasa por una columna rellena de un material poroso, normalmente un gel orgánico. Las moléculas pequeñas consiguen penetrar en los poros, mientras que las gruesas quedan excluidas. El recorrido de las moléculas grandes es, pues, más corto y se eluyen antes. Las moléculas de tamaño medio penetran en algunos poros y se eluyen posteriormente. Las moléculas más pequeñas, con un radio hidrodinámico medio más pequeño que los poros del gel, pueden penetrar en todos ellos y se eluyen posteriormente. Las moléculas más pequeñas, con un radio hidrodinámico medio más pequeño que los poros del gel, pueden penetrar en todos ellos y se eluyen al final,
En una situación ideal, la separación depende completamente del tamaño de las distintas moléculas, pero en la práctica es difícil evitar la interferencia al menos de algunos efectos de absorción. La situación empeora en caso de relleno irregular de la columna o de volúmenes muertos (2).
Se procede a la detección basándose, por ejemplo, en el índice de refracción o en la absorción UV para llegar a una curva de distribución simple. Sin embargo, para precisar sobre la curva valores efectivos de los pesos moleculares, es necesario calibrar la columna haciendo pasar polímeros de peso molecular conocido y, a ser posible, de estructura globalmente idéntica, por ejemplo, diversos patrones de poliestireno. Resulta de manera característica una curva de Gauss, a veces distorsionada por una pequeña cola hacia el lado de los pesos moleculares bajos; el eje vertical indica la cantidad, en peso, de las especies de distintos pesos moleculares eluidas, y el eje horizontal el logaritmo del peso molecular.
El contenido en sustancias de bajo peso molecular se obtiene de esta curva. El cálculo solo puede ser preciso si las especies de peso molecular bajo responden del mismo modo, en función del peso, que el polímero en su conjunto.
1.5. CRITERIOS DE CALIDAD
La repetibilidad (desviación típica relativa: RSD) del volumen de elución deberá sobrepasar el 0,3 %. La repetibilidad requerida del análisis estará garantizada por corrección mediante patrón interno si se evalúa un cromatograma en función del tiempo y no corresponde al criterio previamente mencionado (1). Las polidispersidades dependen de los pesos moleculares de los patrones. En el caso de los patrones de poliestireno, ios valores característicos son.
|
Mp < 2 000 |
Mw/Mn < 1,20 |
|
2 000 ≤ Mp ≤ 106 |
Mw/Mn < 1,05 |
|
Mp > 106 |
Mw/Mn < 1,20 |
(Mp es el peso molecular del patrón en el máximo del pico)
1.6. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
1.6.1. Preparación de las soluciones patrón de poliestireno
Se disuelven los patrones de poliestireno mezclándolos cuidadosamente con el eluyente elegido. Se tendrán en cuenta las recomendaciones del fabricante para la preparación de las soluciones.
Las concentraciones de los patrones elegidos dependen de distintos factores como, por ejemplo, el volumen de inyección, la viscosidad de la solución y la sensibilidad del detector analítico. El volumen máximo de inyección debe adaptarse a la longitud de la columna, con el fin de evitar las sobrecargas. Generalmente, los volúmenes inyectados para las separaciones analíticas por CPG en una columna de 30 cm × 7,8 mm se sitúan entre 40 y 100 μl. Es posible utilizar volúmenes mayores, pero no deben sobrepasar 250 μl. La relación óptima entre el volumen de inyección y la concentración debe determinarse antes de la calibración efectiva de la columna.
1.6.2. Preparación de la solución de muestra
En principio, las mismas exigencias se aplican a la preparación de las soluciones de muestras. La muestra se disuelve en un disolvente conveniente como, por ejemplo, retrahidrofurano (THF), agitando con cuidado. No debe nunca disolverse en baño de ultrasonidos. En caso necesario, la solución de muestra se purifica con un filtro de membrana cuyo tamaño de poro se sitúa entre 0,2 y 2 μm.
Debe señalarse en el informe final la presencia de partículas no disueltas, puesto que puede tratarse de sustancias de elevado peso molecular. Se utilizará un método conveniente para determinar el porcentaje en peso de partículas no disueltas. Las disoluciones se utilizarán en el plazo de 24 horas.
1.6.3. Corrección del contenido en impurezas y aditivos
Es generalmente necesario proceder a una corrección del contenido de las sustancias de M < 1 000 para tener en cuenta la contribución de los componentes específicos no poliméricos presentes (por ejemplo, impurezas o aditivos), salvo si el contenido medido es ya < 1 %. Esto se hace mediante el análisis directo de la solución polimérica o del eluido de la CPG.
Cuando el eluido, después de su paso a través de la columna, está demasiado diluido para el análisis posterior, hay que concentrarlo. Puede ser necesario evaporarlo hasta estado seco para disolverlo de nuevo a continuación. La concentración del eluido debe hacerse en condiciones que garanticen que no sufre ningún cambio. El tratamiento del eluido después de la CPG depende del método analítico utilizado para las determinaciones cuantitativas.
1.6.4. Equipo
La CPG exige el siguiente equipo:
|
— |
depósito de disolvente, |
|
— |
desgasificador (en su caso), |
|
— |
bomba, |
|
— |
amortiguador de pulsaciones (en su caso), |
|
— |
sistema de inyección, |
|
— |
columnas de cromatografía, |
|
— |
detector, |
|
— |
caudalímetro (en su caso), |
|
— |
conjunto de registro-tratamiento de los datos, |
|
— |
receptáculo de residuos. |
Es necesario garantizar que el sistema de CPG es inerte respecto a los disolventes utilizados (por ejemplo, utilizando capilares de acero para el disolvente THF).
1.6.5. Sistema de inyección y de distribución del disolvente
Un volumen definido de la solución de muestra se pasa a la columna con ayuda de un muestreador automático o bien manualmente, en una zona estrictamente definida. La liberación o la depresión demasiado rápida del émbolo de la jeringuilla pueden causar modificaciones en la distribución de pesos moleculares observada. El sistema de distribución del disolvente deberá, en la medida de lo posible, estar libre de pulsaciones, y lo ideal es que incluya un amortiguador de pulsaciones. El caudal es del orden de 1 ml/mm.
1.6.6. Columna
Según la muestra, el polímero se caracterizará con ayuda de una columna simple o de varias columnas conectadas en secuencia. Está disponible en el comercio diverso material para columnas porosas, de propiedades definidas (por ejemplo, tamaño de poro, límites de exclusión). La elección del gel de separación o de la longitud de la columna depende tanto de las propiedades de la muestra (volúmenes hidrodinámicos, distribución de los pesos moleculares) como de las condiciones específicas de la separación, como disolvente, la temperatura y el caudal (1) (2) (3).
1.6.7. Platos teóricos
La columna (o la combinación de columnas) utilizada para la separación debe caracterizarse por el número de platos teóricos. En el caso de tomar el THF como disolvente de elución, es necesario cargar una solución de etilbenceno u otro soluto homopolar adecuado en una columna de longitud conocida. El número de platos teóricos viene dado por la siguiente ecuación:
|
|
o |
|
donde
|
N |
= |
número de platos teóricos |
|
Vc |
= |
volumen de elución en el máximo del pico |
|
W |
= |
anchura del pico en la línea de base |
|
W1/2 |
= |
anchura del pico a la mitad de la altura. |
1.6.8. Capacidad de separación
Además del número de platos teóricos, cantidad que determina la anchura de la banda, la capacidad de separación desempeña también un papel y está determinada por la pendiente de la curva de calibración. La capacidad de separación de una columna viene dada por la siguiente relación:

donde
|
Ve, Mx |
= |
volumen de elución de un poliestireno de peso molecular Mx |
|
Ve,(10.Mx) |
= |
volumen de elución de un poliestireno de peso molecular diez veces superior. |
La resolución del sistema se define generalmente del siguiente modo:
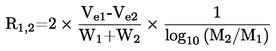
donde
|
Ve1 Ve2 |
= |
volúmenes de elución de los dos patrones de poliestireno en el máximo del pico |
|
W1 W2 |
= |
anchuras de los picos en la línea de base |
|
M1, M2, |
= |
pesos moleculares en el máximo del pico (deberán diferir en un factor de 10) |
El valor R del sistema de columna debería sobrepasar 1,7 (4).
1.6.9. Disolvente
Todos los disolventes deben ser de gran pureza (para el THF, se exigirá una pureza del 99,5 %). El depósito de disolvente (en caso necesario, en una atmósfera de gas inerte) debe ser suficientemente grande para la calibración de la columna y varios análisis de muestras. El disolvente debe desgasificarse antes de su transporte a la columna por la bomba.
1.6.10. Ajuste de la temperatura
La temperatura de los componentes internos esenciales (circuito de inyección, columnas, detector y conducciones) será constante y en coherencia con la elección del disolvente.
1.6.11. Detector
De detector tiene por objeto registrar cuantitativamente la concentración de muestra eluida de la columna. Para evitar el ensanchamiento inútil de los picos, el volumen de la célula deí detector debe ser lo más bajo posible. No debería exceder de 10 μl excepto en caso de detectores de dispersión de luz y de viscosidad. La detección se hace generalmente por refractometría diferencial. Sin embargo, si alguna propiedad de la muestra o del disolvente de elución lo impone, se puede recurrir a otros tipos de detectores, como por ejemplo, detectores de UV/visible, IR, viscosidad, etc.
2. RESULTADOS E INFORMES
2.1. DATOS
Hay que remitirse a la norma DIN (1) respecto al detalle de los criterios de evaluación así como para los imperativos relativos a la recogida y tratamiento de los datos.
De cada muestra deben realizarse dos experiencias independientes, que se analizaran separadamente. En todos los casos, es absolutamente necesario determinar también los datos procedentes de controles tratados en las mismas condiciones que la muestra.
Es necesario indicar explícitamente que los valores medidos son valores relativos equivalentes a los pesos moleculares del patrón utilizado.
Después de la determinación de los volúmenes de retención o del tiempo de retención (eventualmente corregidos con ayuda de un patrón interno), los valores log Mp (Mp es el máximo del pico del patrón de calibración) se representan gráficamente en función de una de estas cantidades. Son necesarios al menos dos puntos de calibración por década de pesos moleculares y cinco puntos de medida al menos para la curva entera, que debe cubrir el peso molecular estimado de la muestra. El peso molecular más bajo de la curva de calibración se define con n-hexilbenceno u otro soluto homopolar adecuado. La porción de la curva que corresponde a los pesos moleculares inferiores a 1 000 se determina y se corrige según sea necesario en función de las impurezas y aditivos. Las curvas de elución se evalúan generalmente por tratamiento informático de los datos. Si se procede a una digitalización manual, se puede consultar el documento ASTM D 3536-91 (3).
Si se retiene cualquier polímero insoluble en la columna, su peso molecular será probablemente superior al de la fracción soluble y. si no se tiene en cuenta, conducirá a sohrestimar el contenido en pesos moleculares bajos. El anexo propone principios de corrección del contenido en pesos moleculares bajos en función de los polímeros insolubles.
La curva de distribución debe darse en forma de cuadro o de gráfico (frecuencia diferencial o porcentajes de las sumas frente al log M). Para la representación gráfica, una década de pesos moleculares debería normalmente ocupar unos 4 cm de anchura y el máximo del pico debería encontrarse a unos 8 cm de altura. En el caso de curvas de distribución integral, la diferencia de ordenadas entre 0 y 100 % debería ser de cerca de 10 cm.
2.2. INFORME DEL ENSAYO
El informe del ensayo debe contener la siguiente información:
2.2.1. Sustancia de ensayo
|
— |
información disponible sobre la sustancia de ensayo (identidad, aditivos, impurezas), |
|
— |
descripción del tratamiento de la muestra, observaciones, problemas. |
2.2.2. Equipo
|
— |
Depósito de eluyente, gas inerte, desgasificación del eluyente, composición del eluyente, impurezas. |
|
— |
Bomba, amortiguador de pulsaciones, sistema de inyección. |
|
— |
Columnas de separación (fabricante, toda la información sobre las características de las columnas como tamaño del poro, naturaleza del material de separación, etc., número, longitud y orden de las columnas utilizadas). |
|
— |
Número de platos teóricos de la columna (o combinación), capacidad de separación (resolución del sistema). |
|
— |
Información sobre la simetría de los picos. |
|
— |
Temperatura de la columna, naturaleza del ajuste de la temperatura. |
|
— |
Detector (principio de medida, volumen de la célula). |
|
— |
Caudalímetro si se utiliza (fabricante, principio de medida). |
|
— |
Sistema de registro y tratamiento de los datos (material y programas informáticos). |
2.2.3. Calibración del sistema
|
— |
Descripción precisa del método empleado para construir la curva de calibración. |
|
— |
Información sobre los criterios de calidad de este método (por ejemplo, coeficiente de correlación, suma de los cuadrados de los errores, etc.). |
|
— |
Explicaciones sobre todas las extrapolaciones, hipótesis y aproximaciones hechas durante el procedimiento experimental y la evaluación y tratamiento de los datos. |
|
— |
Todas las medidas utilizadas para construir la curva de calibración deben precisarse en un cuadro que incluya la información siguiente de cada punto de calibración:
|
2.2.4. Información sobre el contenido en polímeros de bajo peso molecular
|
— |
Descripción del método utilizado para el análisis y manera de llevar las experiencias. |
|
— |
Información sobre el porcentaje de contenido en especies de bajo peso molecular (p/p) en relación con la muestra total. |
|
— |
Información sobre las impurezas, aditivos y otras especies no poliméricas en porcentaje en peso en relación con la muestra total. |
2.2.5. Evaluación
|
— |
Evaluación en función del tiempo: métodos utilizados para garantizar la reproducibilidad requerida (método de corrección, patrón interno, etc.). |
|
— |
Información sobre sí la evaluación se ha realizado basándose en el volumen de elución o en el tiempo de retención. |
|
— |
Información sobre los límites de la evaluación si no se analiza completamente un pico. |
|
— |
Descripción de los métodos de suavizado, si se utilizan. |
|
— |
Procedimientos de preparación y de tratamiento preliminar de la muestra. |
|
— |
Posible presencia de partículas no disueltas. |
|
— |
Volumen de inyección (μl) y concentración de inyección (mg/ml). |
|
— |
Observaciones sobre los efectos que llevan a divergencias en relación con el perfil ideal de la CPG. |
|
— |
Descripción detallada de todas las modificaciones de procedimientos de ensayo. |
|
— |
Precisiones sobre las gamas de errores. |
|
— |
Toda la información y observaciones pertinentes para la interpretación de los resultados. |
3. REFERENCIAS
|
(1) |
DIN 55672 (19.95) Gelpermeationschrornatographie (CPG) mit Tetrahydrofuran (THF) als Elutionsmittel, Teil 1. |
|
(2) |
Yau, W.W., Kirkland, J.J., y Bly, D.D. eds, (1979). Modern Size Exclusión Liquid Chromato- graphy, J. Wiley and sons. |
|
(3) |
ASTM D 3536-91, (1991). Standard Test Method for Molecular Weight Averages and Molecular Weight Distribution by Liquid Exclusion Cbromatography (Gel Permeation Chromato-grapby-GPC). American Society for Testing and Materials, Philadelphia, Pennsylvania. |
|
(4) |
ASTM D 5296-92, (1992). Standard Test Method for Molecular Weight Averages and Molecular Weight Distribution of Polystirene by High Performance Size-Exclusion Chromatography. American Society for Testing and Materials, Philadelphia, Pennsylvania. |
Anexo
Orientaciones para la corrección del contenido en bajos pesos moleculares en presencia de polímeros ínsolubles
Cuando está presente en una muestra un polímero insoluble, ocasiona una pérdida de masa durante el análisis por CPG. El polímero insoluble se retiene irreversiblemente en la columna o filtro de la muestra mientras que la porción soluble de la muestra pasa a través de la columna. Cuando puede estimarse o medirse el aumento del índice de refracción (dn/dc) del polímero, se puede estimar la pérdida de masa de la muestra en la columna. En este caso, se procede a una corrección mediante calibración externa con materiales patrón de concentraciones y dn/dc conocidas, para calibrar la respuesta del refractómetro. En el siguiente ejemplo, se utiliza un patrón de poli (metilmetacrilato) (pMMA).
En la calibración externa para el análisis de polímeros acrílicos, se analiza por CPG un patrón de pMMA de concentración conocida en tetrahidrofurano y los datos resultantes sirven para establecer la constante del refractómetro según la ecuación:
K = R/(C x V x dn/dc)
donde:
|
K |
= |
constante del refractómetro (en microvoltios; segundos/ml) |
|
R |
= |
respuesta del patrón de pMMA (en microvoltios; segundos) |
|
C |
= |
concentración del patrón de pMMA (en mg/ml) |
|
V |
= |
volumen de inyección (en ml) |
|
dn/dc |
= |
aumento del índice de refracción del pMMA en tetrahidrofurano (en ml/mg). |
El patrón de pMMA presenta los siguientes datos característicos.
|
R |
= |
2 937 891 |
|
C |
= |
1,07 mg/ml |
|
V |
= |
0,1 ml |
|
dn/dc |
= |
9 x 10-5 ml/mg. |
El valor K resultante 3,05 x 10", se utiliza entonces para calcular la respuesta teórica del detector si el 100 % del polímero inyectado eluye a través del detector.
A.20. COMPORTAMIENTO DE DISOLUCIÓN/EXTRACCIÓN
1. MÉTODO
El método descrito es copia de la versión revisada del documento TG 120 de la OCDE (1997). Se encontrará más información técnica en la referencia 1.
1.1. INTRODUCCIÓN
Para algunos polímeros como los polímeros emulsionados, puede ser necesario un trabajo preparatorio antes de que pueda aplicarse el método indicado. El método no es aplicable a los polímeros líquidos ni a los que reaccionan con el agua en las condiciones del ensayo.
Cuando el proceso es poco práctico o inaplicable, el comportamiento de disolución/extracción se puede estudiar por medio de otros métodos. Deberán entonces indicarse los detalles y la justificación del método empleado.
1.2 SUSTANCIAS DE REFERENCIA
Ninguna.
1.3. PRINCIPIO DEL MÉTODO DE ENSAYO
El comportamiento de disolución/extracción de los polímeros en medio acuoso se determina con ayuda del método de frasco (véase A.6, «Hidrosolubilidad, método de frasco») con las modificaciones siguientes.
1.4. CRITERIOS DE CALIDAD
Ninguno.
1.5. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
1.5.1. Equipo
Es necesario el equipo siguiente:
|
— |
material de trituración, por ejemplo, triturador que produzca partículas de tamaño conocido, |
|
— |
agitador, con posibilidad de ajuste de la temperatura, |
|
— |
sistema de filtro de membrana, |
|
— |
equipo analítico adecuado, |
|
— |
tamices normalizados, |
1.5.2. Preparación de la muestra
En primer lugar, una muestra representativa debe reducirse a partículas de tamaño incluido entre 0,125 y 0,25 mm con ayuda de los tamices convenientes. Puede ser necesario enfriar para garantizar la estabilidad de la muestra o para el proceso de trituración. Los materiales de carácter gomoso pueden triturarse a la temperatura del nitrógeno líquido (1).
Si no se puede llegar al tamaño de partículas requerido, se intentará reducir el tamaño de las partículas en la medida de lo posible y se indicará el resultado. Es necesario que en el informe se indique cómo se ha conservado antes del ensayo la muestra triturada.
1.53. Procedimiento
Se pesan tres muestras de 10 g de sustancia de ensayo en cada uno de los tres frascos equipados de tapones de vidrio; se añaden 1 000 ml de agua a cada uno. Si resulta difícil manipular una cantidad de 10 g de polímero, se utilizará la cantidad inmediata superior que pueda manipularse y se ajustará en consecuencia el volumen de agua.
Los frascos se tapan firmemente y se agitan después a 20 oC. El dispositivo utilizado para sacudir o agitar será capaz de funcionar a temperatura constante. Después de 24 h, se centrifuga o filtra el contenido de cada frasco y se determina por un método de análisis conveniente la concentración de polímero en la fase acuosa clara. Si no se dispone de ningún método de análisis adecuado para la fase acuosa, se podrá considerar la solubilidad/extractividad total a partir del peso seco del residuo filtrado o del precipitado centrifugado.
Es generalmente necesario diferenciar cuantitativamente las impurezas y aditivos por una parte, y las especies de bajo peso molecular por otra parte. En caso de determinación gravimétrica, es importante también realizar un ensayo en blanco, sin utilizar la sustancia de ensayo, para poder tener en cuenta los residuos derivados del procedimiento experimental.
El comportamiento de disolución/extracción de los polímeros en el agua a 37 oC a pH2 y pH9 puede determinarse como se ha descrito para el experimento a 20 oC. Los valores de pH pueden obtenerse por adición de amortiguadores convenientes o de ácidos o bases adecuados, como ácido clorhídrico, ácido acético, hidróxido de potasio o de sodio de grado analítico, o NH3.
Según el método de análisis empleado, se procederá a uno o dos ensayos. Cuando se disponga de métodos suficientemente específicos para un análisis directo en la fase acuosa del componente polimérico, bastará con un ensayo tal como se describe arriba. Sin embargo, cuando no se disponga de tales métodos y la determinación del comportamiento de disolución/extracción del polímero se límite a un análisis indirecto que determina solamente el contenido total en carbono orgánico (COT) del extracto acuoso, será necesario efectuar un segundo ensayo. Este se realizará también por triplicado, con muestras de polímeros diez veces más pequeñas y las mismas cantidades de agua que en el primer ensayo.
1.5.4. Análisis
1.5.4.1. Ensayo realizado con un único tamaño de muestra
Se dispondrá quizá de métodos de análisis directo de los componentes poliméricos en la fase acuosa. Si no, se podrá considerar también un análisis indirecto de los componentes poliméricos disueltos/extraídos determinando el contenido total de las partes solubles y aportando las correcciones necesarias que tendrán en cuenta los componentes no específicamente poliméricos.
Se puede proceder al análisis del conjunto de las especies poliméricas de la fase acuosa:
con ayuda de un método suficientemente sensible como, por ejemplo:
|
— |
COT, con digestión por persulfato o bicromato para liberar CO2, seguida de una estimación por IR o análisis químico, |
|
— |
espectrometría de absorción atómica (AAS) o su equivalente de emisión de plasma acoplado por inducción (ICP) para polímeros que contienen silicio o metales, |
|
— |
absorción UV o espectrofluorometría para los polímeros arílicos, |
|
— |
espectrometría de masas con cromatografía líquida (LC-MS) para las muestras de bajo peso molecular, |
o por evaporación al vacío hasta estado seco del extracto acuoso y análisis espectroscopio (IR, UV, etc.} o análisis AAS/ICP del residuo.
Si el análisis de la fase acuosa como tal no es realizable, el extracto acuoso se someterá a extracción con ayuda de un disolvente orgánico inmiscible con el agua, por ejemplo, un hidrocarburo clorado. El disolvente se evapora a continuación y el residuo se analiza como se índica arriba para determinar su contenido en el polímero correspondiente. Todos los componentes de este residuo identificados como impurezas o aditivos deben restarse para determinar el grado de disolución/extracción del propio polímero.
Cuando estén presentes cantidades relativamente importantes de estos materiales, puede ser necesario someter los residuos a un análisis por CLAR o CG, por ejemplo, para diferenciar las impurezas presentes de las especies monoméricas y derivadas de los monómeros, de modo que pueda determinarse el contenido auténtico de estas últimas.
En algunos casos, pueden bastar la simple evaporación del disolvente orgánico hasta el estado seco y el peso del residuo seco.
1.5.4.2. Ensayo realizado con dos tamaños de muestras diferentes
El COT se analizará en todos los extractos acuosos.
Se procederá a una determinación por gravimetría de la parte no disuelta/no extraída de la muestra. Si después de centrifugación o filtrado del contenido de cada frasco, sigue habiendo residuos poliméricos pegados a las paredes de este, se enjuagará con el filtrado hasta que el frasco quede libre de todos los residuos visibles. Luego se filtrará o se centrifugará de nuevo el filtrado. Los residuos que permanezcan sobre el filtro o en el tubo de centrifugación se secan a 40 oC al vacío y se pesan. La operación de secado se prolonga hasta llegar a un peso constante.
2. RESULTADOS
2.1. ENSAYO REALIZADO CON UN ÚNICO TAMAÑO DE MUESTRA
Se darán los resultados individuales de cada uno de los tres frascos y los valores medios, expresados en unidades de masa por volumen de solución (normalmente en mg/1) o de masa por masa de muestra de polímero (normalmente en mg/g). Además, se indicará también la pérdida de peso de la muestra (calculada como el peso de soluto dividido por el peso de la muestra inicial). Se calculará la desviación típica relativa (RSD). Se darán cifras individuales de la sustancia total (polímero + aditivos esenciales, etc.) y del polímero solo (es decir, después de sustracción de la participación de estos aditivos).
2.2. ENSAYO REALIZADO CON DOS TAMAÑOS DIFERENTES DE MUESTRAS
Se darán los valores de COT individuales de los extractos acuosos de las dos experiencias por triplicado y el valor medio de cada experiencia, en unidades de masa por volumen de solución (normalmente en mg C/l), así como en unidades de masa por peso de la muestra inicial (normalmente en mg C/g).
Sí no hay diferencia entre los resultados con elevada proporción muestra/agua y con baja proporción muestra/agua, se podrá suponer que todos los componentes, susceptibles de extraerse se han extraído realmente. En este caso, el análisis directo no sería necesario en principio.
Se darán los pesos individuales de los residuos y se expresarán en porcentaje de los pesos inicíales de las muestras. Las medias se calcularán para cada experiencia. Las diferencias entre 100 y los porcentajes encontrados representan los porcentajes de materiales solubles y extraíbles en la muestra original.
3. INFORME
3.1 INFORME DEL ENSAYO
El informe del ensayo debe contener la siguiente información:
3.1.1 Sustancia de ensayo
|
— |
Información disponible sobre la sustancia de ensayo (identidad, aditivos, impurezas, contenido en especies de bajo peso molecular). |
3.1.2. Condiciones experimentales
|
— |
Descripción de los procedimientos utilizados y de las condiciones experimentales. |
|
— |
Descripción de ios métodos de análisis y de detección. |
3.1.3 Resultados
|
— |
Resultados de solubilidad/extractividad en mg/1; valores individuales y medios para los ensayos de extracción en las distintas soluciones, desglose del contenido en polímeros e impurezas, aditivos, etc. |
|
— |
Resultados de solubilidad/extractividad en mg/g de polímero. |
|
— |
Valores de COT de los extractos acuosos, peso del soluto y porcentajes calculados, si se miden. |
|
— |
pH de cada muestra. |
|
— |
Información sobre los valores en blanco. |
|
— |
En caso necesario, referencias a la inestabilidad química de la sustancia de ensayo, tanto durante el procedimiento de ensayo como durante los análisis. |
|
— |
Toda la información importante para la interpretación de los resultados. |
4. REFERENCIAS
|
(1) |
DIN 53733 (1976) Zerkleinerung von Kunststofferzeugnissen für Prüfzwecke. |
A.21. PROPIEDADES COMBURENTES (LÍQUIDOS)
1. MÉTODO
1.1 INTRODUCCIÓN
El presente método se ha diseñado para medir el potencial de una sustancia líquida de incrementar la velocidad o la intensidad de combustión de una sustancia combustible, o de provocar la inflamabilidad espontánea cuando se mezcla homogéneamente con una sustancia combustible. Se funda en el ensayo de la ONU de líquidos comburentes (1) y ambos son equivalentes. No obstante, dado que el método A.21 se ha diseñado ante todo para cumplir los requisitos establecidos en la Directiva 67/548/CEE, es suficiente efectuar la comparación con una sola sustancia de referencia. Si está previsto emplear los resultados del ensayo para otros fines (9), puede ser necesario realizar el ensayo y la comparación con varias sustancias de referencia.
Si el análisis de la fórmula estructural excluye toda duda razonable de que la sustancia pueda reaccionar exotérmicamente con un material combustible, no será preciso realizar el ensayo.
Antes de llevar a cabo el ensayo, conviene disponer de información preliminar sobre las posibles propiedades explosivas de la sustancia.
Este ensayo no puede realizarse con sólidos, gases, sustancias explosivas o altamente inflamables ni peróxidos orgánicos.
Si ya se dispone de los resultados del ensayo de la ONU de líquidos comburentes (1) para la sustancia en cuestión, no será necesario realizar el presente ensayo,
1.2 DEFINICIONES Y UNIDADES
Tiempo medio de aumento de la presión: media de los tiempos medidos para que la presión de la mezcla de ensayo aumente de 690 kPa a 2 070 kPa (presión manométrica).
1.3 SUSTANCIA DE REFERENCIA
La sustancia de referencia ha de ser ácido nítrico acuoso (pureza de grado analítico) al 65 % (p/p) (10).
Si el experimentador prevé que los resultados del ensayo pueden emplearse para otros fines, cabe realizar el ensayo con otras sustancias de referencia (11).
1.4 PRINCIPIO DEL MÉTODO DE ENSAYO
Se mezcla el líquido que va a estudiarse en proporción de 1/1, en masa, con celulosa fibrosa y se introduce en un recipiente a presión. Si se produce una combustión espontánea durante la mezcla o la introducción, no es necesario proseguir el ensayo.
En caso contrario, se lleva a cabo el ensayo completo. Se calienta]a mezcla en el recipiente a presión y se mide el tiempo medio que tarda la presión en aumentar de 690 kPa a 2 070 kPa (presión manométrica). Se compara el resultado con el tiempo medio de aumento de la presión de una mezcla 1/1 de la sustancia o sustancias de referencia y celulosa.
1.5 CRITERIOS DE CALIDAD
En una serie de cinco pruebas con la misma sustancia ningún resultado debe diferir en más del 30 % de la media aritmética. En caso contrario, es preciso descartar los resultados que difieran en más del 30 % de la media aritmética, mejorar la técnica de mezcla e introducción y repetir el ensayo.
1.6 DESCRIPCIÓN DEL MÉTODO
1.6.1 Preparación
1.6.1.1 Sustancia combustible,
Se emplea como material combustible celulosa fibrosa deshidratada, cuyas fibras tengan Una longitud comprendida entre 50 y 250 μm y un diámetro medio de 25 μm (12). La celulosa se deshidrata hasta peso constante en una capa de espesor máximo de 25 mm a 105 oC durante 4 horas y se conserva en un desecador, con desecante, hasta que sé enfríe y vaya a utilizarse. El contenido acuoso de la celulosa deshidratada ha de ser inferior al 0,5 % de la masa deshidratada (13). Si es preciso, se prolonga el tiempo de deshidratación (14). Debe emplearse el mismo lote de celulosa para todo el ensayo.
1.6.1.2 Equipo
1.6.1.2.1 Recipiente a presión
Se necesita un recipiente a presión, formado por un cilindro de acero de 89 mm de largo y 60 mm de diámetro exterior (véase la figura 1). Se practican dos rebajes planos diametralmente opuestos (que reducen esa sección transversal del recipiente a 50 mm) para inmovilizarlo mientras se ajustan la pieza de encendido y la de sujeción del diafragma de seguridad. El recipiente ha de tener una luz de 20 mm, que se ensanchará a ambos extremos del cilindro rebajando las paredes internas en una longitud de 19 mm y se fileteará de manera que pueda alojar una rosca del tipo British Standard Pipe (BSP) de 1" o su equivalente métrico. En la superficie convexa del recipiente, a 35 mm de un extremo y a 90° de los rebajes planos, se enrosca un tubo lateral que servirá para medir la presión. El extremo del tubo estará provisto de una rosca de 1/2" BSP (o su equivalente métrico), que se ajustará en una cavidad de 12 mm de profundidad practicada en la pared del recipiente y fileteada debidamente. Si es necesario, se sella con un material inerte para evitar la fuga de gas. El tubo lateral sobresale 55 mm del recipiente a presión y tiene una luz de 6 mm. La cara interna del extremo libre del tubo se rebaja y filetea para que pueda alojar un transductor de presión de tipo diafragmático. Puede emplearse cualquier medidor de presión que no sufra alteraciones debido a los gases calientes ni a los productos de descomposición y que pueda detectar aumentos de presión de 690 a 2 070 kPa en menos de 5 ms.
Se cierra el extremo del recipiente más alejado del tubo lateral con una pieza de encendido provista de dos electrodos, uno de ellos aislado del cuerpo de la pieza y el otro unido a masa con este. El otro extremo del recipiente se obtura con un diafragma de seguridad (presión aproximada de rotura: 2 200 kPa), que se mantiene en su lugar mediante una pieza de sujeción que presenta una luz de 20 mm. Si es necesario, se sella la pieza de encendido con material inerte para evitar la fuga de gas. Para mantener el conjunto en la posición correcta durante la utilización, se emplea un soporte de acero suave (figura 2), que suele estar formado por una base de 235 mm × 184 mm × 6 mm y un tubo hueco de sección cuadrada (70 mm × 70 mm × 4 mm) de 185 mm de longitud.
En uno de los extremos del tubo cuadrado se seccionan dos caras opuestas, de manera que se obtenga un tubo de 86 mm de largo prolongado por dos lados planos a modo de patas, que se cortan para que, una vez soldado a la base, el tubo forme un ángulo de 60o con la horizontal. En el extremo superior de una de las caras del tubo se practica una muesca de 22 mm de ancho y 46 mm de profundidad, de manera que, al colocar el recipiente a presión en el soporte con la pieza de encendido hacia abajo, el tubo lateral para medir la presión quede alojado en ella. En la cara interna de la pared del tubo orientada hacia abajo se suelda una pieza de acero de 30 mm de ancho y 6 mm de grueso para que sirva de separador. En la pared opuesta se hacen dos orificios y se enroscan dos tornillos de palomilla de 7 mm para fijar el recipiente a presión. Para sujetar el recipiente desde abajo, se sueldan dos rebordes de acero de 12 mm de ancho y 6 mm de grueso a los laterales de la parte inferior de la sección cuadrada.
1.6.1.2.2 Dispositivo de ignición
El dispositivo de ignición está integrado por un hilo de Ni/Cr de 25 cm de largo y 0,6 mm de sección, con una resistencia de 3,85 ohm/m. El hilo se enrolla en forma de bobina con una varilla de 5 mm de diámetro y se une a los electrodos de la pieza de encendido. La bobina ha de ajustarse a uno de los esquemas de la figura 3. La distancia entre la cara superior de la pieza de encendido y. la cara inferior de la bobina de ignición debe ser de 20 mm. Si los electrodos no son regulables, los segmentos de hilo que se encuentran entre la bobina y la cara superior de la pieza de encendido deben aislarse con un revestimiento cerámico. El hilo se calienta mediante una alimentación eléctrica-estable que proporcione una intensidad de 10 A como mínimo.
1.6.2 Realización del ensayo (15)
Se coloca en el soporte el recipiente montado con el transductor de presión y el dispositivo de ignición, pero sin el diafragma de seguridad, con la pieza de encendido hacia abajo. Se introducen 2,5 g del líquido que vaya a someterse a ensayo y 2,5 g de celulosa deshidratada en una copa de vidrio y se mezclan con un agitador de cristal (16). Por motivos de seguridad, la persona que realice la mezcla debe interponer una pantalla protectora. Si se produce una combustión espontánea' durante la mezcla o la introducción, no es necesario proseguir el ensayo. Se introduce la mezcla en pequeñas cantidades en el recipiente a presión dando ligeros golpecitos y se comprueba que la mezcla rodea bien y está en contacto con la bobina de ignición sin dejar hueco. La bobina no debe deformarse durante este proceso, pues ello podría falsear los resultados (17). Se coloca en su lugar el diafragma de seguridad y se enrosca al máximo la pieza de sujeción. Se coloca el recipiente cargado en el soporte con el disco de seguridad hacia arriba y se introduce el conjunto en una campana extractora blindada o una cámara de fuego. Se conectan los bornes externos de la pieza de encendido a una alimentación eléctrica y se aplica una corriente de 10 A. No deben transcurrir más de 10 minutos entre el inicio de la preparación de la mezcla y el momento en que se aplica la corriente.
La señal emitida por el transductor de presión se recoge en un sistema que permita el registro continuo de la curva presión/tiempo y su análisis (por ejemplo, un registrador de señales transitorias acoplado a un registrador sobre cinta de papel). Se calienta la mezcla hasta que se rompa el diafragma de seguridad o durante un tiempo mínimo de 60 s. Si no se ha roto el diafragma, debe esperarse a que la mezcla se enfríe antes de abrir el recipiente con prudencia y tomando las debidas precauciones por si estuviera aún a presión. Se realizan cinco ensayos con la mezcla y con la sustancia o sustancias de referencia. Se registra el tiempo que tarda la presión en aumentar de 690 kPa a 2 070 kPa (presión manométrica). Se calcula la media de los tiempos registrados.
En algunos casos, las sustancias pueden generar un aumento de presión (demasiado elevado o demasiado escaso) debido a reacciones químicas que no son características de las propiedades comburentes de dichas sustancias. Puede entonces ser necesario repetir el ensayo sustituyendo la celulosa por un material inerte, como la diatomita (kieselguhr), para cerciorarse de que no se trata de un efecto no comburente.
2. RESULTADOS
Tiempos de aumento de la presión con la sustancia de ensayo y con la sustancia o sustancias de referencia. Tiempos de aumento de la presión con una sustancia inerte, si procede.
2.1. TRATAMIENTO DE LOS RESULTADOS
Se calcula el tiempo medio de aumento de la presión con la sustancia de ensayo y con la sustancia o sustancias de referencia.
Se calcula el tiempo medio de aumento de la presión con una sustancia inerte, si procede.
El cuadro 1 recoge algunos ejemplos de resultados.
Cuadro 1
Ejemplos de resultados (18)
|
Sustancia (19) |
Tiempo medio de aumento de la presión con una mezcla de 1/1 de celulosa (ms) |
|
Ácido nítrico, 65 % |
4 767 (20) |
|
Ácido perclórico, 50 % |
121 (20) |
|
Ácido perclórico, 55 % |
59 |
|
Bicromato de amonio, en solución acuosa saturada |
20 800 |
|
Clorato de sodio, en solución acuosa al 40 % |
2 555 (20) |
|
Nitrato de calcio, en solución acuosa saturada |
6 700 |
|
Nitrato férrico, en solución acuosa saturada |
4 133 |
|
Nitrato de níquel, en solución acuosa saturada |
6 250 |
|
Nitrato de plata, en solución acuosa saturada |
(21) |
|
Nitrato de potasio, en solución acuosa al 30 % |
26 690 |
|
Nitrato de sodio, en solución acuosa al 45 % |
4 133 |
|
Perclorato de litio, en solución acuosa saturada |
1 686 |
|
Perclorato de magnesio, en solución acuosa saturada |
777 |
|
Sustancia inerte |
|
|
Agua/celulosa |
(21) |
3. INFORME
3.1 INFORME DEL ENSAYO
En el informe del ensayo debe figurar la información siguiente:
|
— |
identidad, composición, pureza, etc. de la sustancia de ensayo, |
|
— |
concentración de la sustancia de ensayo, |
|
— |
procedimiento empleado para deshidratar la celulosa, |
|
— |
contenido acuoso de la celulosa empleada, |
|
— |
resultados de las mediciones, |
|
— |
resultados de los ensayos con la sustancia inerte, si procede, |
|
— |
tiempos medios calculados de aumento de la presión, |
|
— |
cualquier desviación respecto al presente método y justificación correspondiente, |
|
— |
cualquier información u observación complementaria importante para interpretar los resultados; |
3.2 INTERPRETACIÓN DE LOS RESULTADOS (22)
La evaluación de los resultados del ensayo se fundará en los siguientes criterios:
|
a) |
la mezcla de sustancia de ensayo y celulosa se inflama espontáneamente o no; |
|
b) |
la comparación del tiempo medio de aumento de la presión de 690 kPa a 2 070 kPa con el tiempo medio correspondiente a la sustancia o sustancias de referencia. |
Se considerará que una sustancia líquida es comburente si:
|
a) |
una mezcla de 1/1 en masa de dicha sustancia y celulosa se inflama espontáneamente, o |
|
b) |
el tiempo medio de aumento de la presión de una mezcla de 1/1 en masa de dicha sustancia y celulosa es inferior ó igual al tiempo medio de una mezcla de 1/1 en masa de ácido nítrico acuoso al 65 % (p/p) y celulosa. |
Con el fin de evitar falsos positivos, a la hora de interpretar los resultados deben tomarse en consideración los resultados del ensayo con la sustancia y un material inerte.
4. BIBLIOGRAFÍA
|
(1) |
Recommendations on the Transport of Dangerous Goods, Manual of Tests and Criteria. 2- edición revisada. Publicación de la ONU no ST/SG/AC.10/11/Rev. 3, 1999, p. 342. Test O.2: Test for oxidizing liquids. |
Figura 1
Recipiente a presión

Figura 2
Soporte

Figura 3
Dispositivo de ignición
Nota: cualquiera de estas dos configuraciones puede ser utilizada.
(1) Valor dependiente del tipo de instrumento y del grado de pureza de las sustancias.
(2) Valor dependiente del tipo de instrumento y del grado de pureza de las sustancias.
(3) Valor dependiente del tipo de instrumento y del grado depureza de las sustancias.
(4) Valor dependiente del tipo de instrumento y del grado de pureza de las sustancias.
(5) Esta precisión solo es válida para aparatos sencillos, como el descrito en la norma ASTM D 1120-72; es posible mejorarla utilizando ebullómetros más perfeccionados.
(6) Válido solamente para sustancias puras. El uso en otras circunstancias debe justificarse.
(7) Según el grado de pureza.
(8) Estos métodos pueden utilizarse también en el intervalo de 1 a 10 Pa siempre que se tenga cuidado.
(9) Por ejemplo, en el marco de las normas de transporte de la ONU.
(10) Debe valorarse el ácido antes del ensayo para confirmar su concentración.
(11) Por ejemplo, en la referencia bibliográfica 1 se emplean ácido perclórico al 50 % (p/p) y clorato de sodio al 40 % (p/p).
(12) Por ejemplo, celulosa en polvo para columna de cromatografía Whatman CF 11, no de catálogo 4021 050.
(13) Confirmado, por ejemplo, mediante valoración de Karl-Fisher.
(14) También puede obtenerse ese contenido acuoso (por ejemplo) calentando la celulosa a 105 oC al vacío durante 24 h.
(15) Las mezclas de sustancias comburentes y celulosa deben tratarse como potencialmente explosivas y manipularse con las debidas precauciones.
(16) En la práctica, puede prepararse una mezcla 1/1 de líquido problema y celulosa en cantidad superior a la necesaria para el ensayo, tomar 5 ±0,1 g e introducirlos en el recipiente a presión. La mezcla debe prepararse justo antes de cada ensayo.
(17) Debe evitarse sobre todo que entren en contacto las circunvoluciones adyacentes de la bobina.
(18) Véase la referencia bibliográfica (1) para la clasificación según el régimen de transporte de la ONU.
(19) Las soluciones saturadas deben prepararse a 20 oC.
(20) Valor medio de ensayos interlaboratorios comparados.
(21) No se alcanza la presión máxima de 2 070 kPa.
(22) Véase la referencia bibliográfica 1 para la interpretación de los resultados conforme a las normas de transporte de la ONU en caso de que se utilicen varias sustancias de referencia.
---
ANEXO
PARTE B: MÉTODOS PARA LA DETERMINACIÓN DE LA TOXICIDAD Y OTROS EFECTOS SOBRE LA SALUD
ÍNDICE
INTRODUCCIÓN GENERAL DE LA PARTE B
|
B.1 bis. |
TOXICIDAD ORAL AGUDA. MÉTODO DE DOSIS FIJAS |
|
B.1 ter. |
TOXICIDAD ORAL AGUDA. MÉTODO DE LAS CLASES DE TOXICIDAD AGUDA |
|
B.2. |
TOXICIDAD AGUDA POR INHALACIÓN |
|
B.3. |
TOXICIDAD AGUDA POR VÍA CUTÁNEA |
|
B.4. |
TOXICIDAD AGUDA: IRRITACIÓN/CORROSIÓN CUTÁNEA |
|
B.5. |
TOXICIDAD AGUDA: IRRITACIÓN/CORROSIÓN OCULAR |
|
B.6. |
SENSIBILIZACIÓN DE LA PIEL |
|
B.7. |
TOXICIDAD POR ADMINISTRACIÓN CONTINUADA (28 DÍAS) POR VÍA ORAL |
|
B.8. |
TOXICIDAD POR ADMINISTRACIÓN CONTINUADA (28 DÍAS) POR INHALACIÓN |
|
B.9. |
TOXICIDAD POR ADMINISTRACIÓN CONTINUADA (28 DÍAS) VÍA CUTÁNEA |
|
B.10. |
MUTAGENICIDAD — ENSAYO DE ABERRACIONES CROMOSÓMICAS IN VITRO EN MAMÍFEROS. |
|
B.11. |
MUTAGENICIDAD — ENSAYO DE ABERRACIONES CROMOSÓMICAS IN VIVO EN MÉDULA ÓSEA DE MAMÍFEROS |
|
B.12. |
MUTAGENICIDAD — ENSAYO DE MICRONÚCLEOS EN ERITROCITOS DE MAMÍFERO IN VIVO |
|
B.13/14. |
MUTAGENICIDAD — ENSAYO DE MUTACIÓN INVERSA EN BACTERIAS |
|
B.15. |
ENSAYOS DE MUTAGÉNESIS Y DETECCIÓN DE CARCINOGÉNESIS -MUTACIÓN GÉNICA — SACCHAROMYCES CEREVISIAE |
|
B.16. |
RECOMBINACIÓN MITÓTICA — SACCHAROMYCES CEREVISIAE |
|
B.17. |
MUTAGENICIDAD — ENSAYO DE MUTACIÓN GÉNICA DE CÉLULAS DE MAMÍFERO IN VITRO |
|
B.18. |
LESIÓN Y REPARACIÓN DE DNA — SÍNTESIS DE DNA NO PROGRAMADA — CÉLULAS DE MAMÍFEROS IN VITRO |
|
B.19. |
ENSAYO IN VITRO DE INTERCAMBIO DE CROMÁTIDAS HERMANAS |
|
B.20. |
ENSAYO DE LETALIDAD RECESIVA LIGADA AL SEXO EN DROSOPHILA MELANOGASTER |
|
B.21. |
ENSAYO DE TRANSFORMACIÓN DE CÉLULAS DE MAMÍFERO IN VITRO |
|
B.22. |
ENSAYO DE LETALIDAD DOMINANTE EN ROEDORES |
|
B.23. |
ENSAYO DE ABERRACIONES CROMOSÓMICAS EN ESPERMATOGONIAS DE MAMÍFERO |
|
B.24. |
ENSAYO DE LA MANCHA EN EL RATÓN |
|
B.25. |
TRANSLOCACIÓN HEREDITARIA EN EL RATÓN |
|
B.26. |
ENSAYO DE TOXICIDAD ORAL SUBCRÓNICA — TOXICIDAD ORAL POR ADMINISTRACIÓN CONTINUADA (90 DÍAS) EN ROEDORES |
|
B.27. |
ENSAYO DE TOXICIDAD ORAL SUBCRÓNICA — TOXICIDAD ORAL POR ADMINISTRACIÓN CONTINUADA (90 DÍAS) EN NO ROEDORES |
|
B.28. |
TOXICIDAD DÉRMICA SUBCRÓNICA — ENSAYO DE 90 DÍAS EN ROEDORES |
|
B.29. |
TOXICIDAD SUBCRÓNICA POR INHALACIÓN — ENSAYO DE 90 DÍAS EN ROEDORES |
|
B.30. |
ENSAYO DE TOXICIDAD CRÓNICA |
|
B.31. |
ESTUDIO DE TOXICIDAD PARA EL DESARROLLO PRENATAL |
|
B.32. |
ENSAYO DE CARCINOGÉNESIS |
|
B.33. |
ENSAYO COMBINADO DE TOXICIDAD CRÓNICA Y CARCINOGÉNESIS |
|
B.34. |
ENSAYO DE REPRODUCCIÓN EN UNA GENERACIÓN |
|
B.35. |
ESTUDIO DE TOXICIDAD PARA LA REPRODUCCIÓN EN DOS GENERACIONES |
|
B.36. |
TOXICOCINÉTICA |
|
B.37. |
NEUROTOXICIDAD RETARDADA DE SUSTANCIAS ORGANOFOSFORADAS POR ADMINISTRACIÓN ÚNICA |
|
B.38. |
NEUROTOXICIDAD RETARDADA DE SUSTANCIAS ORGANOFOSFORADAS. ESTUDIO POR ADMINISTRACIÓN CONTINUADA DE 28 DÍAS |
|
B.39. |
ENSAYO DE SÍNTESIS DE ADN NO PROGRAMADA (UDS) EN HEPATOCITOS DE MAMÍFERO IN VIVO |
|
B.40. |
CORROSIÓN CUTÁNEA IN VITRO: ENSAYO DE RESISTENCIA ELÉCTRICA TRANSCUTÁNEA (RET) |
|
B.40 BIS. |
CORROSIÓN CUTÁNEA IN VITRO: ENSAYO CON MODELO DE PIEL HUMANA |
|
B.41. |
ENSAYO DE FOTOTOXICIDAD IN VITRO 3T3 ARN (ABSORCIÓN DE ROJO NEUTRO) |
|
B.42. |
SENSIBILIZACIÓN CUTÁNEA: PRUEBA CON GANGLIOS LINFÁTICOS LOCALES |
|
B.43. |
ESTUDIO DE NEUROTOXICIDAD EN ROEDORES |
|
B.44. |
ABSORCIÓN CUTÁNEA: MÉTODO IN VIVO |
|
B.45. |
ABSORCIÓN CUTÁNEA: MÉTODO IN VITRO |
INTRODUCCIÓN GENERAL DE LA PARTE B
A. CARACTERIZACIÓN DE LA SUSTANCIA DE ENSAYO
Antes de iniciar cualquier estudio de toxicidad debe conocerse la composición de la sustancia de ensayo, con inclusión de las impurezas principales, y sus propiedades fisicoquímicas pertinentes, incluida la estabilidad.
Las propiedades fisicoquímicas de la sustancia de ensayo proporcionan datos importantes para la selección de la vía de administración, el diseño de cada estudio particular y el manejo y almacenamiento de la sustancia.
Antes de iniciarse el estudio debe, desarrollarse un método analítico para la determinación cualitativa y cuantitativa de la sustancia de ensayo (con inclusión de sus principales impurezas, siempre que sea posible) en el medio de administración y en el material biológico.
Debe incluirse en el informe del ensayo toda la información a la identificación, propiedades fisicoquímicas, pureza y comportamiento de la sustancia estudiada,
B. CUIDADOS DE LOS ANIMALES
En los estudios de toxicidad es fundamental controlar de forma estricta las condiciones ambientales y aplicar técnicas adecuadas de cuidado de los animales.
i) Condiciones de alojamiento
Las condiciones ambientales de los ¡ocales o recintos destinados a los animales de experimentación deben ser adecuadas para la especie que se utilice. Para ratas, ratones y cobayas, la temperatura del local debe ser de 22 ± 3 oC, con una humedad relativa del 30 al 70 %; para los conejos, la temperatura debe ser de 20 ± 3 oC, con una humedad relativa del 30 al 70 %.
Algunas técnicas experimentales son particularmente sensibles a ¡os efectos de la temperatura; en tales casos, en la descripción del método de ensayo se incluyen indicaciones detalladas sobre las condiciones apropiadas. En todas las investigaciones sobre efectos tóxicos deben medirse y registrarse la temperatura y la humedad, y estos parámetros deben consignarse en el informe final del estudio.
La iluminación debe ser artificial, con una alternancia de 12 horas de luz y 12 horas de oscuridad, Los datos relativos al programa de alumbrado deberán registrarse y consignarse en el informe final del estudio.
Salvo que el método contenga alguna indicación en contra, los animales se alojarán individualmente, o estarán en jaulas de pequeños grupos del mismo sexo, En caso de que se utilicen jaulas colectivas, no habrá más de 5 animales en cada jaula.
En los informes sobre los experimentos con animales, es importante indicar el tipo de jaula utilizada, así como el número de animales alojados en cada una, tanto durante la exposición a la sustancia química como durante el período posterior de observación.
ii) Condiciones de alimentación
La dieta debe cubrir todas ¡as necesidades nutricionales de la especie sometida a experimentación. Cuando se incorporen sustancias de ensayo a la dieta de los animales, el valor nutricional puede verse reducido por interacciones entre la sustancias y algún componente de la dieta. A la hora de interpretar los resultados de los ensayos debe estudiarse la posibilidad de que se dé una interacción de este tipo. Pueden utilizarse dietas convencionales de laboratorio junto con un aporte ilimitado de agua potable. La elección de la dieta puede verse influida por la necesidad de garantizar una mezcla conveniente de la sustancia estudiada si se administra por este método.
Ningún contaminante de la dieta con influencia conocida sobre la toxicidad podrá estar presente en concentraciones que puedan interferir.
C. ENSAYOS ALTERNATIVOS
Constituye un objetivo científico de la Unión Europea la elaboración y validación de técnicas alternativas que puedan proporcionar el mismo nivel de información que los actuales ensayos con animales, pero utilizando menos animales, provocando menos sufrimiento o evitando completamente el uso de animales.
Tales métodos, según vayan existiendo, deben tenerse en cuenta siempre que sea posible para la caracterización de riesgos y la correspondiente clasificación y etiquetado en función de los riesgos intrínsecos.
D. EVALUACIÓN E INTERPRETACIÓN
A la hora de evaluar e interpretar los ensayos, deben tenerse en cuenta las limitaciones de la extrapolación directa al hombre de los resultados de los estudios realizados in vitro o con animales y, en consecuencia, deben utilizarse pruebas de la aparición de efectos adversos en personas, siempre que sea posible, a fin de confirmar los resultados de los ensayos.
E. REFERENCIAS
La mayoría de estos métodos se han elaborado en el marco del programa de la OCDE de líneas directrices de ensayo, y deben realizarse de conformidad con los principios de buenas prácticas de laboratorio, a fin de garantizar la «aceptación mutua de datos» lo más amplia posible.
Puede encontrarse información adicional en las referencias que se dan en las directrices de la OCDE y en otras publicaciones relacionadas.
B.1 BIS. TOXICIDAD ORAL AGUDA. MÉTODO DE DOSIS FIJAS
1. MÉTODO
El presente método reproduce las directrices de ensayo de la OCDE TG 420 (2001).
1.1. INTRODUCCIÓN
Los métodos tradicionales para evaluar la toxicidad aguda utilizan la muerte de los animales como parámetro. En 1984 la British Toxicology Society propuso un nuevo enfoque de los ensayos de toxicidad aguda basado en la administración de una serie de dosis fijas (1) Este enfoque evitaba recurrir a la muerte de los animales como parámetro y se basaba, en cambio, en la observación de signos claros de toxicidad a un nivel determinado de una serie de dosis fijas. Tras una serie de estudios de validación in vivo británicos (2) e internacionales (3), este procedimiento fue adoptado como método de ensayo en 1992. Posteriormente, se han evaluado las propiedades estadísticas del método de dosis fijas utilizando modelos matemáticos en una serie de estudios (4) (5) (6). Conjuntamente, los estudios in vivo y los basados en modelos han demostrado que el procedimiento es reproducible, requiere menos animales y causa menos sufrimiento que los métodos tradicionales. Al mismo tiempo, permite clasificar las sustancias de manera parecida a los otros métodos de ensayo de toxicidad aguda.
En el Guidance Document on Acute Oral Toxicity Testing (7) se dan directrices sobre la selección del método de ensayo más adecuado en función del objetivo. Además, en este documento orientativo se facilita información complementaria sobre la realización e interpretación del método de ensayo B.1bis.
Es un principio del método que en el estudio principal solo se empleen dosis moderadamente tóxicas y que se evite la administración de dosis que se prevean letales. Asimismo, no es necesario administrar dosis de las que se sepa que producen dolor y sufrimiento acusados por sus efectos corrosivos o muy irritantes. Los animales moribundos o que den muestras claras de dolor o muestren signos de sufrimiento intenso y continuo serán sacrificados de forma compasiva y en la interpretación de los resultados serán considerados de la misma manera que los que hayan muerto durante el ensayo. En otro documento orientativo (8) se dan criterios para tomar la decisión de matar animales moribundos o sometidos a sufrimiento intenso y directrices para reconocer cuándo la muerte es previsible o inminente.
El método aporta información sobre las propiedades peligrosas y permite clasificar las sustancias de acuerdo con el Sistema Armonizado Mundial (SAM) [Globally Harmonised System (GHS)] de clasificación de sustancias químicas que causan toxicidad aguda (9)
El laboratorio que haga los ensayos debe tener en cuenta toda la información disponible sobre la sustancia estudiada antes de llevar a cabo la prueba. Tal información incluirá la identidad y la estructura química de la sustancia, sus propiedades fisicoquímicas, los resultados de otros ensayos de toxicidad in vitro o in vivo, los datos toxicológicos sobre otras sustancias estructuralmente relacionadas, y el uso o usos previstos de la sustancia. Esta información es necesaria para que todos los afectados puedan estar seguros de que el ensayo es pertinente para la protección de la salud humana y ayuda a la selección de una dosis inicial adecuada.
1.2. DEFINICIONES
Toxicidad oral aguda: efectos nocivos que se manifiestan tras la administración oral de una dosis única de la sustancia o de dosis múltiples dadas dentro de un período de 24 horas.
Muerte retardada significa que el animal no muere ni parece moribundo en el plazo de 48 horas sino que muere más tarde durante el período de observación de 14 días.
Dosis: cantidad de sustancia de ensayo administrada. La dosis se expresa en peso de la sustancia de ensayo por unidad de peso del animal sometido al experimento (por ejemplo, mg/kg).
Toxicidad manifiesta es un término general que describe signos claros de toxicidad tras la administración de una sustancia [véanse ejemplos en (3)]. de tal manera que a la dosis fija inmediatamente superior pueden preverse en la mayoría de los animales dolores fuertes y signos continuados de sufrimiento grave, agonía [véanse los criterios al respecto en Humane Endpoints Guidance Document (8)] o muerte probable.
SAM: Sistema Armonizado Mundial [Globally Harmonised System (GHS)] de clasificación y etiquetado de productos químicos. Actividad conjunta de la OCDE (salud humana y medio ambiente), el Comité de Expertos en Transporte de Mercaderías Peligrosas de las Naciones Unidas (propiedades físico-químicas) y la OIT (comunicación de peligros), coordinada por el Programa Interorganismos para la Gestión Racional de Sustancias Químicas (IOMC, en sus siglas inglesas).
Muerte inminente: fase en la que se prevé que el animal muera o entre en la agonía antes del siguiente momento de observación previsto. Entre los signos que indican esta situación en los roedores cabe citar: convulsiones, posición lateral, posición yacente y temblores. [Véase el Humane Endpoint Guidance Document (8) para más información].
DL50 (dosis letal mediana): dosis única, obtenida por estadística, de una sustancia capaz de provocar la muerte del 50 % de los animales a los que se haya administrado por vía oral. El valor de la DL50 se expresa en peso de la sustancia por unidad de peso del animal (mg/kg).
Dosis límite: dosis del límite máximo del ensayo (2 000 o 5 000 mg/kg).
Agonía: situación en la que el animal se está muriendo o es incapaz de sobrevivir aunque reciba tratamiento. [Véase el Humane Endpoint Guidance Document (8) para más información].
Muerte previsible: presencia de signos clínicos que indican la muerte en un momento conocido del futuro antes de la terminación prevista del experimento, por ejemplo: incapacidad de alcanzar alimentos o agua. [Véase el Humane Endpoint Guidance Document (8) para más información].
1.3. PRINCIPIO DEL MÉTODO DE ENSAYO
Se administran de manera gradual a grupos de animales de un solo sexo dosis fijas de 5, 50, 300 y 2 000 mg/kg (excepcionalmente podría considerarse una dosis adicional de 5 000 mg/kg, véase el punto 1.6.2). La dosis inicial se determina basándose en un estudio preliminar y consiste en la que se prevé que produzca ciertos signos de toxicidad sin causar efectos tóxicos graves ni mortalidad. En un documento orientativo aparte de la OCDE (8) se describen de manera detallada las afecciones y los signos clínicos que denotan dolor, sufrimiento y muerte inminente. Pueden darse dosis más altas o más bajas a otros grupos de animales según la presencia o ausencia de signos de toxicidad o mortalidad. Este procedimiento continúa hasta que se determina la dosis que causa toxicidad manifiesta o solo una muerte o cuando no se observan efectos a la dosis más alta o cuando ocurren muertes a la dosis más baja.
1.4. DESCRIPCIÓN DEL MÉTODO
1.4.1. Selección de la especie animal
La especie de preferencia entre los roedores es la rata, aunque pueden utilizarse otras especies de roedores. Normalmente se emplean hembras (7). Se hace así porque la bibliografía sobre los ensayos convencionales de DL50 muestra que normalmente hay poca diferencia en cuanto a sensibilidad entre los sexos, pero, en los casos en que se observan diferencias, las hembras son generalmente un poco más sensibles (10). Sin embargo, si se conocen propiedades toxicológicas o toxicocinéticas de sustancias químicas estructuralmente relacionadas que indiquen que es probable que los machos sean más sensibles, deberá utilizarse este sexo. Cuando el ensayo se haga con machos, deberá justificarse adecuadamente.
Hay que utilizar animales adultos jóvenes y sanos de una cepa de laboratorio corriente. Las hembras deben ser nulíparas y no grávidas. El animal, al inicio de la administración de la sustancia, debe tener entre 8 y 12 semanas de edad y su peso debe estar en un intervalo del ± 20 % del peso medio de los animales a los que previamente se haya administrado la sustancia.
1.4.2. Alojamiento y alimentación
El cuarto de experimentación ha de estar a una temperatura de 22 oC (± 3 oC). Aunque la humedad relativa debe ser como mínimo del 30 % y preferiblemente no superior al 70 %, salvo durante la limpieza del local, lo ideal es que esté comprendida entre el 50 y el 60 %. Se aplicará una iluminación artificial en una secuencia de 12 horas de luz y 12 horas de oscuridad. Puede darse una dieta alimentaria corriente para animales de laboratorio y agua potable a voluntad. Los animales pueden alojarse en jaulas agrupados por dosis, pero el número de animales de cada jaula no debe obstaculizar la realización de observaciones claras de cada animal.
1.4.3 Preparación de los animales
Se seleccionan al azar los animales, se marcan para permitir su identificación individual y se mantienen en sus jaulas durante al menos 5 días antes de iniciar la administración de la sustancia, a fin de que se aclimaten a las condiciones del laboratorio.
1.4.4 Preparación de las dosis
En general, las sustancias estudiadas deben administrarse en un volumen constante para toda la gama de dosis que deban ensayarse, variando la concentración del preparado administrado. Sin embargo, cuando deba administrarse una mezcla o producto final líquidos, puede resultar más adecuado para la posterior evaluación de riesgos utilizar la sustancia no diluida, es decir, a una concentración constante. Algunas autoridades reguladoras establecen este criterio con carácter obligatorio. En cualquiera de los dos casos, no debe superarse el volumen de dosis máximo. El volumen máximo de líquido que puede administrarse de una sola vez depende del tamaño del animal. En los roedores, el volumen no debe sobrepasar 1 ml/100 g de peso corporal, excepto en el caso de las soluciones acuosas, de las que se pueden usar 2 ml/100 g de peso corporal. En cuanto a la formulación del preparado que se administre, se recomienda el uso de una solución/suspensión/emulsión acuosa siempre que sea posible, seguida, en orden de preferencia, por una solución/suspensión/emulsión oleosa (por ejemplo, en aceite de maíz) y luego posiblemente por la solución en otros vehículos. Si se emplean vehículos distintos del agua, deben conocerse sus características tóxicas. Las dosis tienen que prepararse poco antes de la administración a menos que se conozca la estabilidad del preparado durante el período en que se vaya a utilizar y conste que esta sea aceptable.
1.5 PROCEDIMIENTO
1.5.1 Administración de las dosis
La sustancia estudiada se administra en una dosis única por alimentación forzada mediante sonda gástrica o cánula adecuada de intubación. Cuando se dé el caso, poco habitual, de que no pueda administrarse una dosis única, se podrá dividir esta en partes más pequeñas durante un período que no sobrepase las 24 horas.
Los animales se mantendrán en ayunas antes de la administración de la dosis (por ejemplo, las ratas no deberán recibir alimento alguno durante la noche, pero sí agua). Los ratones no deberán ser alimentados durante 3-4 horas, pero podrán beber agua). Tras el período de ayuno, los animales se pesarán antes de la administración de la sustancia estudiada. Una vez administrada esta, podrá continuarse el ayuno durante unas 3 o 4 horas en el caso de las ratas y 1 o 2 horas cuando se trate de ratones. Cuando una dosis se administre en fracciones a lo largo de un período, podrá ser necesario proporcionar a los animales alimento y agua, en función de la duración del período.
1.5.2 Estudio preliminar
La finalidad del estudio preliminar es determinar la dosis inicial adecuada para el estudio principal. La sustancia estudiada se administra a cada animal de manera secuencial según lo indicado en los gráficos del anexo 1. El estudio preliminar se considera terminado cuando puede tomarse una decisión sobre la dosis inicial para el estudio principal (o si se observa una muerte a la dosis fija más baja)
La dosis inicial para el estudio preliminar se selecciona a partir de los niveles de dosis fija de 5, 50, 300 y 2 000 mg/kg y consiste en la dosis que se prevé que produzca toxicidad manifiesta, basándose, cuando sea posible, en datos obtenidos in vivo e in vitro referentes a la misma sustancia química y a otras sustancias estructuralmente relacionadas. A falta de esta información, la dosis inicial será de 300 mg/kg.
Se dejará transcurrir un plazo mínimo de 24 horas entre las administraciones de dosis a cada animal. El período de observación de todos los animales debe ser de, al menos, 14 días.
Excepcionalmente, y solo cuando esté justificado por determinados imperativos legales, puede considerarse el uso de otra dosis fija superior de 5 000 mg/kg (véase el anexo 3). Por razones de bienestar animal, se desaconseja el ensayo de sustancias de la categoría 5 del Sistema Armonizado Mundial (DL50 entre 2 000-5 000 mg/kg) con animales. Este ensayo solo debe plantearse cuando sea muy probable que sus resultados sirvan directamente para la protección de la salud humana o animal o del medio ambiente.
En los casos en que se ensaye en un animal la dosis fija más baja (5 mg/kg) en un estudio preliminar y el animal muera, el procedimiento normal es poner fin al estudio y asignar la sustancia a la categoría 1 del SAM (tal como se indica en el anexo 1). Sin embargo, si se requiere confirmar la clasificación, puede seguirse un procedimiento opcional complementario, que se indica a continuación. Se administra a otro animal una dosis de 5 mg/kg. Si este muere, queda confirmada la categoría 1 del SAM y se pone fin al estudio inmediatamente. Si el segundo animal sobrevive, se administra una dosis de 5 mg/kg a otros tres animales. Dado que habrá un riesgo elevado de mortalidad, los animales recibirán la dosis de manera secuencial para proteger el bienestar animal. El intervalo entre la administración de la dosis a cada animal debe ser suficiente para que quede claro que el animal anterior tiene probabilidades de sobrevivir. Si ocurre una segunda muerte, se pondrá fin inmediatamene a la administración de dosis, y no se dará la sustancia a ningún otro animal. Dado que el hecho de que se produzca una segunda muerte (independientemente del número de animales sometidos a ensayo en el momento de la terminación) corresponde al resultado A (2 o más muertes), se seguirá la norma de clasificación del anexo 2 a la dosis fija 5 mg/kg (categoría 1 si hay dos o más muertes o categoría 2 si solo hay una muerte). Además, el anexo 4 da orientaciones sobre la clasificación en el sistema comunitario hasta que se aplique el nuevo SAM.
1.5.3 Estudio principal
1.5.3.1 Número de animales y dosis
La actuación que debe seguirse tras el ensayo al nivel de dosis inicial se indica en los gráficos del anexo 2. Deberá optarse por una de las tres actuaciones siguientes: terminar el ensayo y asignar a la sustancia la categoría de peligro adecuada, ensayar a una dosis fija superior o ensayar a una dosis fija inferior. Sin embargo, para proteger a los animales, no se repetirá en el estudio principal ningún nivel de dosis que haya causado un resultado de muerte en el estudio preliminar (véase el anexo 2). La experiencia ha mostrado que el resultado más probable al nivel de dosis inicial es que pueda clasificarse la sustancia y que no sean necesarios más ensayos.
Para cada nivel de dosis investigado se utilizará normalmente un total de cinco animales de un mismo sexo. Este grupo de cinco animales estará compuesto de un animal del estudio preliminar al que se haya administrado la sustancia al nivel de dosis seleccionado más otros cuatro (excepto cuando un nivel de dosis utilizado en el estudio principal no estuviera incluido en el estudio preliminar, situación que es poco frecuente).
El intervalo de tiempo entre la administración de dosis a cada nivel se determina según la aparición, duración y gravedad de los signos tóxicos. El tratamiento de animales con la dosis siguiente no se realizará hasta que haya seguridad sobre la supervivencia de los animales previamente tratados. Se recomienda, si es necesario, un período de tres o cuatro días entre la administración de cada nivel de dosis para poder observar la toxicidad retardada. Este intervalo puede ajustarse según convenga, por ejemplo en caso de respuestas no concluyentes.
Cuando se emplee una dosis fija superior de 5 000 mg/kg, se seguirá el procedimiento indicado en el anexo 3 (véase también el punto 1.6.2).
1.5.3.2 Ensayo límite
El ensayo límite se utiliza principalmente en situaciones en las que el experimentador tiene información de que la sustancia estudiada es probable que no sea tóxica, es decir, solo resulta tóxica por encima de las dosis límite reglamentarias. Puede obtenerse información acerca de la toxicidad de la sustancia estudiada a partir de los conocimientos disponibles sobre compuestos, mezclas o productos ensayados semejantes, teniendo en cuenta cuáles son los componentes que se sabe que son importantes desde el punto de vista toxicológico y en qué porcentaje aparecen. En situaciones en que se dispone de poca o ninguna información sobre la toxicidad de la sustancia o en la que se prevé que esta será tóxica, se llevará a cabo el ensayo principal.
Utilizando el procedimiento normal, una dosis inicial en el estudio preliminar de 2 000 mg/kg (o excepcionalmente 5 000 mg/kg) seguida por la administración de este nivel de dosis a cuatro animales sirve de ensayo límite para las presentes directrices.
1.6 OBSERVACIONES
Tras la administración de la dosis, se observan los animales uno por uno, al menos una vez durante los primeros 30 minutos y periódicamente durante las primeras 24 horas, prestando especial atención durante las primeras 4 horas, y a continuación diariamente, a lo largo de un total de 14 días, excepto cuando tengan que eliminarse del estudio y sacrificarse de manera compasiva por razones de bienestar animal o bien cuando resulten muertos. Sin embargo, no debe fijarse rígidamente la duración de la observación, sino que esta debe determinarse según las reacciones tóxicas, su momento de aparición y la longitud del período de recuperación, por lo que podrá ampliarse cuando se considere necesario. Son importantes los momentos en que aparezcan y desaparezcan los signos de toxicidad, especialmente si hay tendencia a una aparición retardada de los signos tóxicos (11). Todas las observaciones se registrarán sistemáticamente en una ficha de cada animal.
Será necesario proceder a observaciones adicionales si los animales siguen presentando signos de toxicidad. Entre las observaciones deben incluirse los cambios de la piel y del pelaje, ojos y membranas mucosas, y también de los sistemas respiratorio, circulatorio y nervioso (central y autónomo), así como la actividad locomotriz y las pautas de comportamiento. Debe prestarse especial atención a la observación de temblores, convulsiones, salivación, diarrea, letargo, sueño y coma. Se tendrán en cuenta los principios y criterios resumidos en el Humane Endpoints Guidance Document (8). Los animales moribundos y los que muestren dolor intenso o signos continuos de sufrimiento intenso deberán sacrificarse de forma compasiva. Cuando se encuentre algún animal muerto o se sacrifique por razones compasivas, deberá registrarse con la mayor precisión posible el momento de la muerte.
1.6.1 Peso corporal
Cada animal se pesará justo antes de la administración de la sustancia, y después al menos una vez por semana. Se calcularán y registrarán los cambios de peso. Al final de la prueba, los animales supervivientes se pesarán antes de sacrificarse de forma compasiva.
1.6.2 Patología
Todos los animales sometidos al ensayo, incluidos los que mueran durante su realización y los que se eliminen del estudio por razones de bienestar animal, se someterán a necropsia macroscópica. Se registrarán los cambios patológicos macroscópicos de cada animal. Podrá considerarse también la realización del examen microscópico de los órganos que presenten huellas de patología macroscópica en los animales que sobrevivan un mínimo de 24 horas después de la administración de la dosis inicial, ya que este examen puede proporcionar información útil.
2 RESULTADOS
Los resultados de cada animal deben darse por separado. Además, se reunirán todos los resultados en un cuadro que presente, por cada grupo de ensayo, el número de animales utilizados, el número de animales que presenten signos de toxicidad, el número de animales hallados muertos durante el ensayo o sacrificados por razones compasivas, el momento de la muerte de los distintos animales, la descripción y la evolución temporal de los efectos tóxicos y la reversibilidad, y los resultados de la necropsia.
3 INFORME
3.1 INFORME DEL ENSAYO
El informe del ensayo debe incluir, en su caso, la información siguiente:
|
|
Sustancia estudiada:
|
|
|
Vehículo (si procede):
|
|
|
Animales sometidos a ensayo:
|
|
|
Condiciones de ensayo:
|
|
|
Resultados:
|
|
|
Evaluación e interpretación de los resultados. |
|
|
Conclusiones. |
4 REFERENCIAS
|
(1) |
British Toxicology Society Working Party on Toxicity (1984). Special report: a new approach to the classification of substances and preparations on the basis of their acute toxicity. Human Toxico]., 3, 85-92. |
|
(2) |
Van den Heuvel, M.J., Dayan, A.D. and Shillaker, R.O. (1987). Evaluation of the BTS approach to the testing of substances and preparations for their acute toxicity. Human Toxicol., 6, 279-291. |
|
(3) |
Van den Heuvel, M.J., Clark, D.G., Fielder, R.J., Koundakjian, P.P., Oliver, G.J.A., Pelling, D., Tomlinson, N.J. and Walker, A.P. (1990). The international validation of a fixed-dose procedure as an alternative to the classical LD50 test. Fd. Chem. Toxicol. 28, 469-482 (3). |
|
(4) |
Whitehead, A. and Curnow, R.N. (1992). Statistical evaluation of the fixed-dose procedure. Fd. Chem. Toxicol., 30, 313-324. |
|
(5) |
Stallard, N. and Whitehead, A. (1995). Reducing numbers in the fixed-dose procedure. Human Exptl. Toxicol., 14, 315-323. Human Exptl. Toxicol. |
|
(6) |
Stallard, N., Whitehead, A. and Ridgeway, P. (2002). Statistical evaluation of the revised fixed dose procedure.-Hum. Exp. Toxicol., 21, 183 -196. |
|
(7) |
OECD (2001). Guidance Document on Acute Oral Toxicity Testing. Environmental Health and Safety Monograph Series on Testing and Assessment N. 24. Paris. |
|
(8) |
OECD (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Environmental Health and Safety Monograph Series on Testing and Assessment N. 19. |
|
(9) |
OECD (1998). Harmonised Integrated Hazard Classification for Human Health and Environmental Effects of Chemical Substances as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals in November 1998, Part 2, p. 11 [http://webnetl.oecd.org/oecd/pages/home/displaygenera]/0,3380JEN- documents-521 -14-no-24-no-0,FF.htmI]. |
|
(10) |
Lipnick, R.L., Cotruvo, J.A., Hill, R.N., Bruce, R.D., Stitzel, K.A., Walker, A.P., Chu, I., Goddard, M., Segal, L., Springer, J.A. and Myers, R.C. (1995). Comparison of the Up-and-Down, Conventional LD50, and Fixed-Dose Acute Toxicity Procedures. Fd. Chem. Toxicol. 33, 223-231. |
|
(11) |
Chan P.K and A.W. Hayes (1994) Chapter 16 Acute Toxicity and Eye Irritation, in: Principies and Methods of Toxicology. 3 rd Edition. A.W. Hayes, Editor. Raven Press, Ltd. New York, USA |
ANEXO 1
DIAGRAMA DEL ESTUDIO PRELIMINAR

ANEXO 2
DIAGRAMA DEL ESTUDIO PRINCIPAL

ANEXO 3
CRITERIOS PARA LA CLASIFICACIÓN DE SUSTANCIAS CON VALORES DL50 PREVISTOS SUPERIORES A LOS 2 000 MG/KG SIN NECESIDAD DE ENSAYO
Los criterios de la categoría de peligro 5 tienen por objeto permitir la identificación de sustancias que tengan un peligro de toxicidad aguda relativamente bajo pero que, en determinadas circunstancias, puedan representar un peligro para poblaciones vulnerables. Estas sustancias se prevé que tengan una DL50 oral o dérmica dentro del intervalo 2 000-5 000 mg/kg o dosis equivalentes por otras vías. Las sustancias estudiadas podrían clasificarse en la categoría de peligro definida por: 2 000 mg/kg < DL50 < 5 000 mg/kg (categoría 5 del SAM), en los siguientes casos:
|
a) |
cuando cualquiera de los esquemas de ensayo del anexo 2 lleve a esta categoría, basándose en las cifras de mortalidad; |
|
b) |
cuando se disponga ya de datos fiables que indiquen que la DL50 se sitúa dentro del intervalo de los valores de la categoría 5 o cuando otros estudios con animales o los efectos tóxicos observados en las personas indiquen un peligro agudo para la salud humana; |
|
c) |
mediante extrapolación, estimación o medición de datos si no está justificada la clasificación en una categoría más peligrosa, y
|
ENSAYOS A DOSIS SUPERIORES A 2 000 MG/KG
Excepcionalmente, y solo cuando esté justificado por determinados imperativos legales, puede considerarse el uso de otro nivel de dosis fija superior de 5 000 mg/kg Reconociendo la necesidad de proteger el bienestar animal, se desaconseja el ensayo a la dosis 5 000 mg/kg. Este solo debe plantearse cuando sea muy probable que sus resultados sirvan directamente para la protección de la salud humana o animal (9).
Estudio preliminar
Las normas para la toma de decisiones que rigen el procedimiento secuencial del anexo 1 se amplían de manera que incluyan el nivel de dosis 5 000 mg/kg Por tanto, cuando se emplee una dosis inicial en el estudio preliminar de 5 000 mg/kg, el resultado A (muerte) obligará a que se haga un ensayo con otro animal a 2 000 mg/kg; los resultados B y C (toxicidad manifiesta o no toxicidad) permitirán la selección de la dosis de 5 000 mg/kg como dosis inicial del estudio principal. De la misma manera, si se utiliza una dosis inicial distinta de 5 000 mg/kg se pasará a la de 5 000 mg/kg en caso de que se den los resultados B o C a 2 000 mg/kg; un posterior resultado A a 5 000 mg/kg determinará una dosis inicial para el estudio principal de 2 000 mg/kg y los resultados B y C determinarán una dosis inicial para el estudio principal de 5 000 mg/kg.
Estudio principal
Las normas para la toma de decisiones que rigen el procedimiento secuencial del anexo 2 se amplían de manera que incluyan el nivel de dosis de 5 000 mg/kg. Por tanto, cuando se emplee una dosis inicial en el estudio principal de 5 000 mg/kg, el resultado A (> 2 muertes) obligará a que se haga un ensayo con un segundo grupo a 2 000 mg/kg; el resultado B (toxicidad manifiesta y/o < 1 muerte) o C (no toxicidad) determinará que la sustancia se considere no clasificada con arreglo al SAM. De la misma manera, si se utiliza una dosis inicial distinta de 5 000 mg/kg se pasará a la de 5 000 mg/kg en caso de que se dé un resultado C a 2 000 mg/kg; un posterior resultado A a 5 000 mg/kg dará lugar a que la sustancia se clasifique en la categoría 5 del SAM y los resultados B y C a que se considere no clasificada.
ANEXO 4
MÉTODO DE PRUEBA B.1 bis
Orientaciones sobre clasificación según el plan comunitario para el período de transición hasta la plena aplicación del Sistema Armonizado Mundial (SAM) [tomado de (8)]
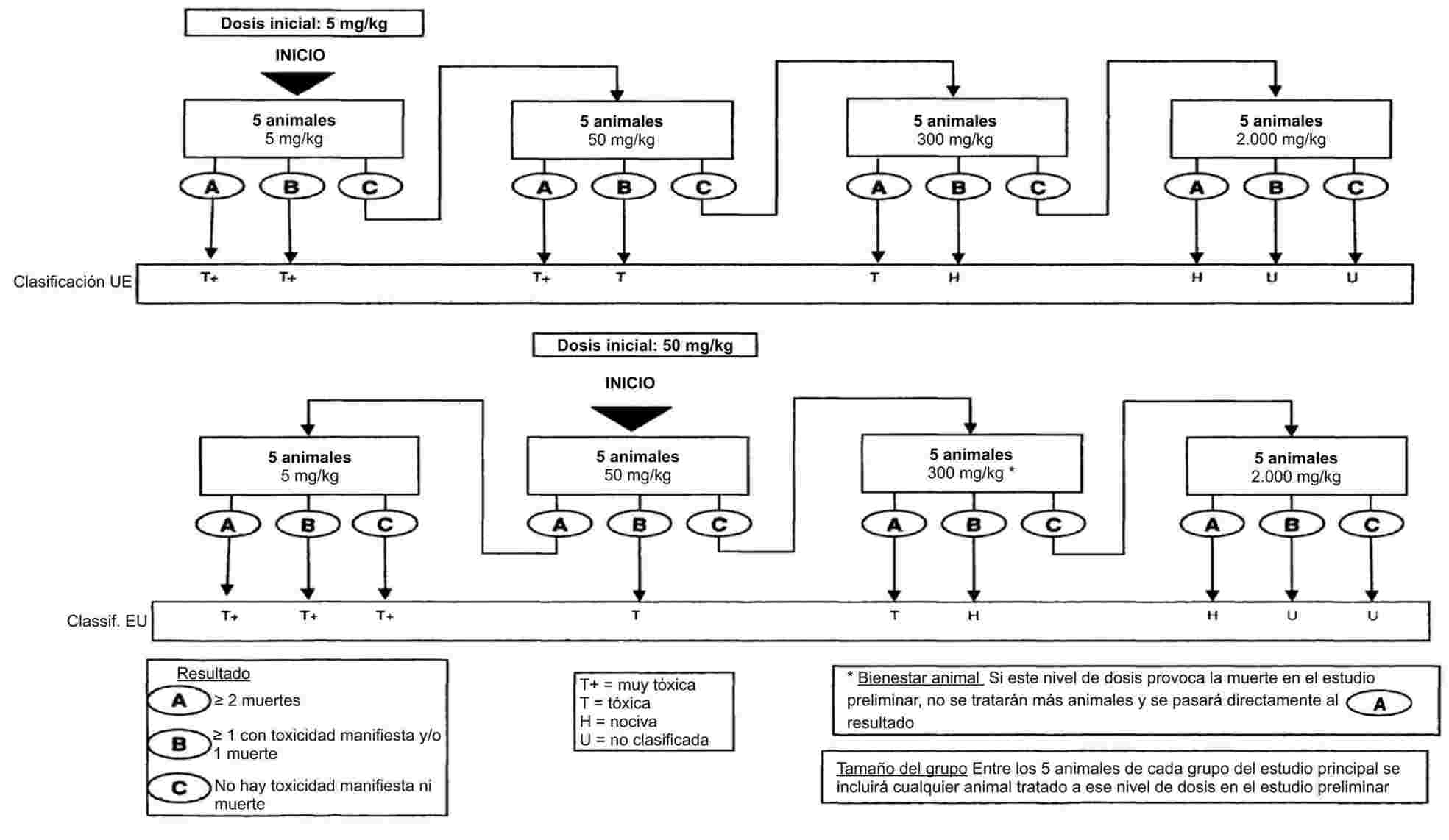

B.1 ter TOXICIDAD ORAL AGUDA. MÉTODO DE LAS CLASES DE TOXICIDAD AGUDA
1. MÉTODO
El presente método reproduce las directrices de ensayo de la OCDE TG 423 (2001).
1.1 INTRODUCCIÓN
El método de las clases de toxicidad aguda (1) establecido en este ensayo es un procedimiento por fases en el que se utilizan 3 animales de un mismo sexo en cada fase. Según la mortalidad y/o la aparición de síntomas de agonía en los animales, pueden requerirse 2-4 fases para juzgar sobre la toxicidad aguda de la sustancia estudiada. Este procedimiento es reproducible, utiliza muy pocos animales y permite clasificar sustancias de manera semejante a los demás métodos de ensayo de toxicidad aguda. El método de las clases de toxicidad aguda se basa en evaluaciones biométricas (2) (3) (4) (5) con dosis fijas, separadas adecuadamente de manera que permitan ordenar una sustancia con fines de clasificación y evaluación de riesgos. Este método en su versión de 1996 fue validado ampliamente in vivo con respecto a datos DL50 tomados de la bibliografía, tanto a nivel nacional (6) como internacional (7).
En el Guidance Document on Acute Oral Toxicity Testing (8) se dan directrices sobre la selección del método de ensayo más adecuado en función del objetivo. Además, en este documento orientativo se facilita información complementaria sobre la realización e interpretación del método de ensayo B.l ter.
No es necesario administrar dosis de las que se sepa que producen dolor y sufrimiento acusados por sus efectos corrosivos o muy irritantes. Los animales moribundos o que den muestras claras de dolor o muestren signos de sufrimiento intenso y continuo serán sacrificados de forma compasiva y en la interpretación de los resultados serán considerados de la misma manera que los que hayan muerto durante el ensayo. En otro documento orientativo (9) se dan criterios para tomar la decisión de matar animales moribundos o sometidos a sufrimiento intenso y directrices para reconocer cuándo la muerte es previsible o inminente.
El método utiliza dosis previamente definidas y los resultados permiten ordenar y clasificar las sustancias de acuerdo con el Sistema Armonizado Mundial (SAM) [Globally Harmonised System (GHS)] de clasificación de sustancias químicas que causan toxicidad aguda (10).
En principio, con este método no se pretende calcular una DL50 precisa, sino determinar unas gamas de exposición definidas en las que puede esperarse la aparición de letalidad, ya que la muerte de una parte de los animales sigue siendo su parámetro principal. El método permite la determinación del valor DL50 solo cuando al menos dos dosis producen una mortalidad superior al 0 % e inferior al 100 %. La utilización de dosis previamente definidas, independientemente de la sustancia que se ensaye, y la relación explícita entre la clasificación y el número de animales observados en diferentes estados, mejora la concordancia entre la información facilitada por distintos laboratorios, así como la repetibilidad.
El laboratorio que haga los ensayos debe tener en cuenta toda la información disponible sobre la sustancia estudiada antes de llevar a cabo el estudio. Tal información incluirá la identidad y la estructura química de la sustancia sus propiedades fisicoquímicas, los resultados de otros ensayos de toxicidad in vitro o in vivo, los datos toxicológicos sobre otras sustancias estructuralmente relacionadas, y el uso o usos previstos de la sustancia. Esta información es necesaria para que todos los afectados puedan estar seguros de que el ensayo es pertinente para la protección de la salud humana y ayuda a la selección de la dosis inicial más adecuada.
1.2 DEFINICIONES
Toxicidad oral aguda: efectos nocivos que se manifiestan tras la administración oral de una dosis única de la sustancia o de dosis múltiples dadas dentro de un período de 24 horas.
Muerte retardada significa que el animal no muere ni parece moribundo en el plazo de 48 horas sino que muere más tarde durante el período de observación de 14 días.
Dosis: cantidad de sustancia de ensayo administrada. La dosis se expresa en peso de la sustancia de ensayo por unidad de peso del animal sometido al experimento (por ejemplo, mg/kg).
SAM: Sistema Armonizado Mundial [Globally Harmonised System (GHS)] de clasificación y etiquetado de productos químicos. Actividad conjunta de la OCDE (salud humana y medio ambiente), el Comité de Expertos en Transporte de Mercaderías Peligrosas de las Naciones Unidas (propiedades físico-químicas) y la OIT (comunicación de riesgos), coordinada por el Programa Interorganismos para la Gestión Racional de Sustancias Químicas (IOMC, en sus siglas inglesas).
Muerte inminente: fase en la que se prevé que el animal muera o entre en la agonía antes del siguiente momento de observación previsto. Entre los signos que indican esta situación en los roedores cabe citar: convulsiones, posición lateral, posición yacente y temblores. [Para más información véase el Humane Endpoint Guidance Document (9)].
DL 50 (dosis letal mediana): dosis única, obtenida por estadística, de una sustancia capaz de provocar la muerte del 50 % de los animales a los que se haya administrado por vía oral. El valor de la DL50 se expresa en peso de la sustancia por unidad de peso del animal (mg/kg).
Dosis límite: dosis del límite máximo del ensayo (2 000 o 5 000 mg/kg).
Agonía: situación en la que el animal se está muriendo o es incapaz de sobrevivir aunque reciba tratamiento. [Para más información véase el Humane Endpoint Guidance Document (9)].
Muerte previsible: presencia de signos clínicos que indican la muerte en un momento conocido del futuro antes de la terminación prevista del experimento, por ejemplo: incapacidad de alcanzar alimentos o agua. [Para más información, véase el Humane Endpoint Guidance Document (9)].
1.3 PRINCIPIO DEL MÉTODO
El principio en el que se inspira el ensayo, basado en un procedimiento por fases en cada una de las cuales se utiliza un número mínimo de animales, es obtener información sobre la toxicidad aguda de la sustancia estudiada que sea suficiente para permitir su clasificación. La sustancia se administra por vía oral a un grupo de animales de experimentación a una de las dosis definidas. En el ensayo se sigue un procedimiento por fases, utilizándose en cada fase tres animales del mismo sexo (normalmente hembras). Según la ausencia o presencia de mortalidad debida a la sustancia en los animales tratados en una fase determinada se decide cuál será la fase siguiente, es decir:
|
— |
terminación del ensayo (no se requieren más ensayos), |
|
— |
administración de la misma dosis de la sustancia a otros tres animales, |
|
— |
administración de la dosis inmediatamente superior o inferior a otros tres animales. |
En el anexo 1 se dan detalles del procedimiento del ensayo. El método permitirá emitir un juicio respecto a la clasificación de la sustancia en una clase de toxicidad dentro de una serie de clases definidas mediante valores límite de DL50.
1.4 DESCRIPCIÓN DEL MÉTODO
1.4.1 Selección de la especie animal
La especie de preferencia entre los roedores es la rata, aunque pueden utilizarse otras especies de roedores. Normalmente se emplean hembras (9). Se hace así porque la bibliografía sobre los ensayos convencionales de DL50 muestra que normalmente hay poca diferencia en cuanto a sensibilidad entre los sexos, pero, en los casos en que se observan diferencias, las hembras son generalmente un poco más sensibles (11). Sin embargo, si se conocen propiedades toxicológicas o toxicocinéticas de sustancias químicas estructuralmente relacionadas que indiquen que es probable que los machos sean más sensibles, deberá utilizarse este sexo. Cuando el ensayo se haga con machos, deberá justificarse adecuadamente.
Hay que utilizar animales adultos jóvenes y sanos de una cepa de laboratorio corriente. Las hembras deben ser nulíparas y no grávidas. El animal, al inicio de la administración de la sustancia, debe tener entre 8 y 12 semanas de edad y su peso debe estar en un intervalo del ± 20 % del peso medio de los animales a los que previamente se haya administrado la sustancia.
1.4.2 Alojamiento y alimentación
El cuarto de experimentación ha de estar a una temperatura de 22 oC (± 3 oC). Aunque la humedad relativa debe ser como mínimo del 30 % y preferiblemente no superior al 70 %, salvo durante la limpieza del local, lo ideal es que esté comprendida entre el 50 y el 60 %. Se aplica una iluminación artificial en una secuencia de 12 horas de luz y 12 horas de oscuridad. Puede darse una dieta alimentaria corriente para animales de laboratorio y agua potable a voluntad. Los animales pueden alojarse en jaulas agrupados por dosis, pero el número de animales de cada jaula no debe obstaculizar la realización de observaciones claras de cada animal.
1.4.3 Preparación de los animales
Se seleccionan al azar los animales, se marcan para permitir su identificación individual y se mantienen en sus jaulas durante al menos 5 días antes de iniciar la administración de la sustancia, a fin de que se aclimaten a las condiciones del laboratorio.
1.4.4 Preparación de las dosis
En general, las sustancias estudiadas deben administrarse en un volumen constante para toda la gama de dosis que deban ensayarse, variando la concentración del preparado administrado. Sin embargo, cuando deba administrarse una mezcla o producto final líquidos, puede resultar más adecuado para la posterior evaluación de riesgos utilizar la sustancia no diluida, es decir, a una concentración constante. Algunas autoridades reguladoras establecen este criterio con carácter obligatorio. En cualquiera de los dos casos, no debe superarse el volumen de dosis máximo. El volumen máximo de líquido que puede administrarse de una sola vez depende del tamaño del animal. En los roedores, el volumen no debe sobrepasar 1 ml/100 g de peso corporal, excepto en el caso de las soluciones acuosas, de las que se pueden usar 2 ml/100 g de peso corporal. En cuanto a la formulación del preparado que se administre, se recomienda el uso de una solución/suspensión/emulsión acuosa siempre que sea posible, seguida, en orden de preferencia, por una solución/suspensión/emulsión oleosa (por ejemplo, en aceite de maíz) y luego posiblemente por la solución en otros vehículos. Si se emplean vehículos distintos del agua, deben conocerse sus características tóxicas. Las dosis tienen que prepararse poco antes de la administración a menos que se conozca la estabilidad del preparado durante el período en que se vaya a utilizar y conste que esta es aceptable.
1.5 PROCEDIMIENTO
1.5.1 Administración de las dosis
La sustancia estudiada se administra en una dosis única por alimentación forzada mediante sonda gástrica o cánula adecuada de intubación. Cuando se dé el caso, poco habitual, de que no pueda administrarse una dosis única, se podrá dividir esta en pequeñas partes durante un período que no sobrepase las 24 horas.
Los animales se mantendrán en ayunas antes de la administración de la dosis (por ejemplo, las ratas no deberán recibir alimento alguno durante la noche, pero sí agua; los ratones no deberán recibir alimento durante 3-4 horas, pero sí agua). Tras el período de ayuno, los animales se pesarán antes de la administración de la sustancia estudiada. Una vez administrada esta, podrá continuarse el ayuno durante unas 3 o 4 horas en el caso de las ratas y 1 o 2 horas cuando se trate de ratones. Si se administra la dosis en pequeñas partes a lo largo de un cierto período podrá ser necesario dar comida y bebida a los animales, en función de la duración de dicho período.
1.5.2 Número de animales y dosis
Se utilizarán tres animales en cada fase. La dosis inicial se seleccionará entre las cuatro dosis fijas, es decir, 5, 50, 300 y 2 000 mg/kg de peso corporal. La dosis inicial será la que produzca con mayor probabilidad la muerte de algunos de los animales a los que se administre. En los diagramas del anexo 1 se describe el procedimiento que debe seguirse para cada una de las dosis iniciales. Además, el anexo 4 da orientaciones sobre la clasificación en el sistema comunitario hasta que se aplique el nuevo SAM.
Cuando la información indique que no es probable la aparición de mortalidad con la dosis inicial máxima (2 000 mg/kg peso corporal), se realizará una prueba límite. Si no hay información sobre una sustancia estudiada, por razones de bienestar animal, se recomienda utilizar como dosis inicial 300 mg/kg peso corporal.
El intervalo de tiempo entre los grupos de tratamiento se determina según la aparición, duración y gravedad de los signos tóxicos. El tratamiento de animales con la dosis siguiente no se realizará hasta que haya seguridad sobre la supervivencia de los animales previamente tratados.
Excepcionalmente, y solo cuando esté justificado por determinados imperativos legales, puede considerarse el uso de otro nivel de dosis superior de 5 000 mg/kg (véase el anexo 2). Por razones de bienestar animal, se desaconseja el ensayo en animales de la categoría 5 del Sistema Armonizado Mundial (2 000-5 000 mg/kg). Este ensayo solo debe plantearse cuando sea muy probable que sus resultados sirvan directamente para la protección de la salud humana o animal o del medio ambiente.
1.5.3 Ensayo límite
El ensayo límite se utiliza en situaciones en las que el experimentador tiene información de que la sustancia estudiada es probable que no sea tóxica, es decir, solo resulta tóxica por encima de las dosis límite reglamentarias. Puede obtenerse información acerca de la toxicidad de la sustancia estudiada a partir de los conocimientos disponibles sobre compuestos, mezclas o productos ensayados semejantes, teniendo en cuenta cuáles son los componentes que se sabe que son importantes desde el punto de vista toxicológico y en qué porcentaje aparecen. En situaciones en que se dispone de poca o ninguna información sobre la toxicidad de la sustancia o en la que se prevé que esta será tóxica, se llevará a cabo el ensayo principal.
Podrá realizarse un ensayo límite con una sola dosis de 2 000 mg/kg peso corporal con seis animales (tres por fase). Podrá realizarse un ensayo límite con una sola dosis de 5 000 mg/kg peso corporal con tres animales (véase el anexo 2). Si aparece mortalidad debida a la sustancia, puede ser necesario realizar una prueba complementaria al nivel inmediatamente inferior.
1.6 OBSERVACIONES
Tras la administración de la dosis, se observan los animales uno por uno, al menos una vez durante los primeros 30 minutos y periódicamente durante las primeras 24 horas, prestando especial atención durante las primeras 4 horas, y a continuación diariamente, a lo largo de un total de 14 días, excepto cuando tengan que eliminarse del estudio y sacrificarse de manera compasiva por razones de bienestar animal o bien cuando resulten muertos. Sin embargo, no se debería fijar rígidamente la duración de la observación sino que esta debe determinarse según las reacciones tóxicas, su momento de aparición y la longitud del período de recuperación, por lo que podrá ampliarse cuando se considere necesario. Son importantes los momentos en que aparezcan y desaparezcan los signos de toxicidad, especialmente si hay tendencia a una aparición retardada de los signos tóxicos (12). Todas las observaciones se registrarán sistemáticamente en fichas individuales de cada animal.
Será necesario hacer más observaciones si los animales siguen presentando signos de toxicidad. Entre las observaciones deben incluirse los cambios de la piel y del pelaje, ojos y membranas mucosas, y también de los sistemas respiratorio, circulatorio y nervioso (central y autónomo), así como la actividad locomotriz y las pautas de comportamiento. Debe prestarse especial atención a la observación de temblores, convulsiones, salivación, diarrea, letargo, sueño y coma. Se tendrán en cuenta los principios y criterios resumidos en el Humane Endpoints Guidance Document (8). Los animales moribundos y los que muestren dolor intenso y signos continuos de sufrimiento intenso deberán sacrificarse de forma compasiva. Cuando se encuentre algún animal muerto o se sacrifique por razones compasivas, deberá registrarse con la mayor precisión posible el momento de la muerte.
1.6.1 Peso corporal
Cada animal se pesará justo antes de la administración de la sustancia, y después al menos una vez por semana. Se calcularán y registrarán los cambios de peso. Al final de la prueba, los animales supervivientes se pesarán antes de sacrificarse de forma compasiva.
1.6.2 Patología
Todos los animales sometidos al ensayo, incluidos los que mueran durante su realización y los que se eliminen del estudio por Tazones de bienestar animal, se someterán a necropsia macroscópica. Se registrarán los cambios patológicos macroscópicos de cada animal. Podrá considerarse también la realización del examen microscópico de los órganos que presenten huellas de patología macroscópica, en los animales que sobrevivan un mínimo de 24 horas, ya que este examen puede proporcionar información útil.
2. RESULTADOS
Los resultados de cada animal deben darse por separado. Además, se reunirán todos los resultados en un cuadro que presente, por cada grupo de ensayo, el número de animales utilizados, el número de animales que presenten signos de toxicidad, el número de animales hallados muertos durante el ensayo o sacrificados por razones compasivas, el momento de la muerte de los distintos animales, la descripción y la evolución temporal de los efectos tóxicos y la reversibilidad, y los resultados de la necropsia.
3. INFORME
3.1 Informe del ensayo
El informe del ensayo debe incluir, en su caso, la información siguiente:
|
|
Sustancia estudiada:
|
|
|
Vehículo (si procede):
|
|
|
especie y cepa utilizadas,
|
|
|
Condiciones de ensayo:
|
|
|
Resultados:
|
|
|
Evaluación e interpretación de los resultados. |
|
|
Conclusiones. |
4. REFERENCIAS
|
(1) |
Roll R., Höfer-Bosse Th. And Kayser D. (1986). New Perspectives in Acute Toxicity Testing of Chemicals. Toxicol. Lett., Suppl. 31, 86 |
|
(2) |
Roll R., Riebschläger M., Mischke U. and Kayser D. (1989). Neue Wege zur Bestimmung der akuten Toxizität von Chemikalien. Bundesgesundheitsblatt 32, 336-341. |
|
(3) |
Diener W., Sichha L., Mischke U., Kayser D. and Schlede E. (1994). The Biometric Evaluation of the Acute- Toxic-Class Method (Oral). Arch. Toxicol. 68, 559-610 |
|
(4) |
Diener W., Mischke U., Kayser D. and Schlede E. (1995). The Biometric Evaluation of the OECD Modified Version of the Acute-Toxic-Class Method (Oral). Arch. Toxicol. 69, 729-734. |
|
(5) |
Diener W., and Schlede E. (1999) Acute Toxicity Class Methods: Alterations to LD/LC50 Tests. ALTEX 16, 129-134 |
|
(6) |
Schlede E., Mischke U., Roll R. and Kayser D. (1992). A National Validation Study of the Acute-Toxic- Class Method-An Alternative to the LD50 Test. Arch. Toxicol. 66, 455-470. |
|
(7) |
Schlede E., Mischke U., Diener W. and Kayser D. (1994). The International Validation Study of the Acute- Toxic-Cíass Method (Oral). Arch. Toxicol. 69, 659-670. |
|
(8) |
OECD (2001) Guidance Document on Acute Oral Toxicity Testing. Environmental Health and Safety Monograph Series on Testing and Assessment N. 24. Paris. |
|
(9) |
OECD (2000) Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Environmental Health and Safety Monograph Series on Testing and Assessment N 19. |
|
(10) |
OECD (1998) Harmonized Integrated Hazard Classification System For Human Health And Environmental . Effects Of Chemical Substances as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals in November 1998, Part 2, p. 11 [http://webnetl.oecd.org/oecd/pages/home/displaygeneral/0,3380,EN-documents-521-14-no-24-no-0,FF.html]. |
|
(11) |
Lipnick R L, Cotruvo, J A, Hill R N, Bruce R D, Stitzel K A, Walker A P, Chu I; Goddard M, Segal L, Springer J A and Myers R C (1995) Comparison of the Up-and Down, Conventional LD50, and Fixed Dose Acute Toxicity Procedures. Fd. Chem. Toxicol 33, 223-231. |
|
(12) |
Chan P.K. and A.W. Hayes. (1994). Chap. 16. Acute Toxicity and Eye Irritancy. Principles and Methods of Toxicology. Third Edition. A.W. Hayes, Editor. Raven Press, Ltd., New York, USA. |
ANEXO 1
PROCEDIMIENTO QUE DEBE SEGUIRSE PARA CADA UNA DE LAS DOSIS INICIALES
OBSERVACIONES GENERALES
Para cada dosis inicial, el procedimiento que debe seguirse se indica en los respectivos esquemas de ensayo que figuran en el presente anexo.
|
— |
Anexo 1 a: la dosis inicial es 5 mg/kg de peso corporal. |
|
— |
Anexo 1 b: la dosis inicial es 50 mg/kg de peso corporal. |
|
— |
Anexo 1 c: la dosis inicial es 300 mg/kg de peso corporal. |
|
— |
Anexo 1 d: la dosis inicial es 2 000 mg/kg de peso corporal. |
En función del número de animales muertos o sacrificados de forma compasiva, el procedimiento de ensayo seguirá las flechas indicadas.
Anexo 1 A
PROCEDIMIENTO DE ENSAYO CON UNA DOSIS INICIAL DE 5 MG/KG DE PESO CORPORAL

ANEXO 1B
PROCEDIMIENTO DE ENSAYO CON UNA DOSIS INICIAL DE 50 MG/KG DE PESO CORPORAL
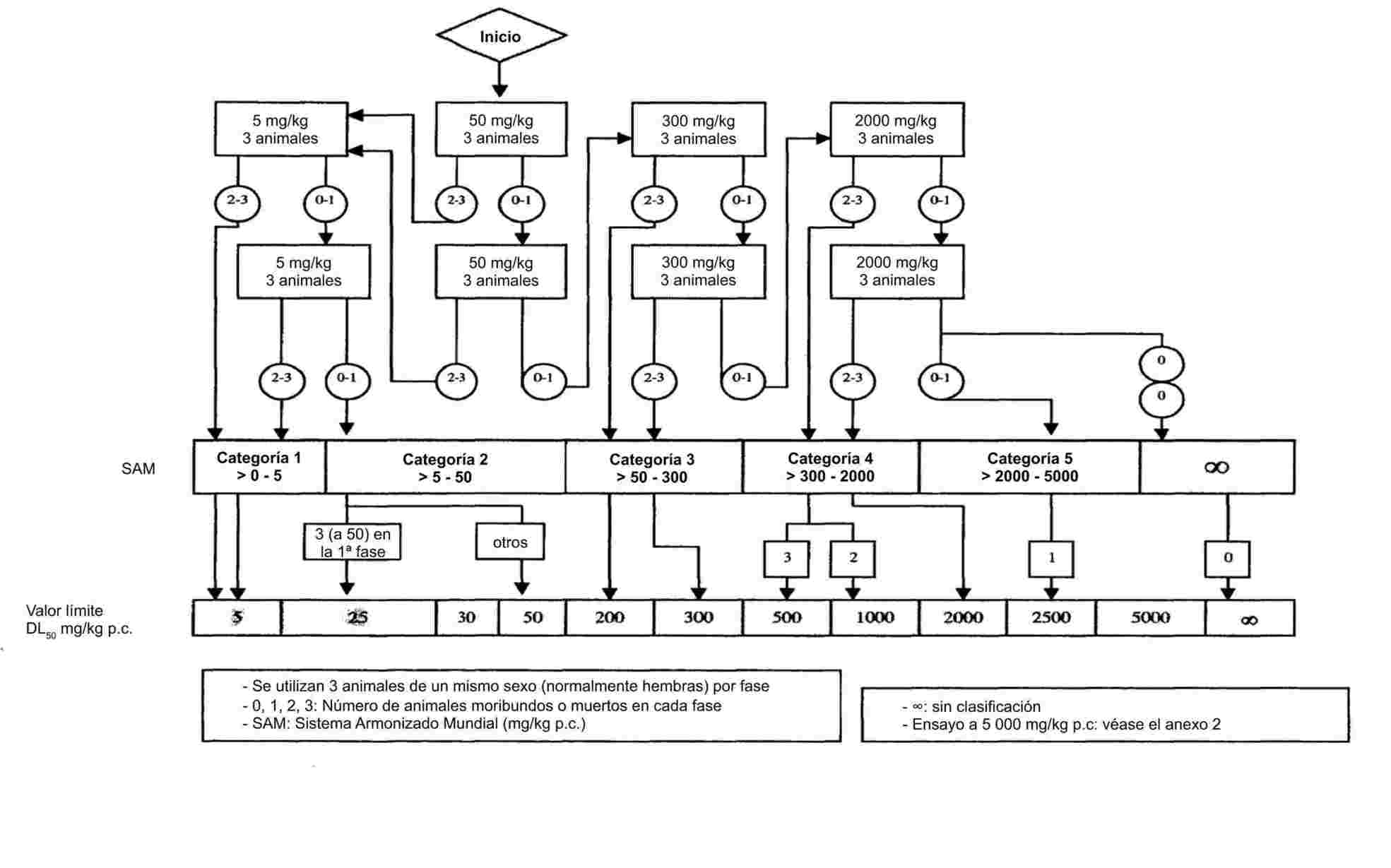
ANEXO 1C
PROCEDIMIENTO DE ENSAYO CON UNA DOSIS INICIAL DE 300 MG/KG DE PESO CORPORAL
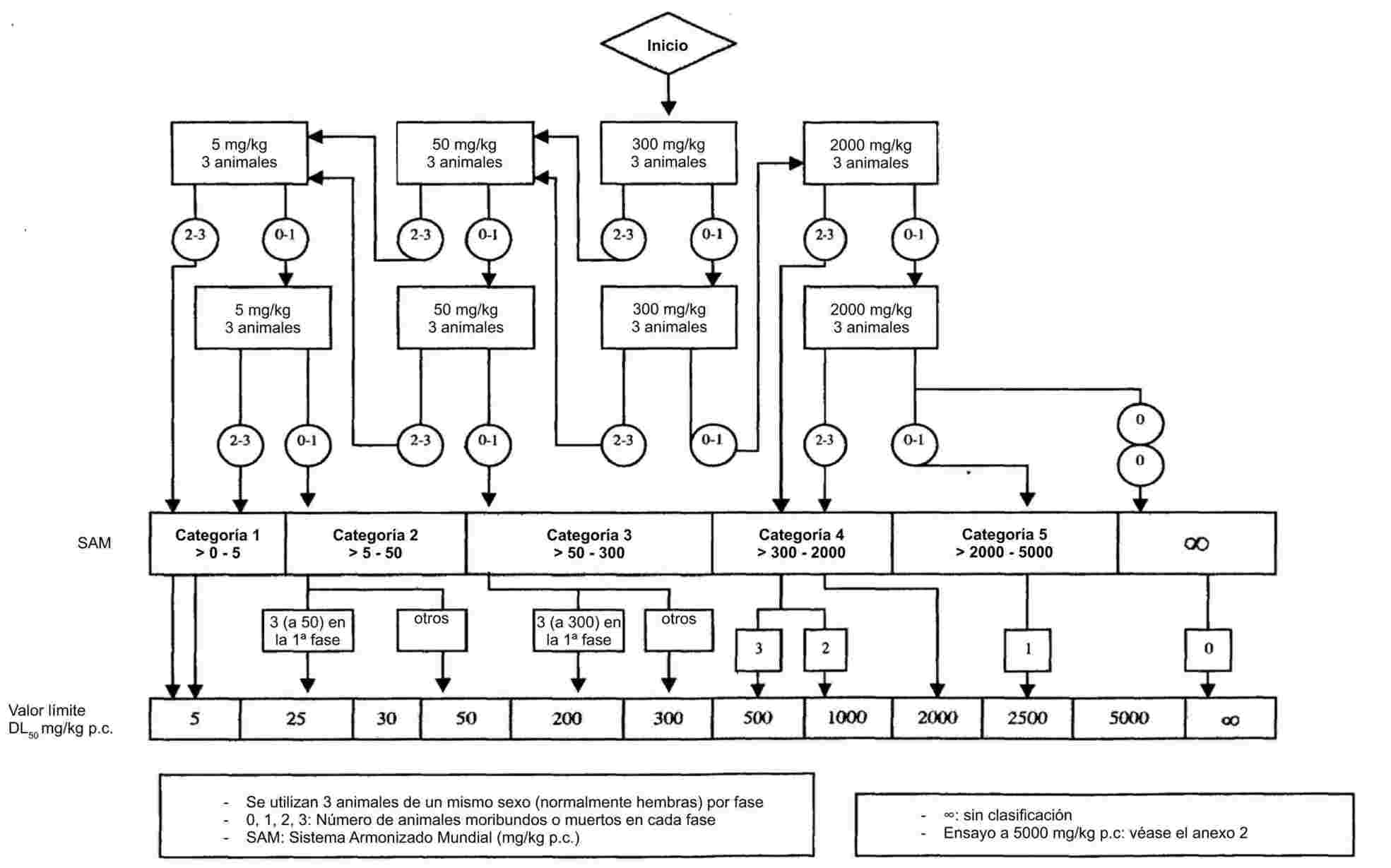
ANEXO 1 D
PROCEDIMIENTO DE ENSAYO CON UNA DOSIS INICIAL DE 2 000 MG/KG DE PESO CORPORAL

ANEXO 2
CRITERIOS PARA LA CLASIFICACIÓN DE SUSTANCIAS CON VALORES DL50 PREVISTOS SUPERIORES A LOS 2 000 MG/KG SIN NECESIDAD DE ENSAYO
Los criterios de la categoría de peligro 5 tienen por objeto permitir la identificación de sustancias que tengan un peligro de toxicidad aguda relativamente bajo pero que, en determinadas circunstancias, puedan representar un peligro para poblaciones vulnerables. Estas sustancias se prevé que tengan una DL50 oral o dérmica dentro del intervalo 2 000-5 000 mg/kg o dosis equivalentes por otras vías. Las sustancias estudiadas deben clasificarse en la categoría de peligro definida por 2 000 mg/kg < DL50 < 5 000 mg/kg (categoría 5 del SAM), en los siguientes casos:
|
a) |
cuando cualquiera de los esquemas de ensayo del anexo 1a-1d lleve a esta categoría, basándose en las cifras de mortalidad; |
|
b) |
cuando se disponga ya de datos fiables que indiquen que la DL50 se sitúa dentro del intervalo de los valores de la categoría 5; o cuando otros estudios en animales o los efectos tóxicos observados en las personas susciten inquietud grave acerca de la salud humana; |
|
c) |
mediante extrapolación, estimación o medición de datos si no está justificada la clasificación en una categoría más peligrosa, y
|
ENSAYOS A DOSIS SUPERIORES A 2 000 MG/KG
Reconociendo la necesidad de proteger el bienestar animal, se desaconseja el ensayo de la categoría 5 del Sistema Armonizado Mundial (2 000-5 000 mg/kg) con animales. Este solo debe plantearse cuando sea muy probable que sus resultados sirvan directamente para la protección de la salud humana o animal (10). No deben hacerse otros ensayos a dosis superiores.
Cuando tenga que hacerse un ensayo a 5 000 mg/kg, solo se requiere una fase (es decir, tres animales). Si el primer animal al que se administre la dosis muere, se pasará a la dosis de 2 000 mg/kg de acuerdo con los diagramas del anexo 1. Si el primer animal sobrevive, se tratará a dos animales más. Si muere solo uno de los tres animales, se prevé que la DL50 supere los 5 000 mg/kg. Si ambos animales mueren, se pasará a la dosis de 2 000 mg/kg.
ANEXO 3
Método de prueba B.1 ter: Orientaciones sobre clasificación según el plan comunitario para el período de transición hasta la plena aplicación del Sistema Armonizado Mundial (SAM) [tomado de (8)]
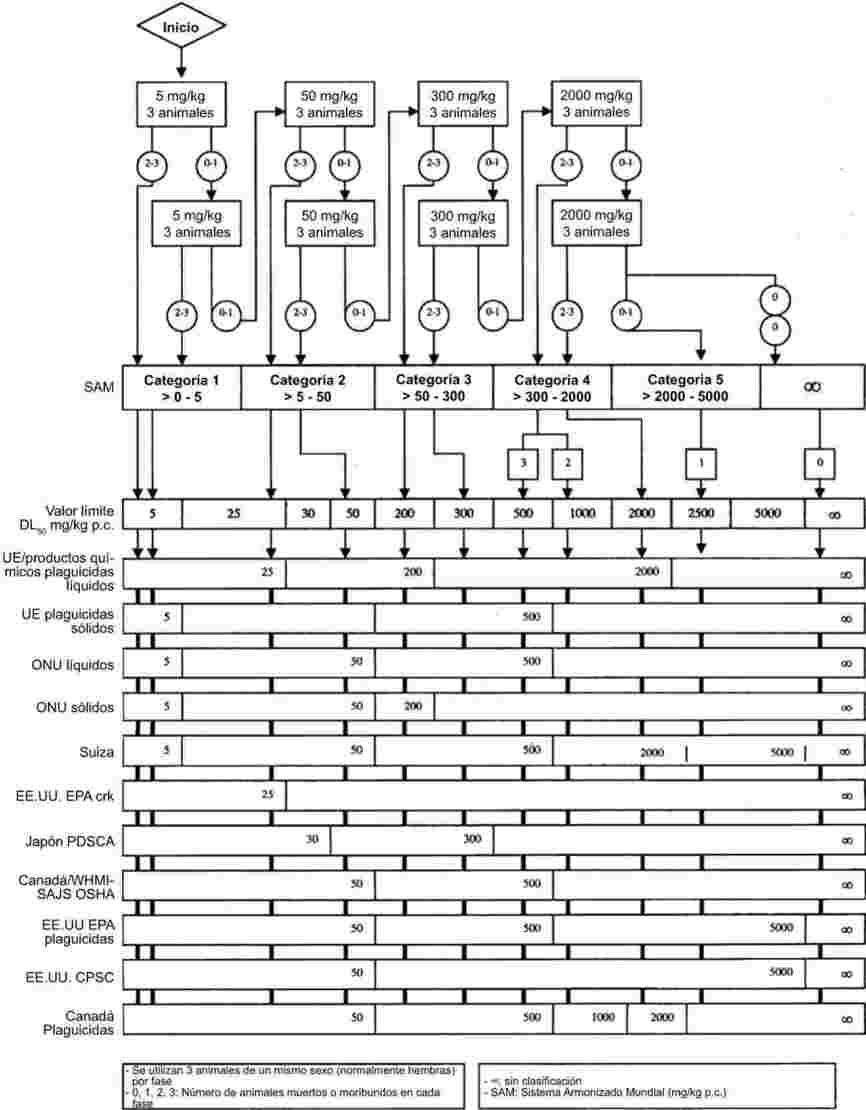


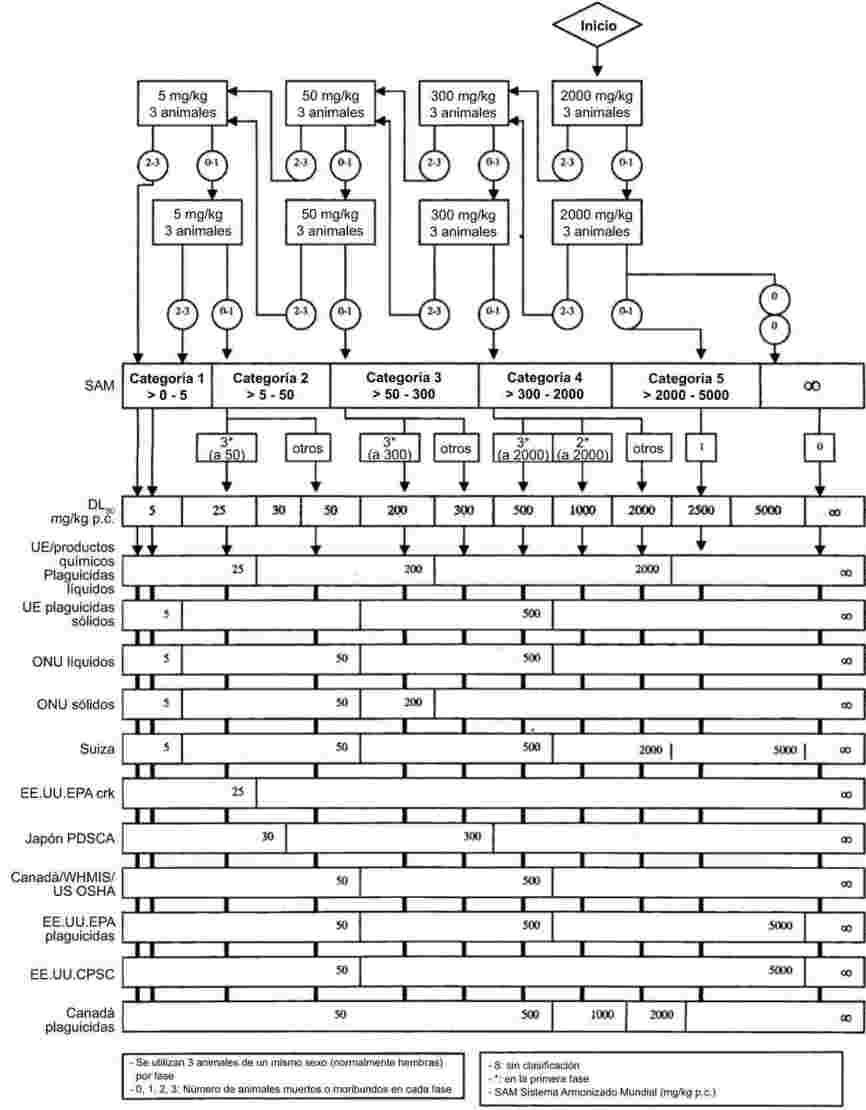
B.2. TOXICIDAD AGUDA POR INHALACIÓN
1. MÉTODO
1.1. INTRODUCCIÓN
Es útil disponer de información preliminar sobre la distribución del tamaño de partícula, la presión de vapor, el punto de fusión, el punto de ebullición, el punto de inflamación y la detonabilidad (si procede) de la sustancia.
Véase también la introducción general de la parte B (letra A).
1.2. DEFINICIONES
Véase la introducción general de la parte B (letra B).
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
Se exponen varios lotes de animales de laboratorio a la sustancia de ensayo a concentraciones diferentes durante un período determinado, utilizándose una sola concentración por lote. Se observan a continuación los efectos y la mortalidad debidos a la sustancia. Se realiza la autopsia a los animales que mueran durante el experimento así como, al final del mismo, a los que hayan sobrevivido.
Los animales que muestren signos de angustia y dolor graves y duraderos deberán sacrificarse de forma humanitaria. No se administrarán sustancias que produzcan dolor y angustia graves debido a sus propiedades corrosivas o irritantes.
1.5. CRITERIOS DE CALIDAD
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Preparación
Se mantiene a los animales en las condiciones de alojamiento y alimentación adecuadas para el experimento por lo menos durante los cinco días anteriores al mismo. Antes de comenzar el ensayo, se eligen al azar animales jóvenes sanos y se reparten entre los diferentes lotes del experimento. No es necesario someterlos a una exposición simulada, a menos que lo exija el dispositivo de exposición utilizado.
Puede ser necesario micronizar las sustancias sólidas para conseguir partículas del tamaño adecuado.
Si es preciso, se puede añadir la sustancia a un vehículo adecuado para obtener una concentración apropiada de aquélla en la atmósfera, en cuyo caso se utilizará un grupo testigo para dicho vehículo. Si para facilitar la dosificación se utiliza un vehículo u otros aditivos, no deberán producir efectos tóxicos. Pueden utilizarse resultados disponibles ya comprobados, siempre que sean apropiados.
1.6.2. Condiciones del ensayo
1.6.2.1. Animales de laboratorio
Salvo indicación contraria, la rata es la especie idónea. Es preciso utilizar cepas de laboratorio corrientes. Para cada sexo, la diferencia de peso al comienzo de la prueba entre los animales utilizados en el ensayo no debe exceder de ± 20 % del valor medio apropiado.
1.6.2.2. Número y sexo
Para cada nivel de concentración se utilizarán al menos diez roedores (5 hembras y 5 machos). Las hembras deberán ser nulíparas y no grávidas.
Nota: Se considerará la posibilidad de utilizar un número menor de animales cuando se realicen ensayos de toxicidad aguda con animales de un orden superior al de los roedores. Las dosis se seleccionarán cuidadosamente y se hará todo lo posible por no sobrepasar dosis moderadamente tóxicas. Se evitará en esos ensayos la administración de dosis letales de la sustancia de ensayo.
1.6.2.3. Concentraciones de exposición
Las concentraciones deben ser en número suficiente, al menos tres, y espaciadas adecuadamente para producir lotes que presenten variedad de efectos tóxicos y de tasas de mortalidad. Los resultados deben ser suficientes para que se pueda trazar una curva concentración/mortalidad y, si fuera posible, permitir una determinación válida de la CL50.
1.6.2.4 Prueba límite
Si una exposición de cinco machos y cinco hembras a una concentración de 20 mg por litro de un gas o 5 mg por litro de un aerosol o de partículas durante cuatro horas (o, si ello no fuera posible debido a las propiedades físicas o químicas, incluso explosivas, de la sustancia de prueba, a la concentración máxima posible) no causa la muerte de ningún animal en 14 días, se podrá considerar innecesaria la prosecución del experimento.
1.6.2.5. Duración de la exposición
La duración de la exposición debe ser de 4 horas.
1.6.2.6. Equipo experimental
Los animales deben exponerse a la sustancia por medio de un dispositivo de inhalación que produzca un flujo dinámico de, por lo menos, 12 renovaciones de aire por hora, para garantizar un contenido de oxígeno suficiente y la distribución uniforme del producto de ensayo en la atmósfera. Si se utiliza una cámara, debe estar concebida de forma que se evite, en lo posible, el amontonamiento de los animales y que su exposición máxima por inhalación de la sustancia quede asegurada. Por regla general, para garantizar la estabilidad de la atmósfera, el «volumen» total de animales de laboratorio no debe sobrepasar el 5 % del volumen de la cámara de ensayo. También se puede recurrir a un sistema de exposición oro-nasal, únicamente de cabeza o de cuerpo entero, en una cámara individual; con los dos primeros tipos de exposición se evita la absorción de la sustancia por otras vías.
1.6.2.7. Período de observación
El período de observación debe ser por lo menos de 14 días. No obstante, su extensión no debe fijarse de forma rígida, sino que debe determinarse en función de las reacciones de toxicidad, de su velocidad de aparición y de la duración del período de recuperación; por lo tanto, en caso de necesidad puede prolongarse. Son importantes el momento en que aparecen y desaparecen los síntomas de toxicidad, así como el momento de la muerte, sobre todo si en la sustancia se observa una tendencia a causar una muerte retardada.
1.6.3. Procedimiento
Poco tiempo antes de la exposición, se pesan los animales y, a continuación, se exponen a la concentración de ensayo en el equipo descrito, durante 4 horas, una vez que se haya estabilizado la concentración en la cámara. La estabilización debe ser rápida. La temperatura durante el ensayo debe mantenerse a 22 ± 3 oC. Lo ideal sería mantener la humedad relativa entre el 30 y el 70 % pero en ciertos casos (por ejemplo, en la prueba de algunos aerosoles) esto puede resultar imposible. El mantener una ligera presión negativa dentro de la cámara (aproximadamente 5 mm de agua) evitará la dispersión de la sustancia de ensayo en la zona circundante. Se privará de alimento y de agua a los animales durante la exposición. Se utilizarán sistemas apropiados para crear y controlar la atmósfera de ensayo. El sistema debe permitir crear condiciones de exposición estables lo más rápidamente posible. La cámara se diseñará y funcionará de forma que se mantenga en su interior una distribución homogénea de la atmósfera del ensayo.
Es conveniente medir o vigilar:
|
a) |
el caudal de aire (permanentemente); |
|
b) |
la concentración real de la sustancia de ensayo en la zona de respiración, a) menos tres veces durante la exposición (algunas atmósferas, por ejemplo, aerosoles a altas concentraciones, necesitarán una vigilancia más frecuente). Durante el período de exposición, la concentración no debe variar en más de ± 15 % respecto al valor medio. Sin embargo, en el caso de ciertos aerosoles puede ser difícil conseguir este control, en cuyo caso se puede aceptar una diferencia mayor. En los aerosoles, debe analizarse el tamaño de las partículas tan a menudo como sea necesario (al menos una vez por grupo de ensayo); |
|
c) |
temperatura y humedad, si es posible continuamente. |
Las observaciones tienen lugar durante y después de la exposición y se registran sistemáticamente; debe abrirse una ficha individual para cada animal. El primer día deben efectuarse con frecuencia las observaciones. Deberá hacerse un examen clínico cuidadoso al menos cada día laborable. Diariamente se harán otras observaciones complementarias actuando de manera que se reduzca la pérdida de animales para el estudio, por ejemplo, mediante autopsia o refrigeración de los animales muertos, así como aislamiento o sacrificio de los animales débiles o moribundos.
La observación incluirá las modificaciones de la piel y del pelo, los ojos, las mucosas, el aparato respiratorio, el sistema circulatorio, los sistemas nerviosos autónomo y central, la actividad somato-motriz y el comportamiento. Deben observarse con especial atención la respiración, los temblores, las convulsiones, la salivación, las diarreas, el letargo, el sueño y el coma. El momento de la muerte debe registrarse con la mayor precisión posible.
El peso de cada animal deberá determinarse semanalmente después de la exposición, así como en el momento de la muerte. Se hará la autopsia a los animales que mueran durante el experimento y, al fina] del mismo, a Jos que hayan sobrevivido. Deben registrarse, especialmente, las modificaciones de las vías respiratorias superiores e inferiores, anotando todos los cambios patológicos importantes. Si es necesario, se extraerán tejidos para un examen histopatológico.
2. RESULTADOS
Los resultados deberán inventariarse en un cuadro que indique, para cada grupo de ensayo, el número de animales al principio del mismo, el momento de la muerte de cada animal, el número de animales que presenten otros síntomas de toxicidad, la descripción de los efectos tóxicos y los resultados de la autopsia. Las variaciones de peso deben calcularse y registrarse cuando la supervivencia del animal supere un día. Los animales sacrificados por razones humanitarias debido a angustia o dolor producidos por la sustancia se registrarán como muertes debidas a la sustancia. La CL50 debe determinarse mediante un método reconocido. La evaluación de los resultados debe incluir la posible relación que existe entre la exposición de los animales a la sustancia y la incidencia y gravedad de todas las anomalías, incluidas las anomalías clínicas y de comportamiento, las lesiones macroscópicas, los cambios de peso corporal, la mortalidad y demás efectos tóxicos.
3. INFORME
3.1. INFORME DEL ENSAYO
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
especie, cepa, origen, condiciones ambientales, régimen alimentario, etc., |
|
— |
condiciones del ensayo: Descripción del dispositivo de exposición, incluidos su concepción, tipo, dimensiones, fuente de aire, sistema generador de aerosoles, método de acondicionamiento del aire y, en su caso, método de alojamiento de los animales en la cámara de ensayo. Debe describirse el equipo utilizado para medir la temperatura y la humedad, así como la concentración de los aerosoles y la granulometría de las panículas. |
Datos relativos a la exposición
Deben presentarse en forma de tabla, indicando los valores medios así como una medida de la variabilidad (por ejemplo desviación típica); a ser posible, deben incluir;
|
a) |
el caudal de aire a través del dispositivo de inhalación; |
|
b) |
temperatura y humedad del aire; |
|
c) |
concentraciones nominales (cantidad total de sustancia de ensayo introducida en el dispositivo de inhalación, dividida por el volumen de aire); |
|
d) |
en su caso, naturaleza del vehículo; |
|
e) |
concentraciones reales en la zona de respiración; |
|
f) |
el diámetro aerodinámico de la mediana de la masa (DAMM) y la desviación geométrica típica (DGT); |
|
g) |
duración de la estabilización, |
|
h) |
duración de la exposición,
|
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la introducción general de la parte B (letra D).
4. REFERENCIAS
Véase la introducción general de la parte B (letra E).
B.3. TOXICIDAD AGUDA POR VÍA CUTÁNEA
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la introducción general de la parte B (letra A).
1.2. DEFINICIONES
Véase la introducción general de la parte B (letra B).
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
Se administran dosis diferentes de la sustancia de ensayo por aplicación cutánea a varios lotes de animales de laboratorio, utilizándose una sola dosis por lote. Se observan a continuación los efectos y la mortalidad causados por la sustancia. Se realiza la autopsia a los animales que mueran durante el ensayo así como al final de esta, a los que hayan sobrevivido.
Los animales que muestren signos de dolor y angustia graves y duraderos deberán sacrificarse de forma humanitaria. No se administrarán sustancias que produzcan dolor y angustia graves debido a sus propiedades corrosivas o irritantes.
1.5. CRITERIOS DE CALIDAD
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Preparación
Se mantiene a los animales en las jaulas para el experimento en las condiciones de alojamiento y alimentación adecuadas para el ensayo por lo menos durante los 5 días anteriores. Antes de comenzar el ensayo se eligen al azar animales adultos jóvenes y sanos y se reparten entre los diferentes lotes del ensayo. Unas 24 horas antes del ensayo se esquila o se rasura el pelo de la región dorsal del tronco de los animales, evitando cualquier lesión de la piel que pueda modificar su permeabilidad. La superficie que hay que preparar para la aplicación de la sustancia no debe ser inferior al 10 % de la superficie corporal. Cuando se sometan a ensayo sólidos, que puedan pulverizarse eventualmente, la sustancia deberá humedecerse con agua o, si es preciso, con un vehículo adecuado, para asegurar un buen contacto con la piel. Si se utiliza un vehículo, se habrá de tener en cuenta su incidencia sobre la penetración de la sustancia en la piel. Las sustancias líquidas, generalmente, se aplican sin diluir.
1.6.2. Condiciones del ensayo
1.6.2.1. Animales de laboratorio
Se pueden utilizar ratas o conejos adultos. Se pueden utilizar igualmente otras especies pero, en tal caso, hay que justificar su utilización. Es preciso utilizar cepas de laboratorio corrientes. Para cada sexo, la diferencia de peso al comienzo del ensayo entre los animales utilizados no debe exceder de ± 20 % del valor medio apropiado.
1.6.2.2. Número y sexo
Para cada dosis se utilizarán al menos 5 animales del mismo sexo. Si se utilizan hembras, deberán ser nulíparas y no grávidas. Cuando se disponga de información que demuestre que un sexo es mucho más sensible, se utilizarán para la prueba animales de ese sexo.
Nota: Se considerará la posibilidad de utilizar un número menor de animales cuando se realicen ensayos de toxicidad aguda con animales de un orden superior al de los roedores. Las dosis se seleccionarán cuidadosamente y se hará todo lo posible por no sobrepasar dosis moderadamente tóxicas. Se evitará en esos ensayos la administración de dosis letales de la sustancia de estudio.
1.6.2.3. Dosis
Las dosis deben ser en número suficiente, al menos tres, y espaciadas adecuadamente para producir lotes que presenten variedad de efectos tóxicos y de mortalidad, Al elegir las dosis debe tomarse en consideración cualquier efecto irritante o corrosivo. Los resultados deben ser suficientes para que se pueda trazar una curva dosis/respuesta y, si fuera posible, permitir una determinación válida de la DL50.
1.6.2.4. Prueba límite
Puede realizarse un ensayo límite, con dosificación única de al menos 2 000 mg/kg de peso corporal, en un grupo de 5 machos y 5 hembras, utilizando los procedimientos descritos anteriormente. Si hay mortalidad debida a la sustancia, habrá que considerar la realización de un estudio completo.
1.6.2.5. Período de observación
El período de observación debe ser de, al menos, 14 días. Sin embargo, su extensión no debe fijarse de forma rígida, sino que debe determinarse en función de las reacciones de toxicidad, su velocidad de aparición y la duración del período de curación; por lo tanto, puede prolongarse en caso de necesidad. Es importante el momento en el que aparecen y en el que desaparecen los síntomas de toxicidad, así como el momento de la muerte, sobre todo si se observa en la sustancia una tendencia a causar una muerte retardada.
1.6.3. Procedimiento
Los animales deben estar alojados en jaulas individuales. La sustancia debe aplicarse sobre una superficie equivalente al 10 % de la superficie total del cuerpo. En el caso de sustancias altamente tóxicas la superficie puede ser menor, pero procurando que en toda la superficie la sustancia forme una película lo más fina y uniforme posible.
Las sustancias de ensayo deben mantenerse en contacto con la piel por medio de un apósito de gasa porosa y un esparadrapo no irritante durante 24 horas. Además, la parte tratada debe estar convenientemente cubierta para mantener en su lugar el apósito de gasa y la sustancia e impedir que los animales puedan ingerir esta última. Se pueden utilizar aparatos de contención para impedir que los animales ingieran la sustancia, pero no se recomienda una inmovilización completa.
Al término del período de aplicación de la sustancia de ensayo, esta deberá eliminarse, a ser posible, con agua o con otro procedimiento adecuado de limpieza de la piel.
Las observaciones se registrarán sistemáticamente a medida que se efectúen, abriendo una ficha individual para cada animal. El primer día las observaciones deben efectuarse con frecuencia. Deberá hacerse un examen clínico atento al menos cada día laborable. Diariamente deberán hacerse otras observaciones actuando de manera que se reduzca el número de animales perdidos para el estudio, por ejemplo mediante autopsia o refrigeración de los animales muertos, así como aislamiento o sacrificio de los animales débiles o moribundos.
Las observaciones deben incluir las modificaciones del pelo, la piel tratada, los ojos y las mucosas, el aparato respiratorio, el sistema circulatorio, los sistemas nerviosos autónomo y central, la actividad somato-motriz y el comportamiento. Deben observarse con especial atención los temblores, las convulsiones, la salivación, las diarreas, el letargo, el sueño y el coma. El momento de la muerte debe anotarse con la mayor precisión posible. Se hará la autopsia a los animales que mueran durante el experimento y, al final del mismo, a los que hayan sobrevivido. Deben registrare todas las modificaciones patológicas macroscópicas. Si es necesario se extraerán te]idos para un examen histopatológico posterior.
Valoración de la toxicidad en el otro sexo
Tras completar el estudio en uno de los sexos, se hará un estudio con al menos un grupo de 5 animales del sexo contrario, de forma que quede establecido que los animales de este sexo no son mucho más sensibles a la sustancia de ensayo. En circunstancias particulares podrá justificarse el uso de menor número de animales. Cuando se disponga de información adecuada que demuestre que los animales del sexo sometido a prueba son mucho más sensibles, podrá prescindirse del ensayo en animales del otro sexo.
2. RESULTADOS
Los resultados deberán inventariarse en un cuadro que indique, para cada lote, el número de animales al principio del ensayo, el momento de la muerte de cada animal, el número de animales que presenten otros síntomas de toxicidad, la descripción de los efectos tóxicos y los resultados de la autopsia. El peso de cada animal debe determinarse y anotarse poco antes de la aplicación de la sustancia y, después de la misma, una vez por semana y en el momento de su muerte; deben calcularse y registrarse las variaciones de peso cuando la supervivencia del animal supere un día. Los animales que se sacrifiquen de forma humanitaria debido a angustia o dolor causados por la sustancia se registrarán como muertes debidas a la sustancia. La DL50 debe determinarse mediante un método reconocido.
La evaluación de los resultados debe incluir la eventual relación que exista entre la exposición de los animales a la sustancia y la incidencia y gravedad de todas las anomalías, incluidas las anomalías clínicas y del comportamiento, las lesiones macroscópicas, los cambios de peso corporal, la mortalidad y cualquier otro efecto tóxico.
3. INFORME
3.1. INFORME DEL ENSAYO
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
especie, cepa, origen, condiciones ambientales, régimen alimentario, etc., |
|
— |
condiciones de ensayo (incluido el procedimiento de limpieza de la piel y el tipo de apósito: oclusivo o no oclusivo), |
|
— |
dosis (indicando las concentraciones y, en su caso, el vehículo), |
|
— |
sexo de los animales utilizados, |
|
— |
tabla de respuestas por sexo y por dosis (número de animales que mueren o son sacrificados durante el ensayo, número de animales con síntomas de toxicidad, número de animales expuestos), |
|
— |
momento de la muerte después de administrar la dosis, motivos y criterios para el sacrificio humanitario de los animales, |
|
— |
todas las observaciones, |
|
— |
valor de la DL50 para el sexo sometido a un estudio completo, determinado el decimocuarto día, indicando con precisión el método de cálculo, |
|
— |
intervalo de confianza estadística del 95 % para la DL50 (si es posible determinarlo), |
|
— |
curva dosis/mortalidad y pendiente de la curva si el método de cálculo lo permite, |
|
— |
resultados de la autopsia, |
|
— |
observaciones histopatológicas, |
|
— |
resultado de cualquier ensayo en el sexo contrario, |
|
— |
discusión de los resultados (prestando especial atención al efecto que puede tener sobre la DL50 calculada el sacrificio humanitario de animales durante el ensayo), |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la introducción general de la parte B (letra D).
4. REFERENCIAS
Véase la introducción general de la parte B (letra E).
B.4. TOXICIDAD AGUDA: IRRITACIÓN/CORROSIÓN CUTÁNEA
1. MÉTODO
Este método de evaluación es equivalente al método TG 404 de la OCDE (2002).
1.1 INTRODUCCIÓN
En el desarrollo la confección de este método actualizado se ha prestado atención especial a las posibles mejoras en relación con las cuestiones del bienestar de los animales y con la evaluación de toda la información existente sobre la sustancia analizada, para no someter a los animales de laboratorio a pruebas innecesarias. En este método se incluye la recomendación de que, antes de llevar a cabo la prueba in vivo que se describe para evaluar la corrosión/irritación de la sustancia, hay que analizar la carga de la prueba que aportan los datos relevantes existentes. Cuando los datos disponibles sean insuficientes, se podrán obtener mediante la aplicación de secuencias de pruebas (1). La estrategia de evaluación recomendada incluye la realización de pruebas in vitro validadas y aceptadas, y se recoge en un anexo de este método. Además, cuando proceda, se recomienda la aplicación sucesiva, no simultánea, de tres aplicaciones de ensayo a los animales en el ensayo in vivo inicial.
Por interés de la ciencia y del bienestar de los animales no se realizarán pruebas in vivo hasta que se hayan evaluado todos los datos relevantes sobre el potencial de corrosión/irritación cutánea de la sustancia en un análisis de la carga de la prueba. Dichos datos incluirán los obtenidos en estudios existentes realizados con seres humanos o con animales de laboratorio, los demostrativos de corrosión/irritación por parte de una o más sustancias relacionadas estructuralmente o por una mezcla de las mismas, los que demuestran la elevada acidez o alcalinidad de la sustancia (2) (3) y los obtenidos en pruebas in vitro o ex vivo validadas y aceptadas (4)(5)(5a). Este análisis debe reducir la necesidad de realizar pruebas in vivo de la corrosión/irritación cutánea de aquellas sustancias de las que ya se disponga de datos de otros estudios efectuados sobre esos dos criterios de valoración.
En un anexo de este método se recoge el orden recomendado para efectuar los ensayos, incluidos los ensayos in vitro o ex vivo validados y aceptados sobre corrosión e irritación. Se trata de una estrategia desarrollada por un taller de la OCDE, cuyos participantes la recomendaron de manera unánime (6), y ha sido adoptada como estrategia de evaluación por el sistema armonizado global para la clasificación de sustancias químicas [Globally Harmonised System for the Classification of Chemical Substances GHS)] (7). Aunque dicha estrategia de evaluación secuencial no forma parte integrante del método de evaluación B.4, se recomienda seguirla antes de llevar a cabo ensayos in vivo. Es el nuevo enfoque de evaluación escalonada recomendado para obtener datos científicamente garantizados sobre la corrosión/irritación de la sustancia. Para sustancias existentes con datos insuficientes sobre su corrosión/irritación cutánea se utilizará la estrategia para obtener los datos que falten. Será necesario justificar el empleo de una estrategia o procedimiento de evaluación diferente, o la decisión de no utilizar un procedimiento escalonado.
Si no fuera posible determinar el poder de corrosión o irritación mediante un análisis de la carga de la prueba compatible con la estrategia secuencial de evaluación, se considerará la posibilidad de realizar un ensayo in vivo (véase el anexo).
1.2 DEFINICIONES
Irritación cutánea: lesión reversible de la piel tras la aplicación de una sustancia de ensayo durante 4 horas.
Corrosión cutánea: lesión irreversible de la piel; en concreto, necrosis visible de la epidermis y la dermis, que se produce tras la aplicación de una sustancia de ensayo durante cuatro horas. Las reacciones corrosivas se caracterizan por úlceras, hemorragias, costras sanguinolentas y, al cabo de 14 días de observación, cambio de coloración por palidez de la piel, zonas completas de alopecia y cicatrices. Para evaluar las lesiones cuestionables se utilizará la histopatología.
1.3 PRINCIPIO DEL MÉTODO DE EVALUACIÓN
La sustancia analizada se aplica en una sola dosis a la piel de un animal de experimentación; las zonas de la piel no tratadas de dicho animal sirven de control. Se comprueba y puntúa el grado de irritación/corrosión a intervalos determinados, y luego se describen para la completa evaluación de los efectos. La duración del estudio ha de ser suficiente para evaluar la reversibilidad o irreversibilidad de los efectos observados.
Los animales que muestren signos continuados de deterioro o dolor graves en cualquier fase del ensayo deben ser sacrificados, con la consiguiente evaluación de la sustancia. Los criterios para sacrificar a los animales moribundos y que sufren intensamente se recogen en la referencia (8).
1.4 DESCRIPCIÓN DEL MÉTODO
1.4.1 Preparación para el ensayo in vivo
1.4.1.1 Selección de las especies de animales
El animal de laboratorio que se prefiere es el conejo albino; se utilizan adultos jóvenes sanos. Si se utilizan otras especies será necesario justificarlo.
1.4.1.2 Preparación de los animales
Aproximadamente 24 horas antes del ensayo se afeitará el pelo con cuidado en la zona dorsal del tronco de los animales. Se tendrá cuidado en no dañar la piel, y solo se utilizarán animales con piel sana e intacta.
Algunas razas de conejo tienen densas placas de pelo que son más llamativas en determinadas épocas del año. Dichas zonas no deben utilizarse para las evaluaciones.
1.4.1.3 Condiciones de alojamiento y alimentación
Los animales deben ser alojados de manera individual. La temperatura de los animalarios debe ser de 20 oC (± 3 oC) para los conejos. Aunque la humedad relativa debe ser del 30 % como mínimo y preferiblemente no superar el 70 %, excepto durante la limpieza del animalario, el objetivo debe ser el 50-60 %. La iluminación será artificial, con 12 horas de luz y 12 de oscuridad. Para la alimentación se podrán utilizar dietas de laboratorio convencionales, con suministro ilimitado de agua para beber.
1.4.2 Procedimiento del ensayo
1.4.2.1 Aplicación de la sustancia estudiada
La sustancia en estudio debe aplicarse a una pequeña zona de piel (de unos 6 cm2), que se cubrirá con una gasa y se sujetará con esparadrapo no irritante. Cuando no sea posible la aplicación directa (por ejemplo, líquidos o algunas pastas), la sustancia en estudio se aplicará primero a la gasa, y a continuación a la piel. La gasa debe quedar en contacto suave con la piel mediante el correspondiente vendaje semioclusivo durante el período de exposición. Si se aplica la sustancia a la gasa, esta debe sujetarse a la piel de forma que el contacto sea bueno y la sustancia se distribuya de manera uniforme por la piel. Se evitará que el animal tenga acceso a la gasa y que pueda comérsela o inhalar la sustancia.
Las sustancias líquidas suelen utilizarse sin diluir para su análisis. Para evaluar sólidos (que se pueden pulverizar si se considera necesario) la sustancia se humedecerá con el mínimo de agua posible (o con otro vehículo adecuado, si se considera necesario), para garantizar un buen contacto con la piel. Si se utilizan vehículos distintos del agua, la posible influencia del vehículo sobre la irritación cutánea que produce la sustancia en estudio ha de ser mínima o ' nula.
Finalizado el período de exposición, que normalmente es de 4 horas, se eliminarán los residuos de la sustancia cuando ello sea posible, con agua o con un disolvente adecuado que no altere la respuesta producida ni la integridad de la epidermis.
1.4.2.2 Nivel de dosis
Se aplicará una dosis de 0,5 ml de líquido o de 0,5 de sólido o pasta en el punto de aplicación.
1.4.2.3 Ensayo inicial (ensayo de irritación/corrosión cutánea in vivo con un animal)
Se recomienda encarecidamente realizar el ensayo in vivo inicialmente con un solo animal, especialmente cuando se sospecha que la sustancia es corrosiva.
Esto coincide con la estrategia de evaluación secuencial (véase el anexo l) diando el análisis de la carga de la prueba determine que una sustancia es corrosiva no se realizarán más ensayos con animales. En la mayor parte de los casos en que se sospecha que una sustancia es corrosiva no es necesario realizar más ensayos in vivo. Pero cuando se crea necesario disponer de datos adicionales por no ser suficientes los obtenidos, pueden hacerse otros ensayos con animales con arreglo a los siguiente: Se hará un máximo de tres aplicaciones secuenciales al animal. La primera se retira a los tres minutos. Si no se observa reacción cutánea grave se aplica una segunda, que se retira al cabo de una hora. Si en este momento las observaciones indican que desde un punto humanitario se puede aumentar la exposición hasta cuatro horas, se hace una tercera aplicación que se retira al cabo de cuatro horas, y se gradúa la respuesta.
Si en alguna de las exposiciones secuenciales se observa un efecto corrosivo se suspende la prueba inmediatamente. Si una vez retirada la última aplicación no se observa efecto corrosivo, se somete a observación al animal durante 14 días, a menos que aparezca corrosión antes.
Si no está previsto que la sustancia analizada sea corrosiva pero sí irritante, se hará una sola aplicación a un solo animal durante cuatro horas.
1.4.2.4 Ensayo de confirmación (ensayo de irritación cutánea in vivo con animales adicionales)
Si en el ensayo inicial no se observa efecto corrosivo se confirmará la respuesta irritante o negativa con un máximo de dos animales más, cada uno con una aplicación, durante un período de exposición de cuatro horas. Si se observa efecto irritante en el ensayo inicial el ensayo de confirmación debe hacerse de forma secuencial, o exponiendo a otros dos animales a la vez. En el caso excepcional de que no se realice el ensayo inicial se podrán tratar dos o tres animales con una sola aplicación, que se retirará al cabo de cuatro horas. Cuando se utilicen dos animales no será necesario realizar más ensayos si ambos muestran la misma respuesta. En caso contrario se analizará el tercer animal. Si la respuesta es equívoca puede ser necesario evaluar a más animales.
1.4.2.5 Período de observación
La duración del período de observación debe ser suficiente para evaluar por completo la reversibilidad de los efectos observados. No obstante se dará por finalizado el experimento si en cualquier momento el animal presenta signos continuados de dolor o malestar graves. Para determinar la reversibilidad de los efectos los animales serán sometidos a observación durante 14 días a partir de la retirada de las aplicaciones. Si se observa la irreversibilidad antes de 14 días el experimento debe concluir en ese momento.
1.4.2.6 Observaciones clínicas y graduación de las reacciones cutáneas
En todos los animales se examinarán los signos de eritema y edema, puntuando las repuestas al cabo de 60 minutos, 24, 48 y 72 horas de la retirada de la aplicación. En el caso de ensayo inicial con un solo animal la zona de ensayo también se examina inmediatamente después de retirar la aplicación. Las reacciones cutáneas se gradúan y registran conforme a los grados de la siguiente tabla. Si al cabo de 72 horas hay una lesión que no se puede clasificar como irritación o corrosión, quizá sea necesario observarla hasta el día 14 para determinar la reversibilidad de los efectos. Además de observar la irritación se realizará una descripción completa de todos los efectos tóxicos, como la destrucción de la grasa de la piel, y de cualquier efecto adverso sistémico (por ejemplo, los efectos sobre los signos clínicos de toxicidad y el peso corporal), que se registrarán. Puede ser necesaria una evaluación histopatológica para aclarar respuestas equívocas.
La graduación de las respuestas cutáneas es subjetiva necesariamente. Para armonizar tal graduación y ayudar a los laboratorios y a quienes efectúan e interpretan las observaciones, el personal encargado recibirá formación adecuada sobre el sistema de puntuación utilizado (véase la tabla siguiente). Puede ser útil disponer de una guía ilustrada para graduar la irritación cutánea y otras lesiones (9). La graduación de las respuestas cutáneas debe hacerse en condiciones ciegas.
2. DATOS
2.1 PRESENTACIÓN DE LOS RESULTADOS
Los resultados del estudio se resumirán en tablas; el informe final debe cubrir todos los apartados enumerados en la sección 3.1.
2.2 EVALUACIÓN DE LOS RESULTADOS
Las puntuaciones de la irritación cutánea deben evaluarse junto con la naturaleza y la intensidad de las lesiones, y el hecho de si son reversibles o no. Las puntuaciones individuales no constituyen una referencia absoluta de las propiedades irritativas del material, pues también se evalúan otros efectos del material evaluado. En su lugar las puntuaciones individuales deben considerarse como valores de referencia, que han de ser evaluadas en combinación con todas las demás observaciones del estudio.
Para evaluar las respuestas irritativas hay que tener en cuenta la reversibilidad de las lesiones cutáneas. Si se producen respuestas como alopecia (zona limitada), hiperqueratosis, hiperplasia y descamación y persisten al final del período de observación de 14 días se considerará que la sustancia analizada es un irritante.
3. NOTIFICACIÓN
3.1 INFORME DEL ENSAYO
El informe del ensayo incluirá la siguiente información:
|
|
Justificación del ensayo in vivo: análisis de la carga de los ensayos realizados anteriormente, incluidos los resultados de la estrategia de evaluación secuencial.
|
|
|
Sustancia analizada:
|
|
|
Vehículo:
|
|
|
Animales de experimentación:
|
|
|
Condiciones del análisis:
|
|
|
Resultados:
|
4. REFERENCIAS
|
(1) |
Barratt, M.D., Castell, J.V., Chamberlain, M., Combes, R.D., Dearden, J.C., Fentem, J.H., Gerner, I., Giuliani, A., Gray, T.J.B., Livingston, D.J., Provan, W.M., Rutten, F.A.J.J.L., Verhaar, H.J.M., Zbinden, P. (1995) The Integrated Use of Alternative Approaches for Predicting Toxic Hazard. ECVAM Workshop Report 8. ATLA 23, 410-429. |
|
(2) |
Young, J.R., How, M.J., Walker, A.P., Worth W.M.H. (1988) Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals. Toxicol. In Vitro, 2, 19-26. |
|
(3) |
Worth, A.P., Fentem, J.H., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Liebsch, M. (1998) Evaluation of the proposed OECD Testing Strategy for skin corrosion. ATLA 26, 709-720. |
|
(4) |
ECETOC (1990) Monograph No. 15, «Skin Irritation», European Chemical Industry, Ecology and Toxicology Centre, Bruselas. |
|
(5) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, 483-524. |
|
(5a) |
Testing Method B.40 Skin Corrosion. |
|
(6) |
OECD (1996) OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Pronunciado en Solna, Suecia, 22-24 de enero de 1996 (http://wwwl.oecd.org/ehs/test/background.htm). |
|
(7) |
OECD (1998) Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, noviembre de 1998 (http://wwwl.oecd.org/ehs/Class/HCL6.htm). |
|
(8) |
OECD (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. OECD Environmental Health and Safety Publications. Series on Testing and Assessment No. 19 (http://wwwl.oecd.org/ehs/test/monos.htm). |
|
(9) |
EPA (1990). Atlas of Dermal Lesions, (20T-2004). United States Environmental Protection Agency, Office of Pesticides and Toxic Substances, Washington, DC, agosto de 1990. [Puede solicitarse a la Secretaría de la OCDE]. |
Tabla I
GRADUACIÓN DE LAS REACCIONES CUTÁNEAS
Eritema y formación de escaras
|
Sin eritema … |
0 |
|
Eritema muy leve (apenas perceptible) … |
1 |
|
Eritema bien definido … |
2 |
|
Eritema moderado a intenso … |
3 |
|
Eritema intenso (enrojecimiento color carne) o formación de escaras que impide graduarlo … |
4 |
Máximo posible 4
Formación de edema
|
Sin edema … |
0 |
|
Edema muy leve (apenas perceptible) … |
1 |
|
Edema ligero (los bordes de la zona están bien definidos por elevaciones concretas) … |
2 |
|
Edema moderado (elevación de 1 mm aproximadamente) … |
3 |
|
Edema intenso (elevación superior a 1 mm y extensión que sobrepasa la zona de exposición) … |
4 |
Máximo posible: 4
Puede ser necesaria una evaluación histopatológica para aclarar respuestas equívocas.
ANEXO
Estrategia de evaluación secuencial de la irritación y la corrosión cutáneas
CONSIDERACIONES GENERALES
Aunque la presente estrategia de evaluación secuencial no forma parte integrante del método de evaluación B.4., sí expresa el enfoque recomendado para la determinación de las características de irritación/corrosión cutáneas. Este enfoque representa la práctica óptima y una piedra angular desde el punto de vista ético para el análisis in vivo de la irritación/corrosión cutáneas. El método de evaluación proporciona instrucciones para la realización del ensayo in vivo, y resume los factores que se han de abordar antes de ponerla en marcha. La estrategia proporciona un enfoque para la evaluación de los datos existentes sobre las propiedades de irritación/corrosión cutáneas de las sustancias analizadas, y un enfoque por partes para generar datos relevantes sobre sustancias que necesitan estudios adicionales o que no han sido estudiadas. También recomienda la realización de ensayos in vitro o ex vivo validados y aceptados para la corrosión/irritación cutáneas en circunstancias concretas.
Es importante evitar el uso innecesario de animales y reducir al mínimo los ensayos que con toda probabilidad producen respuestas graves en los animales, por su bienestar y por motivos científicos. Antes de considerar la realización de ensayos in vivo hay que evaluar toda la información sobre una sustancia en lo que respecta a su posible poder corrosivo/irritativo cutáneo. Puede que existan pruebas suficientes para clasificar el potencial corrosivo o irritativo dérmico de una sustancia analizada, sin necesidad de realizar ensayos con animales de laboratorio. Por eso el uso del análisis de la carga de la prueba y de una estrategia de evaluación secuencial reducirá al mínimo la necesidad de realizar ensayos in vivo, especialmente si es probable que la sustancia produzca reacciones graves.
Se recomienda utilizar un análisis de la carga de la prueba para evaluar la información existente sobre el potencial de irritación y corrosión cutáneas producidas por las sustancias, que permitirá determinar si la realización de estudios adicionales, a parte de los cutáneos in vivo, ayudaría a caracterizar dicho potencial. Cuando sean necesarios otros estudios, se recomienda utilizar la estrategia de evaluación secuencial para obtener los datos experimentales relevantes. Para sustancias no evaluadas anteriormente, se utilizará la estrategia de evaluación secuencial para obtener los datos necesarios para evaluar su potencial corrosivo/irritativo cutáneo. La estrategia de evaluación que se describe en este Anexo fue desarrollada por un taller de la OCDE (1) y posteriormente confirmada y ampliada por el sistema armonizado integrado de clasificación de peligros para la salud humana y efectos ambientales de las sustancias químicas (Harmonised Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances), y fue aprobada enla reunión XXVIII del Comité Conjunto de Productos Químicos (Chemicals Committee and the Working Party on Chemicals) en noviembre de 1998 (2).
DESCRIPCIÓN DE LA ESTRATEGIA DE EVALUACIÓN
Antes de llevar a cabo los ensayos que forman parte de la estrategia de evaluación secuencial (véase la figura), se evaluará toda la información disponible, para determinar la necesidad de practicar estudios cutáneos in vivo. Aunque se puede obtener información significativa a partir de la evaluación de parámetros aislados (por ejemplo, un pH extremo), hay que considerar la totalidad de la información existente. Se evaluarán todos los datos relevantes sobre los efectos de la sustancia en cuestión y de sus análogos para tomar una decisión basada en la carga de la prueba, y se presentará la justificación de dicha decisión, Se hará especial hincapié en los datos existentes sobre la sustancia en seres humanos y animales, seguidos por el resultado de los ensayos in vitro o ex vivo. Siempre que sea posible se evitará realizar estudios in vivo con sustancias corrosivas. Los factores que se tienen en cuenta en la estrategia de evaluación son:
Evaluación de datos existentes en seres humanos y en animales (paso I): En primer lugar se tendrán en cuenta los datos existentes sobre seres humanos, por ejemplo, estudios clínicos u ocupacionales e informes de casos, y los datos de ensayos realizados con animales, por ejemplo, de estudios sobre toxicidad de la exposición cutánea única o reiterada, pues proporcionan información directamente relacionada con los efectos producidos en la piel. No es necesario someter a estudios in vivo a las sustancias irritantes o corrosivas conocidas, ni a las que claramente no son lo uno ni lo otro.
Análisis de las relaciones entre las estructuras (SAR) (paso 2). Hay que tener en cuenta los resultados de los análisis de sustancias relacionadas desde el punto de vista estructural, si es que existen. Cuando se dispone de suficientes datos en seres humanos o animales sobre sustancias relacionadas desde el punto de vista estructural o sobre mezclas de dichas sustancias que indique su potencial corrosivo/irritativo cutáneo, se puede presuponer que la sustancia analizada producirá las mismas respuestas. En estos casos puede que no sea necesario evaluar la sustancia en cuestión. Los datos negativos de los estudios sobre sustancias relacionadas desde el punto de vista estructural o sobre mezclas de estas no constituyen demostración suficiente de que una sustancia no será corrosiva ni irritante en la estrategia de evaluación secuencial. Para identificar el potencial de corrosión e irritación dérmicas hay que utilizar enfoques SAR validados y aceptados.
Propiedades fisicoquímicas y reactividad química (paso 3). Las sustancias con pH extremos, como < 2,0 y > 11,5 pueden tener potentes efectos locales. Si el pH extremo es la base para identificar si una sustancia es corrosiva para la piel, entonces también se tendrá en cuenta la reserva de ácido/álcali (o capacidad amortiguadora) (3) (4). Si la capacidad amortiguadora sugiere la posibilidad de que una sustancia no sea corrosiva para la piel se realizarán mas ensayos para confirmarlo, preferiblemente utilizando un ensayo in vitro o ex vivo validado y aceptado (véanse los pasos 5 y 6).
Toxicidad cutánea (paso 4). Si se demuestra que una sustancia química es muy tóxica por vía cutánea, puede que no sea procedente realizar un estudio sobre irritación/corrosión cutáneas in vivo, habida cuenta de que la cantidad de sustancia que se suele aplicar puede superar una dosis muy tóxica, lo que puede matar a los animales o infligirles graves sufrimientos. Además, cuando ya se hayan realizado estudios de toxicidad cutánea en conejos albinos hasta la dosis límites de 2 000 mg/kg de peso corporal o más sin que se haya observado irritación ni corrosión cutánea, puede que no sea necesario practicar más ensayos de irritación/corrosión cutáneas. Al evaluar la toxicidad cutánea observada en estudios realizados anteriormente hay que tener en cuenta algunas consideraciones. Por ejemplo, puede que la información publicada sobre las lesiones cutáneas no esté completa. Puede que los ensayos y las observaciones se hayan efectuado en una especie distinta del conejo, y la sensibilidad de las respuestas puede variar mucho de unas especies a otras. También puede que la forma de la sustancia analizada que se aplicó a los animales no sea la adecuada para la evaluación de la irritación/corrosión cutáneas (por ejemplo, disolución de las sustancias para los estudios sobre toxicidad cutánea) (5). Pero cuando se hayan realizado estudios sobre toxicidad cutánea bien diseñados y ejecutados en conejos, los hallazgos negativos pueden constituir prueba suficiente de que la sustancia no es corrosiva ni irritativa.
Resultados de los ensayos in vitro o ex vivo (pasos 5 y 6). Cuando se hayan demostrado las propiedades corrosivas o irritantes intensas de una sustancia en un ensayo in vitro o ex vivo validado y aceptado (6) (7) diseñado para la evaluación de estos efectos concretos, no será necesario evaluarla en animales. Se puede dar por supuesto que dichas sustancias producirán similares efectos intensos in vivo.
Ensayo in vivo en conejos (pasos 7 y 8). Si a partir de la carga de la prueba se toma la decisión de realizar un ensayo in vivo, se empezará con un ensayo inicial con un solo animal. Si los resultados indican que la sustancia es corrosiva para la piel no se realizarán más ensayos. Si en el ensayo inicial no se observa efecto corrosivo se confirmará la respuesta irritante o negativa con un máximo de dos animales más, durante un período de exposición de cuatro horas. Si se observa un efecto irritante en el ensayo inicial el ensayo de confirmación debe hacerse de forma secuencial, o exponiendo a otros dos animales a la vez.
REFERENCIAS
|
(1) |
OECD (1996). Test Guidelines Programme: Final Report on the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Held on Solna, Sweden, 22-24 January 1996 (http://wwwl.oecd.org/ehs/tests/background.hrm. |
|
(2) |
OECD (1998). Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, November 1998 (http://wwwl.oecd.org/ehs/Class/HCL6.htm). |
|
(3) |
Worth, A.P., Fentem J.H., Balls M., Botham P.A., Curren R.D., Earl L.K., Esdaile D.J., Liebsch M. (1998). An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosión. ATLA 26, 709-720. |
|
(4) |
Young, J.R., How, M.J., Walker, A.P., Worth, W.M.H. (1988). Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substances, Without Testing on Animals. Toxic In Vitro, 2(1), 19-26. |
|
(5) |
Patil, S.M., Patrick, E., Maibach, H.I. (1996) Animal, Human, and In Vitro Test Methods for Predicting Skin Irritation, in: Francis N. Marzulli and Howard I. Maibach (editors): Dermatotoxicology. Fifth Edition ISBN 1-56032-356-6, Chapter 31, 411-436. |
|
(6) |
Testing Method B.40. |
|
(7) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, 483-524. |
Figura
ESTRATEGIA DE EVALUACIÓN DE LA IRRITACIÓN/CORROSIÓN CUTÁNEA

B.5. TOXICIDAD AGUDA: IRRITACIÓN/CORROSIÓN OCULAR
1. MÉTODO
Este método de evaluación es equivalente al método TG 405 de la OCDE (2002)
1.1 INTRODUCCIÓN
En el desarrollo de este método actualizado se ha prestado atención especial a las posibles mejoras mediante la evaluación de toda la información existente sobre la sustancia analizada, para no someter a los animales de laboratorio a pruebas innecesarias y responder así a la preocupación por el bienestar de los animales. En este método se incluye la recomendación de que antes de llevar a cabo el ensayo in vivo que se describe para evaluar la corrosión/irritación ocular aguda de la sustancia, hay que analizar la carga de la prueba (1) que aportan los datos relevantes existentes. Si los datos disponibles son insuficientes se recomienda obtenerlos mediante la aplicación de secuencias de ensayos (2) (3). La estrategia de evaluación recomendada incluye la realización de ensayos in vitro validados y aceptados, y se recoge en un anexo de este método. Además se recomienda utilizar un ensayo de irritación/corrosión cutánea in vivo para predecir la corrosión ocular ante de realizar un ensayo ocular in vivo.
Por interés de la ciencia y del bienestar de los animales no se realizarán ensayos in vivo hasta que se hayan evaluado todos los datos relevantes sobre el potencial de corrosión/irritación ocular de la sustancia en un análisis de la carga de la prueba. Dichos datos incluirán los obtenidos en estudios existentes realizados con seres humanos o con animales de laboratorio, la demostración de corrosión/irritación por parte de una o más sustancias relacionadas estructuralmente o por una mezcla de las mismas, los que demuestran la elevada acidez o alcalinidad de la sustancia (4) (5) y los obtenidos en ensayos de corrosión e irritación cutánea in vitro o ex vivo validados y aceptados (6) (6a). Los estudios pueden haber sido realizado antes del análisis de la carga de la prueba, o a consecuencia de la misma.
Para ciertas sustancias dicho análisis puede indicar la necesidad de realizar estudios in vivo del potencial de corrosión/irritación ocular de la sustancia. En todos esos casos antes de considerar el uso de un ensayo ocular in vivo es preferible realizar primero un estudio de los efectos cutáneos in vivo de la sustancia, evaluándolo de conformidad con el método de evaluación B.4 (7). La aplicación del análisis de la carga de la prueba y la estrategia de evaluación secuencial debe reducir la necesidad de realizar ensayos in vivo de la corrosión/irritación oculares de aquellas sustancias de las que ya se disponga de datos de otros estudios. Si no fuera posible determinar el potencial de corrosión o irritación ocular con la estrategia de evaluación secuencial, ni siquiera tras realizar un estudio in vivo de la corrosión y la irritación cutáneas, se podrá llevar a cabo una evaluación de la corrosión/irritación oculares in vivo.
En un anexo de este método se recoge el orden recomendado para efectuar los ensayos, incluidos los ensayos in vitro o ex vivo validados y aceptados sobre corrosión e irritación. Se trata de una estrategia desarrollada por un taller de la OCDE, cuyos participantes la recomendaron de manera unánime (8), y ha sido adoptada como estrategia de evaluación recomendada por el sistema armonizado global para la clasificación de sustancias químicas [Globally Harmonised System for the Classification of Chemical Substances (CHS)] (9). Aunque dicha estrategia de evaluación secuencial no forma parte integrante del método de evaluación B.4, se recomienda seguirla antes de llevar a cabo ensayos in vivo. Para sustancia nuevas, es el nuevo enfoque de evaluación escalonada recomendado para obtener datos científicamente garantizados sobre la corrosión/irritación de la sustancia. Para sustancias existentes con datos insuficientes sobre su corrosión/irritación cutánea y ocular se utilizará la estrategia para obtener los datos que falten. Será necesario justificar el empleo de una estrategia o procedimiento de evaluación diferente, o la decisión de no utilizar un procedimiento escalonado.
1.2 DEFINICIONES
Irritación ocular: alteraciones en el ojo tras la aplicación de una sustancia de ensayo en la superficie anterior del ojo, completamente reversibles en los 21 días siguientes a la aplicación.
Corrosión ocular: lesión tisular en el ojo o grave reducción física de la vista, tras la aplicación de una sustancia de ensayo en la superficie anterior del ojo, que no son completamente reversibles en los 21 días siguientes a la aplicación
1.3 PRINCIPIO DEL MÉTODO DE EVALUACIÓN
La sustancia analizada se aplica en una sola dosis a uno de los ojos de un animal de experimentación; el ojo no tratado actúa como control. Se evalúa el grado de irritación/corrosión ocular a intervalos determinados, mediante la puntuación de las lesiones de la conjuntiva, la córnea y el iris. También se describen otros efectos en el ojo y los efectos sistémicos adversos, para proporcionar una evaluación completa de los efectos. La duración del estudio ha de ser suficiente para evaluar la reversibilidad o irreversibilidad de los efectos observados.
Los animales que muestren signos continuados de deterioro o dolor graves en cualquier fase del ensayo deben ser sacrificados, con la consiguiente evaluación de la sustancia. Los criterios para sacrificar a los animales moribundos y que sufren intensamente se recogen en la referencia (10).
1.4 DESCRIPCIÓN DEL MÉTODO DE EVALUACIÓN
1.4.1 Preparación del ensayo in vivo
1.4.1.1 Selección de las especies
El conejo albino es el animal de laboratorio preferido; se emplean individuos adultos jóvenes y sanos. Si se utilizan otras especies será necesario justificarlo.
1.4.1.2 Preparación de los animales
En las 24 horas previas al inicio del ensayo se examinarán los dos ojos de los animales de experimentación provisionalmente seleccionados para la misma. No se utilizarán animales que muestren irritación ocular, defectos oculares o lesión corneal preexistente.
1.4.1.3 Condiciones de alojamiento y alimentación
Los animales deben ser alojados de manera individual. La temperatura de los animalarios debe ser de 20 oC (± 3 oC) para los conejos. Aunque la humedad relativa debe ser del 30 % como mínimo y preferiblemente no superar el 70 %, excepto durante la limpieza del animalario, el objetivo debe ser el 50-60 %. La iluminación será artificial, con 12 horas de luz y 12 de oscuridad. Para la alimentación se podrán utilizar dietas de laboratorio convencionales, con suministro ilimitado de agua para beber
1.4.2 Procedimiento del ensayo
1.4.2.1 Aplicación de la sustancia analizada
La sustancia analizada debe aplicarse a la conjuntiva de un ojo de cada animal, separando suavemente el párpado del globo ocular. A continuación se juntan suavemente los párpados durante un segundo, para que no se pierda el material. El otro ojo no se trata y sirve como control.
1.4.2.2 Irrigación
No se lavarán los ojos de los animales tratados hasta al menos 24 horas después de la instilación de la sustancia ensayada excepto en el caso de sólidos (véase el punto 1.4.2.3.2) y en caso de producirse efectos corrosivos o irritantes inmediatos. Transcurridas 24 horas se podrán lavar los ojos si se considera necesario.
No se recomienda utilizar un grupo satélite de animales para investigar la influencia del lavado, a menos que esté justificado desde el punto de vista estadístico. Si se necesita un grupo satélite estará formado por dos conejos. Las condiciones del lavado deben quedar minuciosamente documentadas, por ejemplo, hora del lavado; composición y temperatura de la solución de lavado; duración, volumen y velocidad de la aplicación.
1.4.2.3 Nivel de dosis
1.4.2.3.1 Evaluación de líquidos
Para evaluar líquidos se emplea una dosis de 0,1 ml. No se deben utilizar aerosoles para instilar la sustancia directamente en el ojo. Antes de aplicarlo, se expulsa el líquido del aerosol en un recipiente, y se instila 0,1 ml en el ojo.
1.4.2.3.2 Evaluación de sólidos
Para evaluar sólidos, cremas y sustancias con partículas, la cantidad utilizada debe tener un volumen de 0,1 ml o no pesar más de 100 mg. El material analizado debe estar reducido a polvo fino. El volumen de material sólido se medirá tras compactarlo con suavidad, por ejemplo, golpeando suavemente con los dedos el envase medidor. Si en el primer punto temporal de observación (1 hora después del tratamiento) la sustancia analizada sólida no ha sido eliminada del ojo del animal de ensayo por mecanismos fisiológicos, se puede lavar el ojo con suero salino o con agua destilada.
1.4.2.3.3 Evaluación de aerosoles
Se recomienda hacer una recogida de los aerosoles antes de instilar el producto en el ojo. Son excepción las sustancias que van en contenedores en aerosol presurizados, que no se pueden recoger debido a la vaporización. En esos casos hay que sujetar el ojo bien abierto, administrando la sustancia analizada en el ojo con una sola pulverización de un segundo aproximadamente, a una distancia de 10 cm directamente delante del ojo. Esta distancia puede variar dependiendo de la presión del aerosol y de su contenido. Hay que tener cuidado para no lesionar el ojo con la presión del aerosol. En determinados casos puede ser necesario evaluar el potencial de lesión «mecánica» del ojo producida por la fuerza del aerosol.
Es posible calcular la dosis de un aerosol, simulando elensayo como se indica a continuación: se pulveriza la sustancia sobre papel de pesar, a través de una abertura del tamaño del ojo de un conejo, colocada directamente delante del papel. El aumento de peso del papel sirve para calcular aproximadamente la cantidad administrada al ojo. Para sustancias volátiles se puede calcular, la dosis pesando un envase antes de aplicar el material de ensayo y después de retirarlo.
1.4.2.4 Ensayo inicial (ensayo de irritación/corrosión ocular in vivo con un animal)
Como ya se ha indicado en la estrategia de evaluación secuencial (véase el anexo 1), se recomienda encarecidamente realizar el ensayo in vivo primero con un solo animal.
Si los resultados de esta indican que la sustancia es muy irritante o corrosiva en el ojo, no se harán más ensayos.
1.4.2.5 Anestésicos locales
El uso de anestésicos locales se considerará caso por caso. Si el análisis de la carga de la prueba indica que la sustancia puede producir dolor, o si la evaluación inicial demuestra que se puede producir una reacción dolorosa, se podrá utilizar un anestésico local antes de aplicar la sustancia en estudio. La elección del tipo, la concentración y la dosis del anestésico local se hará con cuidado, para asegurarse de que su empleo no produce diferencias en la reacción a la sustancia en estudio. El ojo del control debe de recibir la misma anestesia.
1.4.2.6 Ensayo de confirmación (ensayo de irritación ocular in vivo con animales adicionales)
Si en el ensayo inicial no se observa efecto corrosivo se confirmará la respuesta irritante o negativa con un máximo de dos animales más. Si se observa efecto irritante en el ensayo inicial, indicativo de un posible efecto potente (irreversible) en la de confirmación, se recomienda efectuar esta de forma secuencial exponiendo a los animales de uno a uno, en lugar de exponer a dos a la vez. Si el segundo animal presenta efectos corrosivos o irritantes importantes se suspenderá el ensayo. Puede ser necesario utilizar más animales para confirmar respuestas irritantes débiles o moderadas.
1.4.2.7 Período de observación
La duración del período de observación debe ser suficiente para evaluar por completo la reversibilidad de los efectos observados. No obstante se dará por finalizado el experimento si el cualquier momento el animal presenta signos continuados de dolor o malestar graves (9). Para determinar la reversibilidad de los efectos lo normal es someter a los animales a 21 días de observación a partir de la administración de la sustancia analizada. Si se observa la irreversibilidad antes de 21 días el experimento debe concluir en ese momento.
1.4.2.7.1 Observaciones clínicas y graduación de las reacciones oculares
Se examinarán los ojos 1, 24, 48 y 72 horas después de la aplicación de la sustancia analizada. Los animales no permanecerán en el ensayo más tiempo del necesario, una vez obtenida la información definitiva. Los animales que muestren dolor o malestar intenso continuados deben ser sacrificados sin demora, con la consiguiente evaluación de la sustancia. Serán sacrificados los animales que presenten las siguientes lesiones oculares después de la instilación: perforación corneal o ulceración corneal significativa, incluido el estafiloma; presencia de sangre en la cámara anterior del ojo; opacidad corneal de grado 4 que persista durante 48 horas; ausencia de reflejo lumínico (respuesta del iris de grado 2) que persista durante 72 horas; ulceración de la conjuntiva; necrosis de la conjuntiva o de la membrana nictitante, o escarificación. Esto se debe a que tales lesiones no suelen ser reversibles.
Los animales que no presenten lesiones oculares deben permanecer en observación al menos 3 días después de la instilación. Los animales con lesiones leves a moderadas deben permanecer en observación hasta que desaparezcan las lesiones o hasta transcurridos 21 días, que es cuando finaliza el estudio. Las observaciones se efectuarán a los 7, 14 y 21 días, para determinar el estado de las lesiones y si son o no reversibles.
En cada exploración se anotará el grado de reacción ocular (conjuntiva, córnea e iris) (tabla I). También se notificará cualesquiera otras lesiones oculares (por ejemplo, queratitis vascular, manchas oculares) o efectos sistémicos adversos.
Se puede facilitar el examen de las reacciones mediante el uso de una lupa binocular, de una lámpara de hendidura manual, de un biomicroscopio o de otro dispositivo adecuado. Tras registrar las observaciones a las 24 horas, las ulteriores exploraciones pueden hacerse con la ayuda de fluoresceína.
La graduación de las respuestas oculares es subjetiva necesariamente. Para armonizar tal graduación y ayudar a los laboratorios y a quienes efectúan e interpretan las observaciones, el personal encargado recibirá formación adecuada sobre el sistema de puntuación utilizado. La graduación de las respuestas oculares debe hacerse en condiciones ciegas.
2. DATOS
2.2 EVALUACIÓN DE LOS RESULTADOS
Las puntuaciones de la irritación ocular deben evaluarse junto con la naturaleza y la intensidad de las lesiones, y el hecho de si son reversibles o no. Las puntuaciones individuales no constituyen una referencia absoluta de las propiedades irritativas del material, pues también se evalúan otros efectos del material evaluado. En su lugar las puntuaciones individuales deben considerarse como valores de referencia, que solo serán significativos cuando sean respaldados por una descripción completa y la evaluación de todas las demás observaciones.
3. NOTIFICACIÓN
3.1 INFORME DEL ENSAYO
El informe del ensayo incluirá la siguiente información:
|
|
Justificación del ensayo in vivo: análisis de la carga de la prueba de ensayos realizados anteriormente, incluidos los resultados de la estrategia de evaluación secuencial.
|
|
|
análisis de la carga de la prueba para realizar el estudio in vivo.
|
|
|
Vehículo:
|
|
|
Animales de experimentación:
|
|
|
Resultados:
|
|
|
Comentario de los resultados |
3.2 INTERPRETACIÓN DE LOS RESULTADOS
La extrapolación a humanos de los resultados de los estudios de irritación ocular realizados con animales de laboratorio solo es válida en cierto grado. En muchos casos, los conejos albinos son más sensibles a los irritantes o corrosivos oculares que los seres humanos.
Hay que interpretar con cuidado los datos para excluir la irritación resultante de una infección secundaria.
4. REFERENCIAS
|
(1) |
Barrette, M.D., Castell, J.V., Chamberlain, M., Combes, R.D., Dearden, J.C., Fentem, J.H., Gerner, I., Giuliani, A., Gray, T.J.B., Livingston, D.J., Provan, W.M., Rutten, F.A.J.J.L., Verhaar, H.J.M., Zbinden, P. (1995) The Integrated Use of Alternative Approaches for Predicting Toxic Hazard. ECVAM Workshop Report 8. ATLA 23, 410-429. |
|
(2) |
de Silva, O., Cottin, M., Dami, N., Roguet, R., Catroux, P., Toufic, A., Sicard, G, Dossou, K.G., Gerner, I., Schlede, E., Spielmann, H., Gupta, K.C., Hill, R.N. (1997) Evaluation of Eye Irritation Potential: Statistical Analysis and Tier Testing Strategies. Food Chem. Toxicol 35, 159-164. |
|
(3) |
Worth A.P. and Fentem J.H. (1999) A general approach for evaluating stepwise testing strategies ATLA 27, 161-177. |
|
(4) |
Young, J.R., How, M.J., Walker, A.P., Worth W.M.H. (1988) Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals. Toxicol. In Vitro, 2, 19-26. |
|
(5) |
Neun, DJ. (1993) Effects of Alkalinity on the Eye Irritation Potential of Solutions Prepared at a Single pH. J. Toxicol. Cut. Ocular Toxicol. 12, 227-231. |
|
(6) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Edsaile, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, pp.483 - 524. |
|
(6a) |
Testing Method B.40 Skin Corrosion. |
|
(7) |
Testing method B.4. Acute toxicity: dermal irritation/corrosion. |
|
(8) |
OECD (1996) OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Pronunciado en Solna, Suecia, 22-24 de enero de 1996 (http://www.oecd. org/ehs/test/background.htm). |
|
(9) |
OECD (1998) Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, noviembre de 1998 (http://www.oecd.org/ehs/Class/HCL6.htm). |
|
(10) |
OECD (2000) Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. OECD Environmental Health and Safety Publications. Series on Testing and Assessment No. 19 (http://www.oecd.org/ehs/test/monos.htm). |
Tabla I
GRADUACIÓN DE LAS LESIONES OCULARES
Córnea
Opacidad: grado de densidad (se tendrá en cuenta la zona más densa) (1)
|
Sin ulceración ni opacidad … |
0 |
|
Zonas de opacidad diseminadas o difusas (aparte del ligero mate debido a la limpieza habitual); los detalles del iris se aprecian con claridad … |
1 |
|
Zona translúcida fácilmente discernible; los detalles del iris están ligeramente oscurecidos … |
2 |
|
Zona nacarada; no se ven detalles del iris; el tamaño de la pupila apenas es discernible … |
3 |
|
Córnea opaca; no se distingue el iris a su través … |
4 |
Máximo posible: 4
NOTAS
Iris
|
Normal … |
0 |
|
Pliegues notablemente hundidos, congestión, inflamación, moderada hiperemia o inyección circuncorneal; iris reactivo a la luz (si la reacción es defectuosa se considera como efecto): … |
1 |
|
Hemorragia, destrucción visible o ausencia de reacción a la luz … |
2 |
Máximo posible: 2
Conjuntiva
Enrojecimiento (se refiere a la conjuntiva palpebral y bulbar; se excluye la córnea y el iris)
|
Normal … |
0 |
|
Algunos vasos sanguíneos hiperémicos (inyectados) … |
1 |
|
Coloración carmesí difusa; no se distinguen fácilmente los vasos individuales … |
2 |
|
Coloración carne difusa … |
3 |
Máximo posible: 3
Quemosis
Inflamación (se refiere a los párpados o a las membranas nictitantes)
|
Normal … |
0 |
|
Cierta hinchazón superior a lo normal … |
1 |
|
Hinchazón evidente con eversión parcial de los párpados … |
2 |
|
Hinchazón con párpados medio cerrados … |
3 |
|
Hinchazón con párpados más que medio cerrados … |
4 |
Máximo posible: 4
ANEXO
Estrategia de evaluación secuencial de la irritación y la corrosión oculares
CONSIDERACIONES GENERALES
Es importante evitar el uso innecesario de animales y reducir al mínimo las pruebas que con toda probabilidad produce respuestas graves en los animales, por su bienestar y por motivos científicos. Antes de considerar la realización de ensayos in vivo hay que evaluar toda la información sobre una sustancia en lo que respeta a su posible poder corrosivo/irritativo ocular. Puede que existan pruebas suficientes para clasificar el potencial corrosivo o irritativo ocular de una sustancia analizada, sin necesidad de realiza ensayos con animales de laboratorio. Por eso el uso del análisis de la carga de la prueba y de una estrategia de evaluación secuencial reducirá al mínimo la necesidad de realizar ensayos in vivo, especialmente si es probable que la sustancia produzca reacciones graves.
Se recomienda utilizar el análisis de la carga de la prueba para evaluar la información existente sobre el potencial de irritación y corrosión oculares producidas por las sustancias, para determinar si la realización de estudios adicionales, a parte de los oculares in vivo, ayudaría a caracterizar dicho potencial. Cuando sean necesarios otros estudios, se recomienda utilizar la estrategia de evaluación secuencial para obtener los datos experimentales relevantes. Para sustancias no evaluadas anteriormente, se utilizará la estrategia de evaluación secuencial para obtener los datos necesarios para evaluar su potencial corrosivo/irritativo ocular. La estrategia de evaluación que se describe en este Anexo fue redactada en un taller de la OECD (1). Posteriormente fue confirmada y ampliada por el sistema armonizado integrado de clasificación de peligros para la salud humana y efectos ambientales de las sustancias químicas (Harmonised Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances), y fue aprobada en la 1 XXVIII reunión del Comité Conjunto para productos químicos (Chemicals Committee and the Working Party on Chemicals) en noviembre de 1998 (2).
Aunque la presente estrategia de evaluación no forma parte integrante del método de evaluación B.5, sí expresa el enfoque recomendado para la determinación de las características de irritación/corrosión oculares. Este enfoque representa la práctica óptima y una piedra angular desde el punto de vista ético para el análisis in vivo de la irritación/corrosión oculares. El método de evaluación proporciona instrucciones para la realización del ensayo in vivo, y resume los factores que se han de abordar antes de ponerla en marcha. La estrategia proporciona un enfoque basado en la carga de la prueba para la evaluación de los datos existentes sobre las propiedades de irritación/corrosión oculares de las sustancias analizadas, y un enfoque por partes para generar datos relevantes sobre sustancias que necesitan estudios adicionales o que no han sido estudiadas. La estrategia incluye la realización en primer lugar de ensayos in vitro o ex vivo validados y aceptados y a continuación del método de evaluación B.4 estudios de irritación/corrosión cutánea en condiciones concretas (3) (4).
DESCRIPCIÓN DE LA ESTRATEGIA DE EVALUACIÓN ESCALONADA
Antes de llevar a cabo 1 los ensayos que forman parte de la estrategia de evaluación secuencial (véase la figura), se evaluará toda la información disponible, para determinar la necesidad de practicar estudios oculares in vivo. Aunque se puede obtener información significativa a partir de la evaluación de parámetros aislados (por ejemplo, un pH extremo), hay que considerar la totalidad de la información existente. Se evaluarán todos los datos relevantes sobre los efectos de la sustancia en cuestión y de sus análogos estructurales, para tomar una decisión basada en la carga de la prueba, y se presentará la justificación de dicha decisión. Se hará especial hincapié en los datos existentes sobre la sustancia en seres humanos y animales, seguidos por el resultado de los ensayos in vitro o ex vivo. Siempre que sea posible se evitará realizar estudios in vivo con sustancias corrosivas. Los factores que se tienen en cuenta en la estrategia de evaluación son:
Evaluación de datos existentes en seres humanos y en animales (paso 1): En primer lugar se tendrán en cuenta los datos existentes sobre seres humanos, por ejemplo, estudios clínicos u ocupacionales e informes de casos, y los datos de ensayos realizados con animales en estudios oftalmológicos, pues proporcionan información directamente relacionada con los efectos producidos en los ojos. A continuación se evaluarán los datos disponibles de estudios realizados con seres humanos o animales para investigar la corrosión/irritación cutáneas. Las sustancias conocidas por ser corrosivas o intensamente irritantes para el ojo no se aplicarán a los ojos de los animales, ni tampoco aquellas que tengan efectos corrosivos o irritantes para la piel; dichas sustancias debe ser consideradas corrosivas o irritantes para los ojos también. Cuando en estudios oftalmológicos realizados anteriormente se hayan obtenido pruebas suficientes de que una sustancia no es corrosiva ni imitante, tampoco será sometida a estudios oftalmológicos in vivo.
Análisis de las relaciones entre las estructuras (SAR) (paso 2). Hay que tener en cuenta los resultados de los análisis de sustancias relacionadas desde el punto de vista estructural, si es que existen. Cuando se dispone de suficientes datos en seres humanos o animales sobre sustancias relacionadas desde el punto de vista estructural o sobre mezclas de dichas sustancias que indique su potencial corrosivo/irritativo ocular, se puede presuponer que la sustancia analizada producirá las mismas respuestas. En estos casos puede que no sea necesario evaluar la sustancia en cuestión. Los datos negativos de los estudios sobre sustancias relacionadas desde el punto de vista estructural o sobre mezclas de estas no constituyen demostración suficiente de que una sustancia no será corrosiva ni irritante en la estrategia de evaluación secuencial. Para identificar el potencial de corrosión e irritación dérmica y ocular hay que utilizar enfoques SAR validados y aceptados.
Propiedades fisicoquímicas y reactividad química (paso 3). Las sustancias con pH extremos, como ≤ 2,0 y ≥ 11,5 pueden tener potentes efectos locales. Si el pH extremo es la base para identificar si una sustancia es corrosiva o irritante para el ojo, entonces también se tendrá en cuenta la reserva de ácido/álcali (o capacidad amortiguadora) (3) (4). Si la capacidad amortiguadora sugiere la posibilidad de que una sustancia no sea corrosiva para el ojo se realizarán mas pruebas para confirmarlo, preferiblemente utilizando un ensayo in vitro o ex vivo validado y aceptado (véanse los pasos 5 y 6).
Consideración de otras informaciones existentes (paso 4). En esta fase se evaluará toda la información disponible sobre la toxicidad sistémica por vía cutánea. También se tendrá en cuenta la toxicidad cutánea aguda de la sustancia analizada. Si se ha demostrado que la sustancia es muy tóxica por vía cutánea puede no ser necesario estudiarla en el ojo. Aunque no existe necesariamente una relación entre la toxicidad cutánea aguda y la irritación/corrosión ocular, se puede suponer que si un agente es muy tóxico por vía cutánea también lo será si se aplica al ojo. Estos datos también se pueden tener en cuenta entre los pasos 2 y 3.
Resultados de los ensayos in vitro o ex vivo (pasos 5 y 6). Cuando se hayan demostrado las propiedades corrosivas o irritativas intensas de una sustancia en un ensayo in vitro o ex vivo (7) (8) validado y aceptado para la evaluación concreta de la corrosión/irritación ocular o cutánea, no será necesario evaluarla en animales. Se puede dar por supuesto que dichas sustancias producirán similares efectos intensos in vivo. Si no se dispone de ensayos in vitrolex vivo validados y aceptados, debe prescindirse de los pasos 5 y 6 y pasar directamente al paso 7.
Evaluación del poder irritantivo cutáneo in vivo de la sustancia (paso 7). Cuando sean insuficientes los datos para realizar un análisis de la carga de la prueba concluyente sobre el poder irritante/corrosivo ocular de una sustancia a partir de los datos de los estudios que se han enumerado, se procederá en primer lugar a evaluar el potencial de irritación/corrosión cutánea in vivo, utilizando el método de evaluación B.4 (4) y el anexo que le acompaña (9). Si se demuestra que la sustancia produce corrosión o irritación cutánea intensa, se considerará como corrosiva para el ojo, a menos que haya otros datos en apoyo de otra conclusión. En ese caso no será necesario realizar un ensayo ocular in vivo. Si la sustancia no es corrosiva ni intensamente irritante para la piel, se realizará un ensayo cutáneo in vivo.
Ensayo in vivo en conejos (pasos 8 y 9). El ensayo ocular in vivo empezará con un ensayo inicial con un solo animal Si los resultados de esta indican que la sustancia es muy irritante o corrosiva en el ojo, no se harán más ensayos. Si dicho ensayo no muestra efectos corrosivos ni irritantes graves, se llevará a cabo un ensayo de confirmación con otros dos animales.
REFERENCIAS
|
(1) |
OECD (1996) OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Pronunciado en Solna, Suecia, 22-24 de enero de 1996 (http://www1 oecd.org/ehs/test/background.htm). |
|
(2) |
OECD (1998) Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, noviemre de 1998 (http://wwwl.oecd.org/ehs/Class/HCL6.htm). |
|
(3) |
Worth, A.P. and Fentem J.H. (1999). A General Approach for Evaluating Stepwise Testing Strategies. ATLA 27, 161-177. |
|
(4) |
Testing method B.4. Acute Toxicity: dermal irritation/corrosion. |
|
(5) |
Young, J.R., How, M.J., Walker, A.P., Worth W.M.H. (1988) Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals. Toxicol. In Vitro, 2, 19-26. |
|
(6) |
Neun, D.J. (1993) Effects of Alkalinity on the Eye Irritation Potential of Solutions Prepared at a Single pH. J. Toxicol. Cut. Ocular Toxicol. 12, 227-231. |
|
(7) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Edsail, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, 483-524. |
|
(8) |
Testing Method B.40 Skin Corrosion. |
|
(9) |
Annex to Testing method B.4: A Sequential Testing Strategy for Skin Irritation and Corrosion. |
Figura
ESTRATEGIA DE EVALUACIÓN DE LA IRRITACIÓN/CORROSIÓN OCULAR

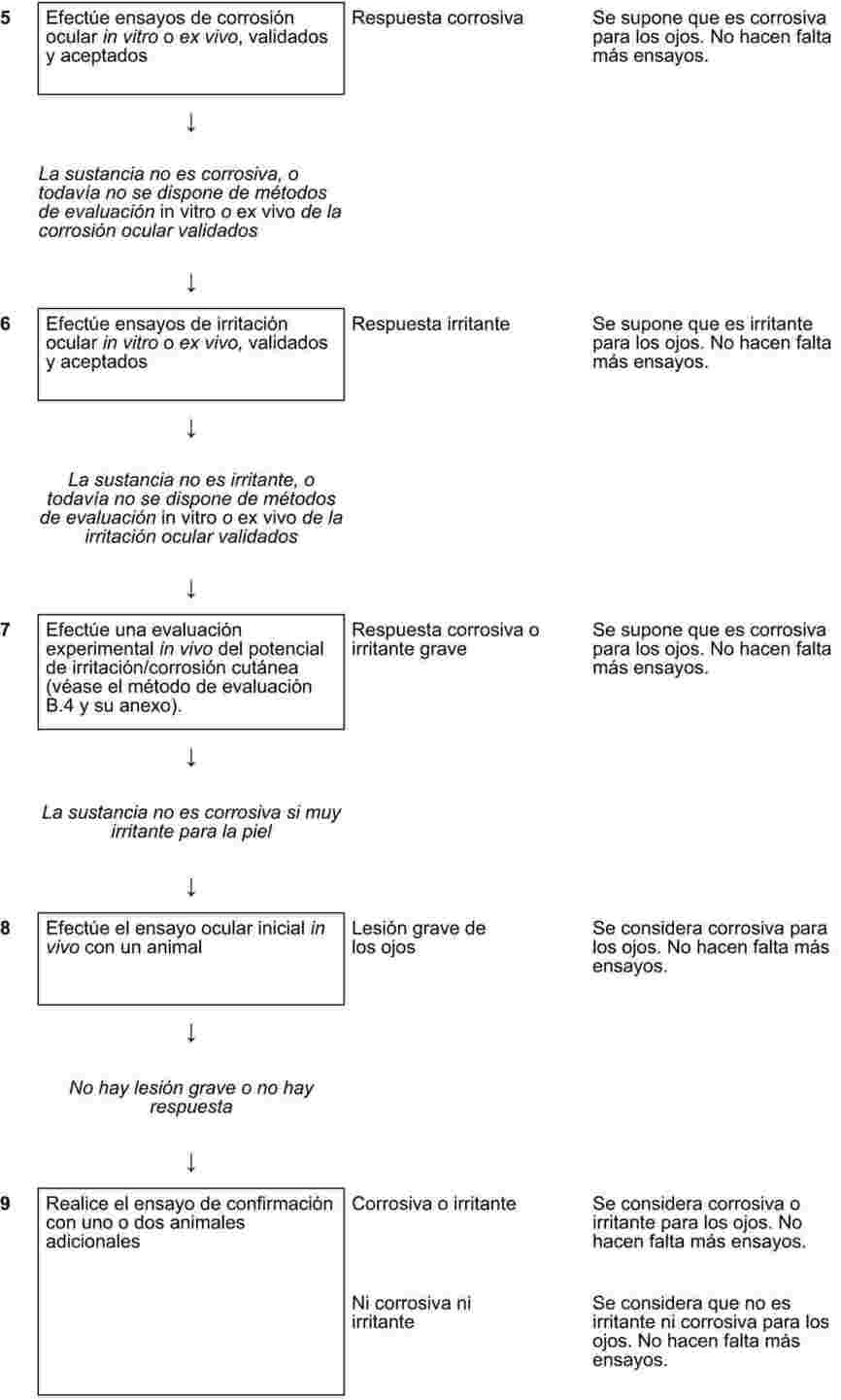
B.6. SENSIBILIZACIÓN DE LA PIEL
1. MÉTODO
1.1. INTRODUCCIÓN
Observaciones:
La sensibilidad y capacidad de los ensayos para detectar sustancias que pueden sensibilizar la piel humana se consideran importantes para un sistema de clasificación de la toxicidad aplicable a la salud pública.
No hay un método único que detecte todas las sustancias con potencial de sensibilización de la piel humana y que sea adecuado para todas las sustancias.
Para seleccionar un ensayo deben tenerse en cuenta factores como las características físicas de la sustancia, incluyendo su capacidad de penetración en la piel.
Se han elaborado dos tipos de ensayos que utilizan cobayas: los ensayos con coadyuvante, en los que se potencia un estado alérgico disolviendo o suspendiendo la sustancia estudiada en coadyuvante completo de Freund (FCA), y los ensayos sin coadyuvante.
Es probable que los ensayos con coadyuvante sean más exactos a la hora de predecir el efecto sensibilizante probable de una sustancia en la piel humana respecto a los métodos que no emplean el coadyuvante completo de Freund, por lo que son los métodos preferidos.
El ensayo de maximización en cobaya (GPMT) es un ensayo con coadyuvante ampliamente utilizado. Aunque se pueden utilizar otros métodos para detectar el potencial sensibilizante de una sustancia, se considera que el GMPT es la técnica con coadyuvante de preferencia.
Los ensayos sin coadyuvantes (suele utilizarse sobre todo el ensayo de Buehler) se consideran menos sensibles con muchas clases de productos químicos.
En ciertos casos puede haber buenas razones para escoger el ensayo de Buehler, que supone una aplicación tópica en vez de la inyección intradérmica utilizada en el ensayo de maximización en cobaya. Cuando se use el ensayo de Buehler deberá justificarse científicamente.
En el presente método se describen el ensayo de maximización en cobaya y el ensayo de Buehler. Se pueden utilizar otros métodos siempre que estén bien validados y se dé su justificación científica.
Si se obtiene un resultado positivo en un ensayo de cribado reconocido, podrá designarse una sustancia de ensayo como sensibilizante potencial y podrá no ser necesario realizar un nuevo ensayo con cobaya. No obstante, si se obtiene un resultado negativo en un ensayo semejante, deberá realizarse un ensayo con cobaya utilizando el procedimiento descrito en este método de ensayo.
Véase también la introducción general de la parte B.
1.2. DEFINICIONES
Sensibilización de la piel (dermatitis alérgicas de contacto): es una reacción cutánea de origen inmunológico ante una sustancia. En los seres humanos las respuestas pueden caracterizarse por prurito, eritema, edema, pápulas, vesículas, ampollas o una combinación de estos fenómenos. En otras especies, las reacciones pueden ser diferentes y apreciarse solo eritema y edema.
Exposición de inducción: exposición experimental de un sujeto a una sustancia con el fin de inducir un estado de hipersensibilidad.
Período de inducción: período de al menos una semana a partir de la exposición de inducción, durante el cual puede aparecer un estado de hipersensibilidad.
Exposición de provocación: exposición experimental de un sujeto previamente tratado a una sustancia después de un período de inducción, a fin de determinar si el sujeto reacciona de forma hipersensible.
1.3. SUSTANCIAS DE REFERENCIA
La sensibilidad y fiabilidad de la técnica experimental utilizada deberá evaluarse cada seis meses mediante el empleo de sustancias conocidas como dermosensibilizantes suaves o moderados.
En un ensayo realizado convenientemente, el empleo de un sensibilizante suave o moderado debe producir una respuesta del 30 % al menos en un ensayo con coadyuvante y del 15 % al menos en un ensayo sin coadyuvante.
Se recomiendan las siguientes sustancias:
|
Número CAS |
Número EINECS |
Denominación EINECS |
Denominación común |
|
101-86-0 |
202-983-3 |
a-hexilcinamaldehído |
ct-hexilcinamaldehído |
|
149-30-4 |
205-736-8 |
benzotiazol-2-tiol (mercaptobenzotiazol) |
kaptax |
|
94-09-7 |
202-303-5 |
benzocaína |
norcaína |
En ciertas circunstancias se podrán utilizar otras sustancias de control que cumplan los criterios citados, justificándose debidamente su elección.
1.4. PRINCIPIO DEL MÉTODO
Los animales utilizados se exponen inicialmente a la sustancia mediante inyecciones intradérmicas o aplicación epidérmica (exposición de inducción). Tras un período de descanso de 10 a 14 días (período de inducción), durante el que puede desarrollarse la respuesta inmunitaria, los animales se someten a una dosis de provocación. La amplitud y el grado de la reacción cutánea de los animales ante la exposición de provocación se compara con la mostrada por animales de control que reciben un tratamiento simulado durante la inducción y se someten a la exposición de provocación.
1.5. DESCRIPCIÓN DE LOS MÉTODOS
Si se considera necesario eliminar la sustancia estudiada, puede hacerse utilizando agua o un disolvente adecuado que no altere la respuesta obtenida ni la integridad de la epidermis.
1.5.1. Prueba de maximización en cobaya (GPMT)
1.5.1.1. Preparación
Durante al menos 5 días antes del inicio del ensayo, se aclimatan a las condiciones del laboratorio cobayas albinas jóvenes y sanas. Antes del ensayo, los animales se eligen al azar y se asignan a los lotes de tratamiento. La eliminación del pelo se hace por corte, afeitado o incluso depilación química, en función del método de ensayo utilizado. Debe evitarse la producción de escoriaciones en la piel. Los animales se pesarán antes del ensayo y al final del mismo.
1.5.1.2. Condiciones del ensayo
1.5.1.2.1. Animales de laboratorio
Se utilizan cepas corrientes de laboratorio de cobaya albina.
1.5.1.2.2. Número y sexo
Se pueden utilizar animales de ambos sexos. Si se utilizan hembras, deben ser nulíparas y no grávidas.
Se utilizan por los menos 10 animales para el lote tratado y, por lo menos, 5 para el lote testigo. Si se utilizan menos de 20 animales tratados y 10 testigos, y no es posible concluir que la sustancia estudiada sea sensibilizante, se recomienda con insistencia realizar ensayos complementarios con otros animales hasta completar un mínimo de 20 cobayas tratadas y 10 cobayas testigos.
1.5.1.2.3. Dosis
La concentración de la sustancia utilizada para cada exposición de inducción debe tolerarse bien sistemáticamente y debe ser la más elevada que produzca irritación cutánea suave o moderada. La concentración utilizada para la exposición de provocación debe ser la mayor dosis no irritante. Si necesario, las concentraciones apropiadas pueden ser determinadas a partir de un estudio piloto en el que se utilicen dos a tres animales. Debe considerarse la posibilidad de utilizar con este fin animales tratados con FCA.
1.5.1.3. Procedimiento
1.5.1.3.1. Inducción
Día 0: lote tratado
Se administran tres pares de inyecciones intradérmicas de 0,1 ml en la región dorsal superior, de la que se habrá eliminado el pelo, de forma que a cada lado de la linea media quede una inyección de cada par.
Inyección 1: mezcla 1:1 (v/v) FCA/agua o suero fisiológico
Inyección 2: la sustancia de ensayo en un vehículo adecuado, en la concentración seleccionada
Inyección 3: la sustancia de ensayo en la concentración seleccionada formulada en una mezcla 1:1 (v/v) FCA/agua o suero fisiológico
En la inyección 3, las sustancias hidrosolubles se disuelven en la fase acuosa antes de mezclarse con el FCA. Las sustancias liposolubles o insolubles se suspenden en la FCA antes de combinarse con la fase acuosa. La concentración final de la sustancia de ensayo será igual a la utilizada en la inyección 2.
Las inyecciones 1 y 2 se ponen próximas entre sí y lo más cerca posible de la cabeza, mientras que la inyección 3 se aplica hacia la parte caudal de la superficie de ensayo.
Día 0: lote testigo
Se ponen tres pares de inyecciones intradérmicas de 0,1 ml en los mismos lugares que en los animales tratados.
Inyección 1: mezcla 1:1 (v/v) FCA/agua o suero fisiológico
Inyección 2: vehículo solo
Inyección 3: formulación al 50 % (p/v) del vehículo en una mezcla 1:1 (v/v) FCA/agua o suero fisiológico.
Días 5-7: lotes tratado y testigo
Aproximadamente 24 horas antes de la aplicación tópica de inducción, si la sustancia no es irritante de la piel, se trata la superficie de ensayo, previo corte al rape del pelo o afeitado del mismo, con 0,5 ml de lauril-sulfato sódico al 10 % en vaselina, a fin de producir irritación local.
Días 6-8: lote tratado
Se elimina de nuevo el pelo de la superficie de ensayo. Se carga totalmente un papel de filtro (2 × 4 cm) con la sustancia de ensayo en un vehículo adecuado, se aplica a la superficie de ensayo y se mantiene con un apósito oclusivo durante 48 horas. La elección del vehículo debe justificarse. Los productos sólidos se pulverizan finamente y se incorporan a un vehículo adecuado. Los líquidos pueden aplicarse sin diluir, en caso de que esto sea apropiado.
Días 6-8: lote testigo
Se elimina de nuevo el pelo de la superficie de ensayo. Solo se aplica el vehículo, de forma similar, en la superficie de ensayo con un apósito oclusivo durante 48 horas.
1.5.1.3.2. Provocación
Días 20-22: lotes tratado y testigo
Se elimina el pelo de los costados de los animales tratados y de los testigos. Se aplica un parche o cámara con la sustancia estudiada sobre un costado de los animales y, cuando sea pertinente, también se puede aplicar en el otro costado un parche o cámara con el vehículo solo. Los parches se mantienen con un apósito oclusivo durante 24 horas.
1.5.1.3.3. Observación y clasificación: lotes tratado y testigo
|
— |
Aproximadamente a las 21 horas de levantar el parche, se limpia la superficie de ensaya y, en caso necesario, se elimina el pelo mediante corte al rape, afeitado o depilación. |
|
— |
Unas 3 horas después (hacia tas 48 horas del inicio de la aplicación de provocación) se observa la reacción cutánea y se registra de acuerdo con la clasificación que se recoge en el apéndice. |
|
— |
Aproximadamente a las 24 horas de esta observación se hace una segunda observación (72 horas), que se registra igualmente. |
Se recomienda la lectura ciega de los animales tratados y testigos.
En caso necesario para aclarar los resultados obtenidos con la primera provocación, debe considerarse la posibilidad de realizar, una semana después de esa primera provocación, una segunda (es decir, una «reprovocación»), cuando convenga con un nuevo lote testigo. La reprovocación también puede aplicarse al lote testigo original.
Deben observarse y registrarse, de acuerdo con la escala de Magnusson y Kligman (véase el apéndice) todas las reacciones cutáneas y cualquier observación extraña, incluidas las reacciones sistémicas, derivadas de los procesos de inducción y provocación. Para aclarar reacciones dudosas, pueden aplicarse otros procedimientos como, por ejemplo, examen histopatológico, medición del espesor del pliegue cutáneo, etc.
1.5.2. Ensayo de Buehler
1.5.2.1. Preparación
Durante al menos 5 días antes del inicio del ensayo, se aclimatan a las condiciones del laboratorio cobayas albinos jóvenes y sanos. Antes del ensayo, los animales se eligen al azar y se asignan a los lotes de tratamiento. La eliminación del pelo se hace por corte, afeitado, o incluso depilación química, en función del método de ensayo utilizado. Debe evitarse la producción de escoriaciones en la piel. Los animales se pesarán antes del ensayo y al final del mismo.
1.5.2.2. Condiciones del ensayo
1.5.2.2.1. Animales de laboratorio
Se utilizan cepas corrientes de laboratorio de cobaya albina.
1.5.2.2.2. Número y sexo
Se pueden utilizar animales de ambos sexos. Si se utilizan hembras, deben ser nulíparas y no grávidas.
Se utilizan por lo menos 20 animales para el lote tratado y, por lo menos, 10 animales para el lote testigo.
1.5.2.2.3. Dosis
La concentración de sustancia de ensayo utilizada en cada exposición de inducción debe ser la más elevada posible que produzca una irritación suave pero no excesiva. La concentración utilizada para la exposición de provocación debe ser la mayor dosis no irritante. Si necesario, las concentraciones apropiadas pueden ser determinadas a partir de un estudio piloto en el que se utilicen dos o tres animales.
En caso de sustancias hidrosolubles, es conveniente utilizar agua o una solución diluida no irritante de un agente tensoactivo como vehículo. Para otros tipos de sustancias, se recomienda etanol/agua al 80 % para la inducción y acetona para la provocación.
1.5.2.3. Procedimiento
1.5.2.3.1. Inducción
Día 0: lote tratado
Se elimina el pelo de un costado (por corte al rape). El sistema del parche de ensayo debe cargarse totalmente con la sustancia en un vehículo adecuado (debe justificarse la elección del vehículo; las sustancias líquidas pueden aplicarse sin diluir, en caso de que sea conveniente).
El sistema del parche de ensayo se aplica a la zona de ensayo y se mantiene en contacto con la piel mediante una cámara o parche oclusivo y un apósito adecuado durante 6 horas.
El sistema del parche de ensayo debe ser oclusivo. Es apropiado utilizar una compresa de algodón, que puede ser circular o cuadrada, con una superficie aproximada de 4 a 6 cm2. Se recomienda sujetarlo utilizando unas bridas adecuadas para garantizar la oclusión. Si se utiliza un envoltorio, puede ser necesario proceder a exposiciones complementarias.
Día 0: lote testigo
Se elimina el pelo de un costado (por corte al rape). Se aplica el vehículo solo de forma similar a la utilizada con el lote tratado. El sistema del parche de ensayo se mantiene en contacto con la piel mediante un parche oclusivo o cámara y un apósito adecuado durante 6 horas. Si puede demostrarse que no es necesario dar placebo al lote testigo, puede omitirse dicho tratamiento.
Días 6-8 y 13-15: lotes tratado y testigo
Se realiza la misma aplicación que en el día 0 en la misma superficie de ensayo (desprovista de pelo en caso necesario) del mismo costado el día 6-8, y de nuevo el día 13-15.
1.5.2.3.2. Provocación
Días 27-29: lotes tratado y testigo
Se elimina el pelo (mediante corte al rape) del costado no tratado de los animales tratados y testigos. Se aplica un parche oclusivo o cámara con la cantidad conveniente de sustancia, a la concentración máxima no irritante, a la parte posterior del costado sin tratar de los animales tratados y testigos.
Cuando sea pertinente, también se aplicará un parche oclusivo o cámara con vehículo solo a la parte anterior del costado sin tratar de los animales tratados y testigos. Los parches o cámaras se mantienen con un apósito adecuado durante 6 horas.
1.5.2.3.3. Observación y clasificación
|
— |
Aproximadamente a las 21 horas de levantar el parche, se elimina el pelo de la superficie de provocación. |
|
— |
Unas 3 horas después (hacia las 30 horas de la aplicación del parche de provocación) se observan las reacciones cutáneas y se registran de acuerdo con la clasificación recogida en el apéndice. |
|
— |
Aproximadamente a las 24 horas después de la observación de las 30 horas (hacia las 54 horas de la aplicación del parche de provocación) se vuelven a observar y registrar las reacciones cutáneas. |
Se recomienda la lectura ciega de los animales tratados y testigos.
En caso necesario para aclarar los resultados obtenidos en la primera provocación, puede considerarse la realización de una segunda provocación (es decir, una «reprovocación»), en su caso con un nuevo lote testigo, a la semana de haber realizado la primera. También puede realizarse una reprovocación con el lote testigo original.
Deben observarse y registrarse con arreglo a la escala de Magnusson y Kligman (véase el apéndice) todas las reacciones cutáneas y cualquier observación extraña, incluidas las reacciones sistémicas, derivadas de los procesos de inducción y provocación. Pueden llevarse a cabo otros procedimientos como, por ejemplo, el examen histopatológico o la medida del espesor de los pliegues cutáneos, a fin de aclarar reacciones dudosas.
2. RESULTADOS (GPMT Y ENSAYO DE BUEHLER)
Los resultados deben resumirse en un cuadro que indique tas reacciones cutáneas de cada animal en cada observación.
3. INFORME (GPMT Y ENSAYO DE BUEHLER)
Si se realiza un ensayo de cribado, por ejemplo, ensayo de ganglios linfáticos locales (LLNA) o prueba de tumefacción de la oreja del ratón (MEST) antes del ensayo con cobayas, deberá darse una descripción o referencia del ensayo, con datos sobre el método, además de los resultados obtenidos con las sustancias estudiadas y de referencia.
Informe del ensayo (GPMT y ensayo de Buehler)
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
|
Animales de ensayo:
|
|
|
Condiciones del ensayo:
|
|
|
Resultados:
|
|
|
Discusión de los resultados |
|
|
Conclusiones |
4. REFERENCIAS
El presente método es análogo a la TG 406 de la OCDE.
Apéndice
CUADRO
Escala de clasificación de Magnusson y Kligman para evaluar las reacciones del ensayo con parche de provocación
0 = sin cambios visibles
1 = eritema ligero o en manchas localizadas
2 = eritema moderado y confluente
3 = eritema intenso y tumefacción.
B.7. TOXICIDAD POR ADMINISTRACIÓN CONTINUADA (28 DÍAS) POR VÍA ORAL
1. MÉTODO
1.1 INTRODUCCIÓN
Véase la introducción general de la parte B.
1.2. DEFINICIONES
Véase la introducción general de la parte B.
1.3. PRINCIPIO DEL MÉTODO
La sustancia de ensayo se administra diariamente por vía oral en dosis graduadas a distintos lotes de animales de experimentación, a razón de una dosis por lote durante un período de 28 días. A lo largo del período de administración, se observa a los animales todos los días atentamente con el fin de descubrir la posible aparición de signos de toxicidad. Se practica la autopsia a los animales que mueran o sean sacrificados durante el ensayo y, al final del mismo, se sacrifican y someten a autopsia los animales supervivientes.
El presente método insiste más en los efectos neurológicos, como parámetro específico, y en la necesidad de hacer observaciones clínicas atentas de los animales, a fin de obtener la máxima información posible. El método sirve para detectar productos químicos con potencial neurotóxico, lo que puede justificar la realización de investigaciones más profundas de este aspecto. Además, el método puede proporcionar una indicación de los efectos inmunológicos y de la toxicidad sobre los órganos reproductores.
1.4. DESCRIPCIÓN DEL MÉTODO
1.4.1. Preparación
Se eligen al azar animales jóvenes y sanos y se reparten en los lotes testigo y tratado. Las jaulas deben disponerse de forma que se reduzcan al mínimo los posibles efectos debidos al enjaulamiento. Los animales se identifican individualmente y se mantienen en sus jaulas durante al menos 5 días antes del inicio del ensayo, a fin de permitir su aclimatación a las condiciones del laboratorio.
La sustancia de ensayo se administra por sonda o con el alimento o el agua de bebida. El método de administración oral depende del objetivo del estudio y de las propiedades fisicoquímicas de la sustancia.
En caso necesario, la sustancia de ensayo se disuelve o suspende en un vehículo adecuado. Se recomienda que, siempre que sea posible, se considere en primer lugar el uso de una solución o suspensión acuosa, después el uso de una emulsión o solución oleosa (por ejemplo, aceite de maíz) y, a continuación, la posible disolución en otros vehículos. En caso de vehículos distintos del agua, deberán conocerse sus características tóxicas. Debe determinarse la estabilidad de la sustancia de ensayo en el vehículo.
1.4.2. Condiciones del ensayo
1.4.2.1. Animales de laboratorio
La especie de preferencia entre los roedores es la rata, aunque pueden utilizarse otras especies de roedores. Hay que utilizar animales jóvenes y sanos de una cepa de laboratorio corriente. Las hembras deben ser nulíparas y no grávidas. La administración debe empezar lo antes posible tras el destete y, en cualquier caso, antes de que los animales tengan nueve semanas de edad.
Al principio del experimento, la diferencia de peso entre los animales utilizados debe ser mínima y no superar el ± 20 % del peso medio de cada sexo.
Cuando se realice un estudio de la toxicidad oral con administración continuada como fase previa de un estudio de toxicidad a largo plazo, es preferible que se utilicen en ambos estudios animales procedentes de la misma cepa y del mismo origen.
1.4.2.2. Número y sexo
Deben utilizarse por los menos 10 animales (5 hembras y 5 machos) para cada dosis. Si se van a sacrificar animales durante el experimento, habrá que añadir el número de animales que se haya previsto sacrificar antes de acabar el estudio.
Además, puede tratarse un grupo satélite de 10 animales (5 de cada sexo) con la dosis más elevada durante 28 días para observar la reversibilidad, la persistencia o la aparición retardada de efectos tóxicos durante los 14 días siguientes al tratamiento. Se utiliza también un lote satélite de 10 animales testigo (cinco de cada sexo).
1.4.2.3. Dosis
Deben utilizarse generalmente como mínimo tres lotes de ensayo y un lote testigo. A excepción de la administración de la sustancia de ensayo, los animales del lote testigo deben ser tratados de la misma manera que los de los lotes de ensayo. Si se utiliza un vehículo para la administración de la sustancia de ensayo, el lote testigo recibirá el mayor volumen utilizado de dicho vehículo.
Si, a partir de la evaluación de otros datos, no cabe esperar ningún efecto con la dosis de 1 000 mg/kg peso corporal/día, puede realizarse un ensayo límite. Si no se dispone de datos apropiados, puede realizarse un estudio de búsqueda de dosis.
Las dosis deben seleccionarse teniendo en cuenta la eventual toxicidad existente y los datos cinéticos o toxicocinéticos disponibles en relación con la sustancia de ensayo o productos afines. La dosis elevada debe seleccionarse con el propósito de inducir efectos tóxicos pero sin llegar a la muerte ni a un sufrimiento intenso. Posteriormente, debe seleccionarse una secuencia descendente de dosis a fin de poner de manifiesto las posibles respuestas en (unción de la dosis; y la dosis mínima será la dosis sin efectos adversos observados (NOAEL). Los intervalos del doble al cuádruple suelen ser óptimos para establecer los niveles descendentes y frecuentemente es preferible añadir un cuarto lote de ensayo antes que utilizar intervalos muy amplios (por ejemplo, con un factor superior a 10) entre dosis.
En el caso de sustancias administradas con los alimentos o el agua de bebida, es importante garantizar que las cantidades de la sustancia de ensayo utilizadas no interfieren con la nutrición normal ni con el equilibrio hídrico. Cuando la sustancia de ensayo se administre con los alimentos, puede utilizarse una concentración constante en la dieta (en ppm) o bien una dosis constante en términos de peso corporal de tos animales; deberá indicarse qué alternativa se ha seguido. En el caso de una sustancia administrada por sonda, la dosis debe darse todos los días a una hora similar, y ajustarse según sea necesario para mantener una dosis constante en términos de peso corporal del animal.
Cuando se utilice un estudio de administración continuada como fase previa de un estudio a largo plazo, deberá utilizarse en ambos estudios una dieta similar.
1.4.2.4. Ensayo límite
Si un ensayo con una sola dosis de, al menos, 1 000 mg/kg peso corporal/día o, en caso de administración con los alimentos o el agua de bebida, con una concentración equivalente en los alimentos o el agua de bebida (según las determinaciones del peso corporal), siguiendo los procedimientos descritos en el presente estudio, no se produce ningún efecto tóxico observable y si, a partir de datos de sustancias estructuralmente afines, no debiera esperarse la aparición de toxicidad, puede considerarse innecesario realizar un estudio completo con tres dosis. El ensayo límite es aplicable salvo cuando la exposición humana indique la necesidad de utilizar una dosis superior.
1.4.2.5. Período de observación
El período de observación debe ser de 28 días. Deben mantenerse animales en un lote satélite previsto para las observaciones de seguimiento durante al menos otros 14 días sin tratamiento, a fin de detectar la aparición retardada o la persistencia o la recuperación de los efectos tóxicos.
1.4.3. Procedimiento
Las dosis de sustancia se administran a los animales todos y cada uno de los días del período de 28; es necesario justificar el uso eventual de una posología de solo cinco días por semana. Cuando la sustancia estudiada se administre por alimentación forzada, deberá hacerse con una sola dosis utilizando una sonda gástrica o una cánula adecuada de intubación. El volumen máximo de líquido que puede administrarse de una sola vez depende del tamaño del animal. El volumen no debe pasar de 1 ml/100 g de peso corporal, excepto en el caso de las soluciones acuosas, en que puede llegarse a 2 ml/100 g de peso corporal. Excepto en el caso de sustancias irritantes o corrosivas, que muestren normalmente efectos exacerbados a concentraciones superiores, la variabilidad del volumen de ensayo deben reducirse al mínimo ajustando la concentración para garantizar un volumen constante en todas las dosis.
1.4.3.1. Observaciones generales
Debe hacerse una observación clínica general al menos una vez al día, preferentemente a la misma hora cada día y teniendo en cuenta el período más agudo de los efectos previstos tras la administración. Se registrará el estado sanitario de los animales. Al menos dos veces el día se observará la posible morbilidad y mortalidad de todos los animales. Cuando se vean, se sacarán los animales moribundos y los que padezcan dolor o sufrimiento grave, se sacrificarán de forma compasiva y se someterán a necropsia.
Se someterán todos los animales a observación clínica detallada antes de la primera exposición (para permitir realizar comparaciones con un mismo sujeto) y al menos una vez por semana después. Estas observaciones deben hacerse fuera de la jaula de alojamiento en un ambiente normal y, de preferencia, siempre a la misma hora. Estas observaciones deben registrarse cuidadosamente, preferentemente mediante sistemas de puntuación definidos explícitamente por el laboratorio de ensayo. Debe procurarse que las variaciones en las condiciones de ensayo sean mínimas y que las observaciones sean realizadas de preferencia por observadores ajenos al tratamiento. Los signos anotados deben incluir, sin ánimo de exhaustividad, los cambios de la piel, ojos, membranas mucosas, presencia de secreciones y excreciones y actividad neurovegetativa (por ejemplo, lagrimeo, piloerección, respiración anómala). Deben registrarse también los cambios observados en la marcha, postura y respuesta a la manipulación, así como la presencia de movimientos clónicos o tónicos, estereotipos (por ejemplo, realización excesiva de movimientos de limpieza, recorridos repetitivos en círculo) o comportamientos anómalos (por ejemplo, automutilación, marcha hacia atrás).
En la cuarta semana de exposición deben evaluarse la receptividad sensorial frente a estímulos de distintos tipos (por ejemplo, auditivos, visuales y propioceptivos), la fuerza de prensión y la actividad motriz. En la bibliografía (véase la introducción general de la parte B) se encuentran más datos sobre los procedimientos que pueden seguirse.
Las observaciones funcionales de la cuarta semana de exposición pueden omitirse cuando el estudio se realice como fase preliminar de un estudio posterior sobre toxicidad subcrónica (de 90 días). En este caso, las observaciones funcionales deberán incluirse en este estudio de continuación. Por otra parte, la disponibilidad de datos sobre las observaciones funcionales en el estudio de administración continuada puede facilitar la selección de las dosis utilizadas en un estudio posterior sobre toxicidad subcrónica.
De forma excepcional, las observaciones funcionales pueden omitirse también en lotes que muestren signos de toxicidad de otro tipo tales que interfieran significativamente con los resultados de las pruebas funcionales.
1.4.3.2. Peso corporal y consumo de alimento y agua
Al menos una vez por semana deben pesarse todos los animales y debe medirse el consumo de alimentos y agua. Si la sustancia estudiada se administra con el agua de bebida, deberá medirse también el consumo de agua al menos semanalmente.
1.4.3.3. Hematología
Los exámenes hematológicos siguientes deben practicarse al final del período de ensayo: hematocrito, concentración de hemoglobina, recuento de eritrocitos, recuento de leucocitos y fórmula leucocitaria, recuento de plaquetas y medida del tiempo o capacidad de coagulación de la sangre.
Las muestras de sangre deben tomarse de un punto indicado, justo antes del sacrificio de los animales (o como parte del método de sacrificio), y conservarse en condiciones adecuadas.
1.4.3.4. Bioquímica clínica
Deben hacerse determinaciones bioquímicas para investigar efectos tóxicos importante sobre los tejidos y, especialmente, en el riñón y el hígado, con muestras sanguíneas obtenidas de todos los animales justo antes de su sacrificio o como parte del método de sacrificio (aparte de los moribundos o sacrificados a lo largo del ensayo). Se recomienda que los animales estén en ayunas desde el día anterior antes de la toma de muestra (2). Los parámetros medidos del plasma o del suero incluirán las concentraciones de sodio, potasio, glucosa, colesterol total, urea, creatinina, proteínas totales y albúminas, al menos dos enzimas indicadoras de los efectos hepatocelulares (alanina aminotransferasa, aspartato aminotransferasa, fosfatasa alcalina, gamma-glutamil transpeptidasa, o sorbitol deshidrogenasa). La medida de otras enzimas (de origen hepático o no) y de ácidos biliares puede proporcionar información útil en ciertas circunstancias.
De forma opcional, pueden realizarse las siguientes determinaciones de orina en la última semana del estudio utilizando la recogida programada de orina: aspecto, volumen, osmolalidad o densidad, pH, proteínas, glucosa y sangre o células sanguíneas.
Además, debe considerarse la realización de estudios para investigar marcadores séricos de lesiones tisulares generales. Otras determinaciones que deben realizarse si se sabe o sospecha que las propiedades conocidas de la sustancia estudiada pueden afectar a las funciones metabólicas correspondientes incluyen las concentraciones de calcio, fosfato, triglicéridos en ayunas, hormonas específicas, metahemoglobina y colinesterasa. Se estudiará la necesidad de hacer estas determinaciones con las sustancias de determinadas clases o bien según cada caso.
En general, es necesario aplicar un enfoque flexible, en función de las especies y los efectos observados o esperados con una sustancia determinada.
Si los datos de que se dispone sobre antecedentes no son adecuados, debe considerarse la determinación de variables hematológicas y bioquímicas antes de iniciar la administración de la sustancia.
1.4.3.5. Necropsia macroscópica
Se debe practicar una necropsia macroscópica completa y detallada a todos los animales utilizados en el estudio, incluyéndose aspectos como un examen atento de la superficie corporal externa, todos los orificios, y las cavidades craneana, torácica y abdominal con su contenido. El hígado, los riñones, cápsulas suprarrenales, testículos, epidídimos, timo, bazo, cerebro y corazón de todos tos animales se limpiarán de los tejidos adherentes, según convenga, y se determinará su peso húmedo lo antes posible tras la disección, a fin de evitar su desecación.
Los tejidos que figuran a continuación deben conservarse en el medio de fijación más adecuado teniendo en cuenta tanto el tipo de tejido como el examen histopatológico posterior a que se vaya a someter todas las lesiones macroscópicas, encéfalo (zonas representativas, con inclusión del cerebro, cerebelo y protuberancia anular), médula espinal, estómago, intestino delgado y grueso (incluidas las placas de Peyer), hígado, riñones, cápsulas suprarrenales, bazo, carazón, timo, tiroides, tráquea y pulmones (conservados mediante inflado con fijador, seguido de inmersión), gónadas, órganos sexuales secundarios (por ejemplo, útero, próstata), vejiga urinaria, ganglios linfáticos (preferiblemente un ganglio relacionado con la vía de administración y otro distante de la misma, para tener en cuenta los efectos sistémicos), nervios periféricos (ciático o tibial), preferentemente muy próximos al músculo, y una sección de la médula ósea (o bien puede optarse por médula ósea aspirada y recién montada). Las observaciones clínicas y de otro tipo pueden indicar la necesidad de examinar otros tejidos. También deben conservarse todos los posibles órganos diana, según las propiedades conocidas de la sustancia estudiada.
1.4.3.6. Examen histopatotógico
Hay que practicar un examen histopatológico completo de los órganos y tejidos conservados de todos los animales del lote expuesto a la dosis elevada y del lote testigo. Estos exámenes deben ampliarse a animales de todos los demás lotes tratados, en caso de que se hayan observado cambios relacionados con el tratamiento en el lote de dosis elevada.
Deben examinarse todas las lesiones macroscópicas.
Cuando se utilice un lote satélite, deberá hacerse un examen histopatológico de los órganos y tejidos que presenten efectos en los lotes tratados.
2. RESULTADOS
Deben proporcionarse datos de cada animal. Además, deben resumirse todos los datos en un cuadro que muestre, respecto a cada lote, el número de animales al inicio del ensayo, el número de animales encontrados muertos durante la prueba o sacrificados por razones compasivas (y el momento de la muerte o sacrificio), el número de animales que presenten signos de toxicidad, una descripción de estos signos de toxicidad observados (con inclusión del momento de aparación, duración y gravedad de los efectos tóxicos), el número de animales que presenten lesiones, el tipo de lesiones y el porcentaje de animales en los que se haya observado cada tipo de lesión.
Siempre que sea posible, los resultados numéricos deberán evaluarse mediante un método estadístico adecuado y de amplia aceptación. Los métodos estadísticos deberán seleccionarse en la fase de diseño del estudio.
3. INFORME
INFORME DEL ENSAYO
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
|
Animales de ensayo:
|
|
|
Condiciones del ensayo;
|
|
|
Resultados:
|
|
|
Discusión de los resultados. |
|
|
Conclusiones. |
4. REFERENCIAS
Este método es análogo a la TG 407 de la OCDE
B.8. TOXICIDAD POR ADMINISTRACIÓN CONTINUADA {28 DÍAS) POR INHALACIÓN
1. MÉTODO
1.1. INTRODUCCIÓN
Es útil disponer de información preliminar sobre la distribución de las partículas por tamaño, la presión de vapor, el punto de fusión, el punto de ebullición, el punto de inflamación y la capacidad explosiva (si procede) de la sustancia.
Véase también la introducción general de la parte B (letra A).
1.2. DEFINICIÓN
Véase la introducción general de la parte B (letra B).
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
Se exponen varios grupos de animales, diariamente, durante un tiempo determinado, a la sustancia de ensayo en concentraciones distintas, utilizándose una sola concentración para cada lote, durante un período de 28 días. Cuando se utilice un vehículo para obtener una concentración adecuada de la sustancia en la atmósfera, se utilizará un lote testigo para el vehículo. Durante el período de administración se observa diariamente a los animales a fin de descubrir los síntomas de toxicidad. Se practica la autopsia a los animales que mueren durante el experimento y, al final del mismo, a los que hayan sobrevivido.
1.5. CRITERIOS DE CALIDAD
Ninguna.
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Preparación
Se mantiene a los animales en las condiciones de alojamiento y alimentación adecuadas para el experimento, por lo menos durante los cinco días anteriores al mismo. Antes de comenzar el ensayo, se eligen al azar animales jóvenes y sanos y se reparten en lotes. Si es necesario, se puede añadir un vehículo apropiado a la sustancia de ensayo para obtener una concentración apropiada de esta en la atmósfera. Si se utiliza un vehículo u otros aditivos para facilitar las dosis, estos no deberán ser tóxicos. Pueden utilizarse datos ya publicados.
16.2. Condiciones de ensayo
1.6.2.1. Animales de laboratorio
Salvo indicación contraria, la rata es la especie idónea. Hay que utilizar animales jóvenes y sanos de una cepa de laboratorio corriente.
Al principio del experimento, la diferencia de peso entre los animales no debe exceder del ± 20 % del valor medio apropiado.
1.6.2.2. Número y sexo
Deben utilizarse diez animales (5 hembras y 5 machos) para cada lote experimental. Las hembras deben ser nulíparas y no grávidas. Si se van a sacrificar algunos animales durante el experimento, habrá que añadir la cantidad de animales que se haya previsto sacrificar, Además, puede tratarse un lote satélite de 10 animales (5 de cada sexo) con la dosis más elevada durante 28 días para observar la reversibilidad, la persistencia o la aparición tardía de efectos tóxicos durante los 14 días siguientes al tratamiento. Se utiliza también un lote satélite de 10 animales testigo (5 de cada sexo).
1.6.2.3. Concentración de exposición
Son necesarias al menos tres concentraciones, así como un lote testigo y, en su caso, un lote testigo para el vehículo (la concentración del vehículo debe ser la del nivel de exposición más elevado). A excepción de la inhalación de la sustancia, los animales del grupo testigo deben ser tratados de la misma forma que los de los grupos experimentales. La dosis más elevada debe producir efectos tóxicos que no originen la muerte de ningún animal (o solo de algunos). La dosis más baja no debe producir ningún efecto tóxico detectable. Si se dispone una estimación aceptable sobre la exposición del hombre, la concentración más baja administrada debe ser superior a este valor. En condiciones ideales, la concentración intermedia debe producir unos efectos tóxicos observables mínimos. Si se utilizan varias concentraciones intermedias, la diferencia entre ellas debe ser suficiente para producir una gradación de los efectos tóxicos. En los lotes correspondientes a las concentraciones bajas e intermedias, así como en los lotes testigo, la mortalidad debe ser baja para que la evaluación de los resultados sea significativa.
1.6.2.4. Período de exposición
La exposición diaria debe ser de 6 horas, pero pueden resultar necesarios otros períodos de exposición para responder a algunas exigencias específicas.
1.6.2.5. Equipo
Los animales deben estar expuestos a la sustancia por medio de un dispositivo de inhalación que asegure un flujo continuo de aire de al menos 12 renovaciones por hora, garantizando un contenido en oxígeno apropiado y un reparto uniforme del producto de ensayo en el aire. Si se utiliza una cámara, debe estar concebida para impedir el amontonamiento de los animales y asegurar su exposición máxima, por inhalación de la sustancia de ensayo. Por regla general, para garantizar la estabilidad de la atmósfera de una cámara se debe procurar que el «volumen» total de animales de laboratorio no sobrepase el 5 % del volumen de la cámara de ensayo. También se puede recurrir a un sistema de exposición oro-nasal, únicamente de la cabeza o de cuerpo entero, en una cámara individual; con los dos primeros tipos de exposición se evita la absorción de la sustancia por otras vías.
1.6.2.6. Período de observación
Deberá observarse diariamente a los animales para descubrir los síntomas de toxicidad durante todo el período de tratamiento y de recuperación. Deberán registrarse el momento de la muerte y el momento en que aparecen y desaparecen los síntomas de toxicidad.
1.6.3. Procedimiento
Los animales están expuestos a la sustancia de ensayo diariamente entre 5 y 7 días por semana, durante un período de 28 días. Los animales de los grupos satélite destinados a observaciones complementarias deberán mantenerse en observación durante 14 días más, sin tratamiento, con el fin de descubrir la desaparición o la persistencia de los efectos tóxicos. La temperatura a la que se efectúa el ensayo debe ser de 22 oC ± 3 oC.
En condiciones ideales, la humedad relativa debe mantenerse entre el 30 y el 70 %, pero, en algunos casos, esto puede resultar imposible (por ejemplo, ensayos con aerosoles). Se puede evitar la pérdida de sustancia por dispersión a la zona circundante manteniendo una ligera presión negativa (≤ 5 mm de agua) dentro de la cámara. Durante la exposición los animales no deben recibir alimento ni agua.
Es conveniente utilizar un sistema de inhalación que funcione en condiciones dinámicas provisto de un dispositivo adecuado para el control analítico de la concentración. Para determinar las concentraciones de exposición adecuadas, se recomienda proceder a un ensayo preliminar. El caudal deberá ajustarse de manera que las concentraciones sean homogéneas en toda la cámara. El sistema debe permitir obtener lo más rápidamente posible unas condiciones de exposición estables.
Se debe medir o controlar:
|
a) |
el caudal de aire (permanentemente); |
|
b) |
la concentración real de la sustancia de ensayo, medida en la zona de respiración. Durante el período de exposición diaria la concentración no debe variar en más de ± 15 % respecto del valor medio. Sin embargo, en el caso de algunos aerosoles, es difícil alcanzar este grado de control, aceptándose en dichos casos una diferencia mayor. Durante todo el ensayo hay que mantener las concentraciones diarias lo más constantes posible. En el caso de los aerosoles, se hará al menos un análisis semanal del tamaño de las partículas en cada lote de ensayo; |
|
c) |
temperatura y humedad, permanentemente si es posible. |
Las observaciones tienen lugar durante y después de la exposición y se registran sistemáticamente; deben conservarse individualmente los datos de cada animal. Deben observarse diariamente todos los animales y registrar los síntomas de toxicidad, así como el momento de su aparición, su intensidad y su duración. La observación incluirá las modificaciones del pelo y la piel, los ojos y las mucosas, el aparato respiratorio, el sistema circulatorio, los sistemas nerviosos autónomo y central, la actividad somato-motriz y el comportamiento. El peso de los animales debe determinarse cada semana. Se recomienda igualmente medir el consumo alimentario cada semana. Es necesario observar regularmente a los animales para procurar que no se pierdan en el ensayo por razones como el canibalismo, autolisis de los tejidos o error de emplazamiento. Al final del ensayo se practicará la autopsia a todos los animales supervivientes de los lotes tratados no satélites. Los animales moribundos y los que muestren angustia o dolor graves deberán ser retirados inmediatamente, se sacrificarán de forma humanitaria y se les practicará la autopsia.
Los exámenes que figuran a continuación se efectuarán en todos los animales (incluidos los testigos) al terminar el ensayo:
|
i) |
análisis hematológico que incluya al menos el hematocrito, la concentración de hemoglobina, el recuento de hematíes y leucocitos, la fórmula leucocitaria, así como un estudio de la coagulación, |
|
ii) |
determinaciones bioquímicas clínicas de la sangre incluyendo al menos un parámetro sobre las funciones hepática y renal: alanina aminotransferasa (antiguamente conocida con el nombre de glutamano-piruvato-transatninasa) y aspartato aminotransferasa (antiguamente conocida con el nombre de glutamato-oxa-lacetato-transaminasa) del suero, nitrógeno ureico, albúmina, creatinina plasmática, bilirrubina total y proteínas séricas totales. |
Otras determinaciones que pueden ser necesarias para una evaluación toxicológica adecuada incluyen el calcio, el fósforo, los cloruros, el sodio, el potasio, la glucosa en ayunas, los lípidos, las hormonas, el equilibrio ácido-básico, la metahemoglobina y la actividad colinesterásica.
Si es necesario pueden realizarse otros análisis bioquímicos para profundizar el estudio de los efectos tóxicos observados.
1.6.3.1. Autopsia general
Se deberá practicar una autopsia general completa a todos los animales del ensayo. Al menos el hígado, los riñones, las glándulas suprarrenales, los pulmones y los testículos se deben pesar húmedos, lo más rápidamente posible después de la disección para evitar que se sequen. Los órganos y tejidos deben conservarse en un medio apropiado para que puedan realizarse exámenes histopatológicos posteriores: las vías respiratorias, el hígado, los riñones, el bazo, los testículos, las glándulas suprarrenales y el corazón, así como todos los órganos que presenten lesiones macroscópicas o modificaciones volumétricas. Los pulmones deben extraerse enteros, pesarse y tratarse con un fijador adecuado para conservar la estructura pulmonar.
1.63.2. Examen histopatológico
Hay que practicar un examen histopatológico de los órganos y tejidos conservados del lote expuesto a la concentración más alta y del lote o lotes testigo. Los órganos y los tejidos que presenten lesiones atribuibles a la sustancia de ensayo en la concentración más elevada deben ser examinados en todos los grupos que hayan estado expuestos a concentraciones más bajas. Deberá someterse a un examen histopatológico a los animales de todos los grupos satélite, centrado especialmente en los órganos y tejidos en los que se hayan comprobado lesiones en los otros lotes tratados.
2. RESULTADOS
Los resultados deben inventariarse en una tabla que indique, para cada lote de ensayo, el número de animales al comienzo del ensayo y el número de animales en los que se haya observado cada tipo de lesión.
Todos los resultados observados deben evaluarse por medio de un método estadístico adecuado. Puede utilizarse cualquier método estadístico reconocido.
3. INFORME
3.1. INFORME DEL ENSAYO
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
especie, cepa, origen, condiciones ambientales, régimen alimentario, etc., |
|
— |
condiciones de ensayo: Hay que describir el aparato de exposición, incluidos diseño, tipo, dimensiones, fuente de aire, sistema generador de aerosoles, método de acondicionamiento del aire, tratamiento del aire de salida y, en su caso, método de alojamiento de los animales en la cámara de ensayo. Debe describirse el equipo utilizado para medir la temperatura, la humedad y, en su caso, la estabilidad de las concentraciones de aerosoles o la granulometría. Datos relativos a la exposición: Deben presentarse en forma de tabla, indicando los valores medios y una medida de la variabilidad (por ejemplo, desviación típica); deben incluir, en la medida de lo posible:
|
|
— |
respuesta tóxica por sexo y concentración, |
|
— |
momento de la muerte durante el ensayo o indicación de que los animales han sobrevivido a la misma, |
|
— |
descripción de los efectos tóxicos o de otro tipo; nivel que no causa ningún efecto, |
|
— |
momento de observación de cualquier síntoma anormal y evolución del mismo, |
|
— |
cantidades de alimento y peso corporal, |
|
— |
análisis hematológicos practicados y resultados de estos, |
|
— |
pruebas bioquímicas clínicas practicadas y resultados de estas, |
|
— |
resultados de la autopsia, |
|
— |
descripción detallada de todas las observaciones histopatológicas, |
|
— |
tratamiento estadístico de los resultados, si es posible, |
|
— |
discusión de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la introducción general de la parte B (letra D).
4. REFERENCIAS
Véase la introducción general de la parte B (letra E).
B.9. TOXICIDAD POR ADMINISTRACIÓN CONTINUADA (28 DÍAS) VÍA CUTÁNEA
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la introducción general de la parte B (letra A).
1.2. DEFINICIÓN
Véase la introducción general de la parte B (letra B).
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
La sustancia de ensayo se aplica diariamente por vía cutánea en dosis distintas a varios lotes de animales de laboratorio, utilizándose una sola dosis para cada lote, durante un período de 28 días. Durante el período de aplicación se observa diariamente a los animales a fin de descubrir los síntomas de toxicidad. Se practica la autopsia a los animales que mueren durante el ensayo así como a los que sobreviven al final del mismo.
1.5. CRITERIOS DE CALIDAD
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Preparación
Se mantiene a los animales en las condiciones de alojamiento y alimentación adecuadas para el experimento, por lo menos, durante los cinco días anteriores al mismo. Antes de comenzar el ensayo, se eligen al azar animales jóvenes y sanos y se reparten en lotes tratados y lotes testigo. Poco tiempo antes del ensayo se esquila la piel de la región dorsal de los animales. Se puede recurrir al rasurado, pero, en este caso, debe efectuarse unas 24 horas antes del ensayo. Suele ser necesario repetir el esquileo o el rasurado aproximadamente cada semana. Durante el esquileo o el rasurado hay que procurar no causar lesiones en la piel. La superficie que hay que despejar para aplicar la sustancia de ensayo no debe ser inferior al 10 % de la superficie corporal. Debe tenerse en cuenta el peso del animal para decidir la zona que hay que despejar y las dimensiones de la superficie que se va a tratar. Cuando la sustancia sea sólida (que, si es preciso, puede pulverizarse), deberá humedecerse con agua o, en su caso, con un vehículo apropiado, de forma que se asegure un buen contacto con la piel. Las sustancias líquidas se utilizan generalmente sin diluir. Se procede a una aplicación diaria, entre 5 y 7 días por semana.
1.6.2. Condiciones de ensayo
1.6.2.1. Animales de laboratorio
Pueden utilizarse ratas, conejos o cobayas adultos. Se pueden utilizar también otras especies pero, en este caso, se habrá de justificar su utilización.
Al principio del ensayo la diferencia de peso entre los animales no debe exceder del ± 20 % del valor medio adecuado.
1.6.2.2. Número y sexo
Deben utilizarse al menos diez animales (5 hembras y 5 machos) de piel sana, para cada dosis. Las hembras deben ser nulíparas y no grávidas. Si se van a sacrificar algunos animales durante el ensayo habrá que añadir la cantidad de animales que se haya previsto sacrificar. Además, puede tratarse un lote satélite de 10 animales (5 de cada sexo) con el nivel de dosis más elevado durante 28 días para observar la reversibilidad, la persistencia o la aparición tardía de efectos tóxicos durante los 14 días siguientes al tratamiento. Se utiliza también un lote satélite de 10 animales testigo (5 de cada sexo).
1.6.2.3. Dosis
Se utilizan al menos tres dosis, así como un lote testigo y, en su caso, un lote testigo para el vehículo. El período mínimo de exposición será de 6 horas diarias. La sustancia de ensayo debe aplicarse todos los días a la misma hora y las dosis deben adaptarse semanalmente o bisemanalmente con objeto de conservar un nivel de dosis constante respecto al peso corporal de los animales. A excepción de la administración de las sustancias de ensayo, debe tratarse a los animales del lote testigo de la misma forma que a los de los lotes experimentales. Cuando para facilitar la administración se utilice un vehículo, este se administrará también al lote testigo, del vehículo en las mismas condiciones que a los lotes tratados; la dosis será la misma que se administre al grupo tratado con la dosis más elevada. La dosis más elevada debe producir efectos tóxicos que no originen la muerte de ningún animal (o solo de algunos). La dosis más baja no debe producir ningún efecto tóxico detectable. Si se dispone de una estimación aceptable sobre la exposición del hombre, la dosis más baja administrada debe superar este valor. En condiciones óptimas la dosis intermedia debe producir unos efectos tóxicos observables mínimos. Si se utilizan varias dosis intermedias, la diferencia entre ellas debe ser suficiente para producir una graduación de los efectos tóxicos. En los lotes correspondientes a dosis bajas o intermedias, así como en los lotes testigo, la mortalidad debe ser baja para que la evaluación de los resultados sea significativa.
Si la aplicación de la sustancia de ensayo provoca una irritación cutánea grave, deben reducirse las concentraciones; esto puede producir una disminución, incluso una desaparición, de otros efectos tóxicos en las dosis más elevadas. Si las lesiones cutáneas son muy graves, puede resultar necesario detener el experimento y comenzar de nuevo con concentraciones más bajas.
1.6.2.4. Prueba límite
Si en un experimento preliminar realizado con una dosis de 1 000 mg/kg, o con una dosis más elevada en función de una posible exposición humana (si se conoce), no se ha observado ningún efecto tóxico, puede ser innecesario continuar el experimento.
1.6.2.5. Período de observación
Se deben observar todos los animales diariamente con el fin de descubrir los síntomas de toxicidad. Debe registrarse el momento de la muerte, así como el momento en que aparecen y desaparecen los síntomas de toxicidad.
1.6.3. Procedimiento
Los animales deben estar alojados en jaulas individuales. En condiciones óptimas, la sustancia se administra a los animales todos los días, durante un período de 28 días. Los animales de todos los grupos satélites destinados a observaciones complementarias deben mantenerse en observación 14 días más, sin tratamiento, para comprobar la curación o la persistencia de los efectos tóxicos. La duración mínima de la exposición debe ser de 6 horas diarias.
La sustancia debe aplicarse uniformemente sobre una superficie equivalente más o menos al 10 % de la superficie total corporal. Cuando se trata de sustancias altamente tóxicas, la superficie cubierta puede ser menor, pero procurando que al aplicar la sustancia se forme en toda la superficie una capa lo más fina y uniforme posible.
Durante la exposición, la sustancia se mantiene en contacto con la piel mediante un apósito de gasa porosa y un esparadrapo no irritante. Además, la superficie tratada debe estar convenientemente cubierta para mantener en su sitio el apósito de gasa y la sustancia e impedir que los animales puedan ingerir esta última. Pueden utilizarse aparatos de contención para impedir la ingestión de la sustancia, pero no se recomienda una inmovilización completa. Puede usarse un dispositivo protector en forma de collar como método alternativo.
Al término del período de exposición, hay que eliminar cualquier residuo de sustancia, a ser posible, con agua, o con cualquier otro procedimiento adecuado de limpieza de la piel.
Deben observarse todos los animales diariamente y registrar los síntomas de toxicidad, así como el momento de su aparición, su intensidad y su duración. La observación incluirá las modificaciones del pelo y la piel, los ojos y las mucosas, el aparato respiratorio, el sistema circulatorio, los sistemas nerviosos autónomo y central, la actividad somato-motriz y el comportamiento. Semanalmente deben determinarse los pesos de los animales, y se recomienda igualmente medir el consumo alimentario cada semana. Es necesario observar regularmente a los animales para procurar que no se pierdan en el experimento por razones como el canibalismo, autolisis de los tejidos o error de emplazamiento. Al término de la experiencia se practica la autopsia a todos los animales supervivientes de los lotes tratados no satélites. Los animales moribundos y los que presenten angustia o dolor graves se retirarán inmediatamente, se sacrificarán de forma humanitaria y se les practicará la autopsia.
Al terminar el ensayo se efectuarán los siguientes análisis en todos los animales (incluidos los del lote testigo):
|
1) |
análisis hematológico que incluya al menos el hematocrito, la concentración de hemoglobina, el recuento de hematíes y leucocitos, la fórmula leucocitaria, así como un estudio de la coagulación; |
|
2) |
determinaciones bioquímicas clínicas de la sangre incluyendo al menos un parámetro sobre las funciones hepáticas y renal: alanina-aminotransferasa (antiguamente conocida con el nombre de glutamato-piruva to-transaminasa) y aspartato aminotransferasa (antiguamente conocido con el nombre de glutamato-oxala- cetato-transaminasa) del suero, nitrógeno ureico, albúmina, creatinina plasmática, bilirrubina total y proteínas séricas totales. |
Otras determinaciones que pueden ser necesarias para una evaluación toxicológica adecuada incluyen el calcio, fósforo, cloruros, sodio, potasio, glucosa en ayunas, lípidos, hormonas, el equilibrio ácido-básico, metahemo-globina y la actividad colinesterásica.
Si es necesario, pueden realizarse otros análisis bioquímicos para profundizar el estudio de los efectos tóxicos observados.
1.6.4. Autopsia general
Se practicará una autopsia general completa a todos los animales sometidos al ensayo. Al menos el hígado, los riñones, las glándulas suprarrenales y los testículos se deben pesar húmedos, lo más rápidamente posible después de la disección, para evitar que se sequen. Los órganos y tejidos, es decir, la piel normal y tratada, el hígado, los riñones, el bazo, los testículos, las glándulas suprarrenales, el corazón y los órganos diana (es decir, los que presentan lesiones importantes o modificaciones volumétricas) deben conservarse en un medio apropiado para que puedan realizarse exámenes histopatológicos posteriores.
1.6.5. Examen histopatológico
Hay que practicar un examen histopatológico de los órganos y tejidos que se conserven del lote expuesto a la dosis más elevada y del lote testigo. Los órganos y los tejidos que presenten lesiones inducidas por la sustancia de ensayo en el nivel de dosis más elevado, deben examinarse en todos los lotes que hayan estado expuestos a dosis más bajas. Los animales del lote satélite deberán someterse a un examen histológico, centrado especialmente en los órganos y tejidos en los que se hayan comprobado lesiones en los demás lotes tratados.
2. RESULTADOS
Los resultados deberán inventariarse en un cuadro que indique, para cada lote de ensayo, el número de animales al comienzo del ensayo y el número de animales en los que se haya observado cada tipo de lesión.
Todos los resultados observados deben evaluarse por medio de un método estadístico adecuado. Puede utilizarse cualquier método estadístico reconocido.
3. INFORME
3.1. INFORME DEL ENSAYO
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
datos relativos a los animales (especie, cepa, origen, condiciones ambientales, régimen alimentario, etc.), |
|
— |
condiciones de ensayo, (incluido el tipo de apósito: oclusivo o no oclusivo), |
|
— |
dosis (en su caso, también el vehículo) y concentraciones, |
|
— |
nivel que no produce ningún efecto, si es posible, |
|
— |
respuesta tóxica por sexo y dosis, |
|
— |
momento de la muerte durante el ensayo o indicación de que los animales han sobrevivido, |
|
— |
efectos tóxicos u otros, |
|
— |
momento de observación de cualquier síntoma anormal y su evolución, |
|
— |
cantidades de alimento y peso corporal, |
|
— |
análisis hematológicos practicados y resultados, |
|
— |
pruebas bioquímicas clínicas practicadas y resultados, |
|
— |
resultados de la autopsia, |
|
— |
descripción detallada de todas las observaciones histopatológicas, |
|
— |
tratamiento estadístico de los resultados, si es posible, |
|
— |
discusión de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la introducción general de la parte B (letra D).
4. REFERENCIAS
Véase la introducción general de la parte B (letra E).
«B.10. MUTAGENICIDAD — ENSAYO DE ABERRACIONES CROMOSOMICAS IN VITRO EN MAMÍFEROS
1. MÉTODO
El presente método reproduce las directrices del documento OCDE TG 473 sobre el ensayo de aberraciones cromosómicas in vitro en mamíferos (1997).
1.1. INTRODUCCIÓN
El ensayo de aberraciones cromosómicas in vitro tiene por objeto detectar agentes que provocan aberraciones cromosómicas estructurales en células de mamífero en cultivo (1) (2) (3). Las aberraciones estructurales pueden ser cromosómicas o cromatídicas. La mayoría de los rnutágenos químicos dan lugar a aberraciones cromatídicas, aunque también pueden producirse aberraciones cromosómicas. Un aumento de la poliploidía puede indicar que una sustancia química es capaz de inducir aberraciones numéricas. No obstante, el presente método no está pensado para medir aberraciones de ese tipo ni se emplea habitualmente con ese fin. Las mutaciones cromosómicas y los efectos relacionados ocasionan numerosas enfermedades genéticas humanas. Además de ello, son muchas las pruebas de que las mutaciones cromosómicas y los efectos relacionados que provocan alteraciones en los oncogenes y los genes supresores de tumores de las células somáticas intervienen en la inducción de cánceres en los seres humanos y los animales de experimentación.
En el ensayo de aberraciones cromosómicas in vitro pueden emplearse cultivos de líneas celulares establecidas, estirpes celulares o cultivos de células primarias. Las células deben seleccionarse sobre la base de su capacidad de crecimiento en cultivo, la estabilidad cariotípica, el número y la diversidad cromosómica y la frecuencia de las aberraciones cromosómicas espontáneas.
En los ensayos in vitro suele ser necesario emplear un sistema de activación metabólica exógena, si bien este no puede reproducir todas las condiciones que se dan in vivo en los mamíferos. Deben evitarse las circunstancias que puedan conducir a resultados positivos que no reflejen la mutagenicidad intrínseca sino que se deban a cambios en el pH, la osmolalidad o altos grados de citotoxicidad (4) (5).
El ensayo se emplea para detectar las posibles sustancias mutágenas o carcinógenas para los mamíferos. Pese a que muchos de los compuestos que dan un resultado positivo en este ensayo son carcinógenos para los mamíferos, no hay una correlación absoluta entre los resultados del mismo y la carcinogenicidad. La correlación depende de la clase química. Además de ello, cada vez hay más pruebas de que existen carcinógenos que el ensayo no detecta, pues, al parecer, estos actúan por mecanismos distintos de la lesión directa del ADN.
Véase asimismo la introducción general de la parte B.
1.2. DEFINICIONES
Aberración cromatídica: lesión cromosómica estructural que consiste en la rotura de una cromátida o la rotura y reunión de cromátidas.
Aberración cromosómica: lesión cromosómica estructural que consiste en la rotura o en la rotura y reunión de ambas cromátidas en el mismo sitio.
Endorreduplicación: proceso en el que, en lugar de entrar en mitosis tras la fase S de replicación del ADN, el núcleo inicia otra tase S, lo cual da lugar a cromosomas con 4, 8, 16, etc. cromátidas.
Gap: lesión acromática más estrecha que la cromátida, con alteración mínima de la alineación de las cromátidas.
Índice mitótico: proporción entre el número de células en metafase y el número total de células de una población celular; indica el grado de proliferación de dicha población.
Aberración numérica: un cambio en el número de cromosomas a partir del número normal propio de las células empleadas.
Poliploidía: un múltiplo del número de dotación cromosómica haploide (n) superior al diploide (3n, 4n, etc.).
Aberración estructural: alteración de la estructura cromosómica (deleciones y fragmentos, intracambios o intercambios) detectable mediante examen microscópico de la metafase de la división celular.
1.3. PRINCIPIO DEL MÉTODO
Se exponen los cultivos celulares a la sustancia de ensayo, con y sin activación metabólica, tras lo cual se tratan a intervalos preestablecidos con una sustancia que detenga la divisón celular en la metafase (por ejemplo, ColcemidR o colchicina). Se recolectan las células, se tiñen y se observan al microscopio aquellas que se encuentren en la metafase con el fin de detectar la presencia de aberraciones cromosómicas.
1.4. DESCRIPCIÓN DEL MÉTODO
1.4.1. Preparación
1.4.1.1. Células
Pueden emplearse líneas celulares, estirpes o cultivos de células primarias, incluidas las células humanas (fibroblastos de hámster chino, linfocitos de la sangre periférica de humanos o de otros mamíferos, etc.).
1.4.1.2. Medios y condiciones de cultivo
Para mantener los cultivos se emplean medios de cultivo y condiciones de incubación adecuados (recipientes de cultivo, concentración de CO2, temperatura y humedad). En el caso de las líneas celulares establecidas y las estirpes, se comprobará regularmente la estabilidad del número cromosómico modal y la ausencia de contaminación por micoplasma. Se desecharán los cultivos contaminados. Debe conocerse la duración del ciclo celular normal de las células empleadas en esas condiciones de cultivo.
1.4.1.3. Preparación de los cultivos
Líneas celulares establecidas y estirpes: las células se obtienen de cultivos madre, se siembran en un medio de cultivo a una densidad a la cual los cultivos no confluyan antes del momento de la recolección y se incuban a 37 oC.
Linofocitos: se añade sangre completa tratada con un antícoagulante (por ejemplo, heparina), o bien linfocitos aislados procedentes de sujetos sanos, a un medio de cultivo que contenga un mitógeno (por ejemplo, fitohemaglutinina) y se incuba a 37 oC.
1.4.1.4. Activación metabólica
Las células deben exponerse a la sustancia de ensayo en presencia y en ausencia de un sistema adecuado de activación metabólica. El sistema más comúnmente empleado es una fracción postmitocondrial (S9) a la que se añaden cofactores y que se obtiene de hígados de roedores tratados con inductores enzimátícos como el Aroclor 12 54 (6) (7) (8) (9) o una mezcla de fenobarbital β-naftoflavona (10) (11) (12).
La fracción postmitocondrial suele emplearse en concentraciones que varían del 1 ai 10 % v/v en el medio de ensayo final. La elección del sistema de activación metabólica puede depender de la clase química de la sustancia estudiada. En algunos casos puede ser oportuno utilizar varias concentraciones distintas de fracción postmitocondrial.
Son varias las técnicas —como la producción mediante ingeniería genética de líneas celulares que expresen enzimas activadoras específicas— que pueden proporcionar el potencial de activación endógena. La elección de las líneas celulares debe justificarse científicamente (por ejemplo, por la importancia de la isoenzima del citocromo P4 50 para el metabolismo de la sustancia de ensayo, etc.).
1.4.1.5. Preparación de la sustancia de ensayo
Las sustancias de ensayo sólidas deben disolverse o suspenderse en disolventes o vehículos adecuados y, si es preciso, diluirse antes de tratar las células. Las sustancias de ensayo líquidas pueden añadirse directamente al sistema de ensayo y/o diluirse antes del tratamiento. Han de emplearse soluciones de la sustancia de ensayo recién preparadas, salvo que los datos relativos a la estabilidad demuestren que es posible conservarlas.
1.4.2. Condiciones del ensayo
1.4.2.1. Disolvente o vehículo
Es preciso cerciorarse de que el disolvente o vehículo no produce reacciones químicas con la sustancia de ensayo y es compatible con la supervivencia de las células y la actividad de la S9. Si se emplean disolventes o vehículos poco conocidos, debe disponerse de información que avale dicha compatibilidad. Siempre que sea posible, se recomienda considerar en primer lugar la utilización de un disolvente o vehículo acuoso. Cuando se estudien sustancias inestables en presencia de agua, deberán emplearse disolventes orgánicos que no contengan agua. Esta podrá eliminarse añadiendo un tamiz molecular,
1.4.2.2. Concentraciones de exposición
Para determinar la mayor concentración de exposición deben considerarse al menos los siguientes criterios: citotoxicidad, solubilidad en el sistema de ensayo y variaciones del pH o la osmolalidad.
La citotoxicidad se determinará con y sin activación metabólica en el experimento principal mediante un indicador adecuado de la integridad y el crecimiento celular, como el grado de confluencia, el recuento de células viables o el índice mitótico. Puede ser útil determinar la citotoxicidad y la solubilidad en un experimento preliminar.
Se emplearán al menos tres concentraciones analizables. Si se produce citotoxicidad, la gama de concentraciones deberá abarcar desde la toxicidad máxima hasta una toxicidad escasa o nula, lo cual suele significar que la separación entre concentraciones corresponde a un factor comprendido entre 2 y 10. En el momento de la recolección, con la concentración mayor debe producirse una reducción considerable del grado de confluencia, el recuento celular o el índice mitótico (en todos los casos superior al 50 %). El índice mitótico solo constituye una medida indirecta de los efectos citotóxicos o citostáticos y está relacionado con el tiempo transcurrido desde el tratamiento. No obstante, cabe utilizar dicho índice para los cultivos en suspensión en los cuales las demás mediciones de toxicidad pueden resultar engorrosas y poco prácticas. Los datos relativos a la cinética del ciclo celular, como el tiempo medio de generación (TMG), pueden servir de información adicional. Sin embargo, el TMG es una media general que no siempre refleja la existencia de subpoblaciones que presentan retraso. Además de ello, incluso un ligero aumento del tiempo medio de generación puede asociarse a un retraso muy importante en el tiempo de mayor producción de aberraciones.
En el caso de sustancias escasamente citotóxicas, la concentración máxima de ensayo será la menor de estas tres: 5 μl/ml, 5 mg/ml o 0,01 M
En el caso de sustancias escasamente solubles que no resultan tóxicas a concentraciones inferiores al límite de solubilidad, la mayor dosis empleada deberá corresponder a una concentración superior a dicho límite en el medio de cultivo final al terminar el período de tratamiento. En algunos casos (por ejemplo, cuando se produce toxicidad únicamente con concentraciones superiores a la concentración mínima de insolubilidad), es aconsejable hacer pruebas con varias concentraciones con precipitación visible. Puede ser útil determinar la solubilidad al principio y al final del tratamiento, pues esta pueda variar durante la exposición en el sistema de ensayo debido a la presencia de células, S9, suero, etc. La insolubilidad puede detectarse por simple observación. El precipitado no debe interferir en la evaluación del resultado.
1.4.2.3. Controles positivos y negativos
En todos los experimentos deberán realizarse controles positivos y negativos (disolvente o vehículo) en paralelo, con y sin activación metabólica. Cuando se aplique esta, la sustancia del control positivo ha de requerir activación para provocar una respuesta mutagénica.
En los controles positivos debe emplearse un clastógeno conocido con grados de exposición en los que quepa esperar un aumento reproducible y detectable respecto a la frecuencia espontánea, que ponga de manifiesto la sensibilidad del sistema de ensayo.
Las concentraciones del control positivo deben ser tales que los efectos sean claros, pero no revelen inmediatamente al lector la identidad de los portaobjetos codificados. A continuación figuran algunos ejemplos de sustancias para los controles positivos:
|
Activación metabólica |
Sustancia |
No CAS |
No Einecs |
|
Ausencia de activación metabólica exógena |
Metanosulfonato de metilo |
66-27-3 |
200-625-0 |
|
Metanosulfonato de etilo |
62-50-0 |
200-536-7 |
|
|
Etil-nitrosourea |
759-73-9 |
212-072-2 |
|
|
Mitomicina C |
50-07-7 |
212-072-2 |
|
|
N-óxido de 4-nitroquinolina |
56-57-5 |
200-281-1 |
|
|
Presencia de activación metabólica exógena |
Bcnzo[a]pireno |
50-32-8 |
200-028-5 |
|
Ciclofosfamida Monohidrato de ciclofosfamida |
50-18-0 6055-19-2 |
200-015-4 |
Pueden emplearse otras sustancias adecuadas para los controles positivos. Deberá considerarse la utilización en dichos controles de sustancias de clases químicas afines, cuando sea posible.
Se realizarán, para cada período de recolección, controles negativos únicamente con el disolvente o vehículo en el medio de tratamiento, y se tratarán de igual manera que los cultivos del ensayo. Se prepararán asimismo controles sin tratar, salvo que exista información anterior sobre controles que demuestre que el disolvente elegido no induce efectos nocivos ni mutágenos.
1.4.3. Procedimiento
1.4.3.1. Tratamiento con la sustancia de ensayo
Las células en crecimiento se tratan con la sustancia de ensayo en presencia y en ausencia de un sistema de activación metabólica. Los linfocitos deben empezar a tratarse unas 48 horas después de la estimulación mitogénica.
|
1.4.3.2. |
Como norma, deben prepararse dos cultivos para cada concentración y se recomienda seriamente hacer lo propio en el caso de los controles negativos o el disolvente. Sí se demuestra, sobre la base de datos anteriores, que la diferencia entre los dos cultivos es mínima (13) (14), puede bastar un solo cultivo para cada concentración. Las sustancias gaseosas o volátiles han de someterse a ensayo con métodos adecuados (recipientes de cultivo herméticos, etc.) (15) (16). |
1.4.3.3. Recolección de las células
En el primer experimento se exponen las células a la sustancia de ensayo, con y sin activación metabólica, durante 3 a 6 horas. Se toma una muestra transcurrido un período, desde el inicio del tratamiento, de aproximadamente 1,5 veces la duración del ciclo celular normal (12). Si este protocolo da resultados negativos con y sin activación, debe efectuarse otro experimento sin activación, pero con tratamiento continuo hasta que se tome la muestra tras un período de aproximadamente 1,5 veces la duración del ciclo celular normal. Algunas sustancias químicas se detectan mejor con un tiempo de tratamiento y toma de muestra superior a dicho período. La obtención de resultados negativos en presencia de activación metabólica debe confirmarse caso por caso. Si dicha confirmación no se considera necesaria, debe justificarse.
1.4.3.4. Preparación de los cromosomas
Se tratan los cultivos celulares con ColcemidR o colchicina por lo general durante un período de 1 a 3 horas antes de la recolección. Los cultivos celulares se recolectan y procesan por separado para preparar los cromosomas. Dicha preparación incluye el tratamiento hipotónico de las células, la fijación y la tinción.
1.4.3.5. Análisis
Antes de analizarlas al microscopio, se asignará un código independiente a cada preparación, incluidos los controles positivos y negativos. Dado que los métodos de fijación provocan con frecuencia la rotura de células en metafase y la pérdida de cromosomas, todos los tipos de células analizadas deben contener un número de centrómeros igual al número modal ± 2. Deben analizarse al menos 200 metafases bien extendidas en cada concentración y control y, en su caso, repartidas de forma equilibrada entre los cultivos dobles. La cantidad puede ser menor si se observan numerosas aberraciones.
Pese a que el objeto del ensayo consiste en detectar aberraciones cromosómicas estructurales, es importante registrar los posibles casos de poliploidía y endorreduplicación.
2. RESULTADOS
2.1. TRATAMIENTO DE LOS RESULTADOS
Dado que la unidad experimental es la célula, se evaluará el porcentaje de estas que presenta aberraciones cromosómicas estructurales. Se hará una relación de los distintos tipos, el número y la frecuencia de dichas aberraciones en los cultivos experimentales y los controles. Los gaps se registrarán por separado y se recogerán en el informe, pero por lo general no se incluirán en la frecuencia total de aberraciones.
Asimismo, se registrarán las determinaciones de citotoxicidad realizadas en paralelo en todos los cultivos tratados y los controles negativos de los principales experimentos sobre aberraciones.
Se proporcionarán los datos relativos a cada cultivo y se resumirán en forma de tabla.
No será preciso comprobar los resultados positivos claros, si bien los resultados dudosos deberán aclararse mediante más ensayos en los que convendría modificar las condiciones experimentales. En el punto 1.4.3.3 se trata la pertinencia de confirmar los resultados negativos. En los experimentos ulteriores se considerará la posibilidad de modificar los parámetros del estudio para ampliar la gama de condiciones analizadas. Entre los parámetros modificables se encuentra la separación entre concentraciones y las condiciones de activación metabólica.
2.2. EVALUACIÓN E INTERPRETACIÓN DE LOS RESULTADOS
Son varios los criterios para determinar si un resultado es positivo (aumento relacionado con la concentración o aumento reproducible del número de células que presentan aberraciones cromosómicas, etc.). Debe considerarse en primer lugar la importancia biológica de los resultados. Pueden emplearse métodos estadísticos como apoyo para evaluar los resultados del ensayo (3) (13). El hecho de que los datos estadísticos sean significativos no ha de ser el único factor para determinar que una respuesta es positiva.
El aumento del número de células poliploides puede indicar que la sustancia de ensayo es capaz de inhibir procesos mitóticos y de producir aberraciones cromosómicas numéricas. El aumento del número de células con cromosomas endorreduplicados puede indicar que la sustancia de ensayo es capaz de inhibir la progresión del ciclo celular (17) (18).
No se considerarán mutágenas en el sistema de ensayo las sustancias que den lugar a resultados que no se ajusten a los criterios anteriores.
Si bien en la mayoría de los experimentos los resultados serán claramente positivos o negativos, en algunos casos el conjunto de datos no permitirá emitir un juicio definitivo sobre la actividad de la sustancia de ensayo. Con independencia del número de veces que se repita el experimento, los resultados pueden seguir siendo ambiguos o dudosos.
Un resultado positivo en el ensayo de aberraciones cromosómicas in vitro indica que la sustancia de ensayo provoca aberraciones cromosómicas estructurales en los cultivos de células somáticas de mamífero. Un resultado negativo indica que, en las condiciones del ensayo, la sustancia de ensayo no provoca aberraciones cromosómicas en los cultivos de células somáticas de mamífero.
3. INFORME
INFORME DEL ENSAYO
El informe del ensayo incluirá la información siguiente:
|
|
Disolvente o vehículo:
|
|
|
Células:
|
|
|
Condiciones del ensayo:
|
|
|
Resultados:
|
|
|
Discusión de los resultados. |
|
|
Conclusiones. |
4. REFERENCIAS
|
(1) |
Evans, H,). (1976), Cytological Methods for Detecting Chemical Mutagens, in: Chemical mutagens, Principies and Methods for their Detection, Vol. 4, Hollaender, A. (ed) Plenum Press, New York and London, pp. 1-29. |
|
(2) |
Ishidate, M. Jr. and Sofuni, T. (1985), The In Vítro Chromosomal Aberration Test Using Chínese Hams ter Lung (CHL) Fibroblast Cells in Culture, in: Progress in Mutation Research, Vol. 5. Ashby, J. et al., (eds) Elsevier Science Publishers, Amsterdam-New York-Oxford, pp. 427–432. |
|
(3) |
Calloway, S. M, Armstrong, M. J., Reuben, C, Colman, S., Brovvn, B., Cannon, C, Bloom, A. D., Nakamura, R. Ahmed, M, Duk, S., Rimpo, J., Margolin, G. H., Resnick, M. A., Andersen, G. and Zeiger, E. (1978), Chromosome aberration and sister chromatic exchanges in Chinese hamster ovary cells: Evaluation of 108 chemicals, Environs. Molec. Mutagen 10 (suppl. 10), pp. 1–175. |
|
(4) |
Scott, D., Galloway, S. M., Marshall, R. R., Ishidate, M. Jr., Brusick, D., Ashby, J. and Myhr, B. C. (1991), Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9, Mutation Res., 257, pp. 147—204. |
|
(5) |
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K. (1992), Clastogenicity of low pH to Various Cultured Mammalian Cells, Mutation Res., 268, pp. 297–305. |
|
(6) |
Ames, B. N., McCann, J. and Yamasaki, E. (1975), Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagcnicity Test, Mutation Res,. 31, pp. 347–364. |
|
(7) |
Maron, D. M. and Ames, B. N. (1983), Revised Methods for the Salmonella Mutagenicity Test, Mutation Res.. 113. pp. 173–215. |
|
(8) |
Natarajan, A. T., Tates, A. D., van Buul, P. P. W., Meijers, M. and de Vogel, N. (1976), Cytogenetic Effects of Mutagen/Carcinogens after Activation in a Microsomal System In Vitro, }. Induction of Chro mosome Aberrations and Sister Chromatid Exchange by Diethylnitrosamine (DEN) and Dimethylnitro- samine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes, Mutation Res., 37, pp. 83-90. |
|
(9) |
Matsuoka, A., Hayashi, M. and Ishidate, M. Jr. {1979), Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro, Mutation Res., 66, pp. 277–290. |
|
(10) |
Elliot, B. M., Combes, R., D., Elcombe, C. R., Gatehouse, D. G., Gibson, G. G., Mackay,]. M. and Wolf, R. C. (1992), Report of UK environmental Mutagen Society Working Party. Alternatives to Aroclar 1254-induced S9 in In Vitro Genotoxicity Assays, Mutagenesis, 7, pp. 175–177. |
|
(11) |
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976), A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems, in: de Serres, F. J., Fouts, J. R., Berid, J. R. and Philpot, R. M. (eds), In Vitro Metabolic Activation in Mutagenesis Testing, Elsevier, North-Holland, pp. 85–88. |
|
(12) |
Galloway, S. M., Aardema, M.]., Ishidate, M. Jr., Iven, J. L, Kirkland, D. J., Morita, T., Mosesso, P., Sofuni, T. (1994), Report from Working Group on in In Vitro Tests for Chromosomal Aberrations, Mutation Res., 312, pp. 241–261. |
|
(13) |
Richardson, C., Williams, D. A., Allen, J. A., Amphlett, G., Chanter, D. O. and Phillips, B. (1989), Analy sis of Data from In Vitro Cytogenetic Assays, in: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D. J., (ed) Cambridge University Press, Cambridge, pp. 141–154. |
|
(14) |
Soper, K. A. and Galloway, S. M. (1994), Replicate Flasks are not Necessary for In Vitro Chromosome Aberration Assays in CHO Cells, Mutation Res., 312, pp. 139–149. |
|
(15) |
Krahn, D. F., Barsky, F. C. and McCooey, K. T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids, in: Tice, R. R., Costa, D. L., Schaich, K. M. (eds), Genotoxic Effects of Airborne Agents, New York, Plenum, pp. 91–103. |
|
(16) |
Zamora, P. O., Benson, J. M., Li, A, P. and Brooks, A. L. (1983), Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Muta tion Assay, Environmental Mutagenesis, 5, pp. 795–801. |
|
(17) |
Locke-Huhle, C. (1983), Endoreduplication in Chinese hamster ceils during alpha-radiation induced G2 arrest, Mutation Res.. 119. pp. 403–413. |
|
(18) |
Huang, Y., Change, C. and Trosko, J. E. (198 3), Aphidicolin-induced endoreduplication in Chinese hamster cells, Cancer Res., 43, pp. 1362–1364.» |
«B.11. MUTAGENICIDAD — ENSAYO DE ABERRACIONES CROMOSÓMICAS IN VIVO EN MÉDULA ÓSEA DE MAMÍFEROS
1. MÉTODO
El presente método reproduce tas directrices del documento OCDE TG 475 sobre el ensayo de aberraciones cromosómicas en médula ósea de mamíferos (1997).
1.1. INTRODUCCIÓN
El ensayo de aberraciones cromosómicas in vivo en mamíferos se realiza para detectar aberraciones cromosómicas estructurales inducidas por la sustancia de ensayo en células de médula ósea de animales —por lo general roedores— (1) (2) (3) (4). Las aberraciones estructurales pueden ser cromosómicas o cromatídicas. Un aumento de la poliploidía puede indicar que sustancia química es capaz de inducir aberraciones numéricas. La mayoría de los mutágenos químicos dan lugar a aberraciones cromatídicas, aunque también pueden producirse aberraciones cromosómicas. Las mutaciones cromosómicas y los efectos relacionados ocasionan numerosas enfermedades genéticas humanas. Además de ello, son muchas las pruebas de que las mutaciones cromosómicas y los efectos relacionados que provocan alteraciones en los oncogenes y los genes supresores de tumores guardan relación con el cáncer en los seres humanos y los sistemas experimentales.
En este ensayo se utilizan habitualmente roedores. El tejido diana es la médula ósea por estar muy vascularizada y por contener una poblicación de células de ciclo corto que pueden aislarse y tratarse con facilidad. En el presente método no se consideran otras especies ni otros tejidos diana.
El presente ensayo de aberraciones cromosómicas está especialmente indicado para evaluar el riesgo mutagenico, pues permite tomar en consideración factores del metabolismo in vivo, de la toxicocinética y de los procesos de reparación del ADN, si bien dichos factores pueden variar según la especie y el tejido. Los ensayos in vivo también resultan útiles para ahondar en el estudio de los efectos mutagénicos detectados en ensayos in vitro.
Si hay pruebas de que la sustancia de ensayo o un metabolito reactivo no llegan al tejido diana, no procede realizar el presente ensayo.
Véase asimismo la introducción general de la parte B.
1.2. DEFINICIONES
Aberración cromatídica: lesión cromosómica estructural que consiste en la rotura de una cromátida o la rotura y reunión de cromátidas.
Aberración cromosómica: lesión cromosómica estructural que consiste en la rotura o la rotura y reunión de ambas cromátidas en el mismo sitio.
Endorreduplicación: proceso en el que, en lugar de entrar en mitosis tras la fase S de replicación del ADN, el núcleo inicia otra fase S, lo cual da lugar a cromosomas con 4, 8, 16, etc. cromátidas.
Gap: lesión acromática más estrecha que la cromátida, con alteración mínima de la alineación de las cromátidas.
Aberración numérica: número de cromosomas distinto del número normal propio de las células empleadas.
Poliploidía: un múltiplo del número de dotación cromosómica haploide (n) superior al díploide (3n, 4n, etc.).
Aberración estructural: alteración de la estructura cromosómica (deleciones y fragmentos, intracambios o intercambios) detectable mediante examen microscópico de la metafase de la división celular.
1.3. PRINCIPIO DEL MÉTODO
Se exponen los animales a la sustancia de ensayo por una vía adecuada y se sacrifican a intervalos apropiados tras el tratamiento. Antes de sacrificarlos, se les administra una sustancia que detenga la división celular en la metafase (por ejemplo, colchicina o ColcemidR). A continuación, se realizan preparaciones de cromosomas de células de médula ósea y se tiñen, tras lo cual se analizan las células en metafase para detectar aberraciones cromosómicas.
1.4. DESCRIPCIÓN DEL MÉTODO
1.4.1. Preparación
1.4.1.1. Selección de la especie animal
Si bien suelen emplearse la rata, el ratón y el hámster chino, puede utilizarse otra especie de mamíferos apropiada. Deben usarse animales adultos jóvenes y sanos de cepas utilizadas habitualmente en laboratorio. La variación del peso de los animales al empezar el estudio ha de ser mínima y no debe exceder de ±20 % del peso medio de cada sexo.
1.4.1.2. Alojamiento y alimentación de los animales
Se aplicarán las condiciones generales recogidas en la introducción general de la parte B. La humedad debe ser del 50 al 60 %.
1.4.1.3. Preparación de los animales
Se reparten al azar animales adultos jóvenes y sanos entre los lotes tratados y los lotes de control. Las jaulas se colocan de manera que los posibles efectos debidos a la posición de las mismas sean mínimos. Se identifica a los animales de forma unívoca y se los acostumbra a las condiciones de laboratorio durante al menos cinco días.
1.4.1.4. Preparación de las dosis
Las sustancias de ensayo sólidas deben disolverse o suspenderse en disolventes o vehículos adecuados y, si es preciso, diluirse antes de su administración a los animales. Las sustancias de ensayo líquidas pueden administrarse directamente o diluirse antes de la administración. Han de emplearse soluciones de la sustancia de ensayo recién preparadas, salvo que los datos relativos a la estabilidad demuestran que es posible conservarlas.
1.4.2. Condiciones del ensayo
1.4.2.1. Disolvente o vehículo
El disolvente o vehículo no deberá producir efectos tóxicos a las dosis empleadas. Es preciso cerciorarse de que el disolvente o vehículo no produce reacciones químicas con la sustancia de ensayo. Si se emplean disolventes o vehículos poco conocidos, debe disponerse de información que avale su compatibilidad. Siempre que sea posible, se recomienda considerar en primer lugar la utilización de un disolvente o vehículo acuoso.
1.4.2.2. Controles
Deberán realizarse controles positivos y negativos (disolvente o vehículo) en paralelo en ambos sexos y en cada ensayo. Salvo por lo que respecta a la exposición a la sustancia de ensayo, los animales de los lotes de control deben tratarse exactamente igual que los de los lotes tratados.
En los controles positivos deberían producirse aberraciones estructurales in vivo con grados de exposición en los que quepa esperar un aumento detectable respecto a la frecuencia espontánea. Las dosis del control positivo deben ser tales que los efectos sean claros, pero no revelen inmediatamente al lector la identidad de los portaobjetos codificados. La sustancia empleada en el control positivo podrá administrarse por una vía distinta de la empleada para la sustancia de ensayo y la toma de muestras podrá realizarse una sola vez. Podrá considerarse la utilización en dichos controles de sustancias de clases químicas afines, cuando sea posible. A continuación figuran algunos ejemplos de sustancias para los controles positivos:
|
Sustancia |
No CAS |
No Einecs |
|
Metanosulfonato de etilo |
62-50-0 |
200-536-7 |
|
Etil-nitrosourea |
759-73-9 |
212-072-2 |
|
Mitomicina C |
50-07-7 |
200-008-6 |
|
Ciclofosfamida Monohidrato de ciclofosfamida |
50-18-0 6055-19-2 |
200-015-4 |
|
Trictilenomelamina |
51-18-3 |
200-083-5 |
Salvo que haya datos anteriores sobre controles que pongan de manifiesto una variabilidad entre animales y una frecuencia de células con aberraciones cromosómicas aceptables, se realizarán, para cada período de muestreo, controles negativos únicamente con el disolvente o vehículo, y se tratarán, por lo demás, de igual manera que los lotes tratados. Si se hace una sola toma de muestras en los controles negativos, el momento más oportuno para hacerla es tras el primer período de muestreo. Se realizarán asimismo controles sin tratar, salvo que exista información anterior o publicada sobre controles que demuestre que el disolvente o vehículo elegido no induce efectos nocivos ni mutágenos.
1.5. PROCEDIMIENTO
1.5.1. Número y sexo de los animales
Los lotes tratados y los controles han de estar compuestos al menos por cinco hembras y cinco machos analizables cada uno. Sí en el momento en que se efectúe el estudio se dispone de datos sobre otros estudios realizados con la misma especie y la misma vía de exposición, que demuestren que no hay diferencia significativa entre sexos en cuanto a la toxicidad, será suficiente el ensayo en un solo sexo. En caso de que la exposición humana a sustancias químicas pueda ser específica de un sexo, como sucede con algunos productos farmacéuticos, el ensayo se realizará con animales del sexo correspondiente.
1.5.2. Pauta de tratamiento
Las sustancias de ensayo se administran preferiblemente en una sola vez, aunque también puede dividirse la dosis —por ejemplo, en dos veces al día separadas por algunas horas como máximo— con el fin de facilitar la administración de grandes cantidades. Si se siguen otras pautas debe justificarse científicamente.
Se toman dos muestras en momentos distintos tras el tratamiento administrado en un día. En el caso de los roedores, la primera muestra se toma transcurrido un período de 1,5 veces la duración del ciclo celular normal (este suele durar de 12 a 18 horas) desde el tratamiento. Dado que el tiempo necesario para que la sustancia de ensayo se asimile y se metabolice, así como el efecto de esta sobre la cinética del ciclo celular pueden modificar el momento óptimo para detectar aberraciones cromosómicas, se recomienda tomar otra muestra 24 horas después de la primera. Si se siguen pautas de más de un día, ha de tomarse una muestra transcurrido un período de 1,5 veces la duración del ciclo celular normal desde la última administración.
Antes de sacrificar los animales, se les inyecta por vía intraperitoneal una dosis adecuada de una sustancia que detenga la división celular en la metafase (por ejemplo, ColcemidR o colchicina). Se extraen las muestras transcurrido un plazo oportuna, que en el caso de los ratones es de unas tres a cinco horas y en el de los hámsters chinos, de unas cuatro a cinco horas. Se toman las células de la médula ósea y se analizan en busca de aberraciones cromosómicas.
1.5.3. Dosis
Si se lleva a cabo un estudio previo de determinación de la gama de dosis por falta de datos adecuados, ha de hacerse en el mismo laboratorio, con la misma especie, cepa, sexo y protocolo de tratamiento que el estudio principal (5). Si se produce toxicidad, se administrarán tres dosis distintas para el primer período de muestreo. La gama de dosis deberá abarcar desde la toxicidad máxima hasta una toxicidad escasa o nula. En el período de muestreo posterior se administrará únicamente la dosis máxima. Se entenderá por «dosis máxima» la que produzca tales signos de toxicidad que si se administrara una dosis superior según el mismo protocolo resultaría probablemente letal. Las sustancias con actividades biológicas específicas a dosis bajas no tóxicas (como las hormonas y los mitógenos) pueden constituir excepciones en cuanto a los criterios de establecimiento de la dosis y han de evaluarse caso por caso. La dosis máxima también puede definirse como la dosis que produce algún signo de toxicidad en la médula ósea (por ejemplo, reducción del índice mitótico superior al 50 %).
1.5.4. Ensayo límite
Si en un ensayo realizado con una dosis de al menos 2 000 mg/kg de peso corporal administrada en una sola vez, o dos veces en el mismo día, no se observan efectos tóxicos y si, sobre la base de datos relativos a sustancias de estructura afín, no cabe esperar genotoxicidad, puede no ser necesario llevar a cabo un estudio completo con tres dosis diferentes. En el caso de estudios de mayor duración, la dosis límite será de 2 000 mg/kg de peso corporal/día cuando el tratamiento dure hasta 14 días y de 1 000 mg/kg de peso corporal/día cuando el tratamiento dure más de 14 días. Si se prevé la exposición humana a la sustancia, puede ser preciso administrar una dosis mayor en el ensayo límite.
1.5.5. Administración de las dosis
La sustancia de ensayo suele administrarse por sonda gástrica o con una cánula de intubación adecuada, o bien por inyección intraperitoneal. Pueden emplearse otras vías de administración, siempre que se justifiquen. El volumen máximo de líquido que puede administrarse de una sola vez por sonda o inyección depende del tamaño del animal de ensayo, pero no excederá de 2 ml/100 g de peso corporal. Deberá justificarse la administración de volúmenes superiores a este. Salvo en el caso de sustancias irritantes o corrosivas, que por lo general producen efectos exacerbados con concentraciones mayores, deberá reducirse al mínimo la variabilidad del volumen de ensayo ajustando la concentración de manera que el volumen sea el mismo en todas las dosis.
1.5.6. Preparación de los cromosomas
Se extrae la médula ósea inmediatamente después del sacrificio, se trata con una solución hipotónica y se fija. A continuación, se extienden las células en portaobjetos y se tiñen.
1.5.7. Análisis
Se evalúa la citotoxícidad determinando el índice mitótico en un mínimo de 1 000 células por animal, en todos los animales tratados (incluidos los controles positivos) y en los controles negativos sin tratar.
Deben analizarse al menos 100 células de cada animal. La cantidad puede ser menor si se observan numerosas aberraciones. Antes de analizarlas al microscopio, se asigna un código independiente a cada portaobjetos, incluidos los controles positivos y negativos. Dado que los métodos de preparación de los portaobjetos provocan con frecuencia la rotura de células en metafase y la pérdida de cromosomas, las células analizadas deben contener un número de centrómeros igual a 2n + 2.
2. RESULTADOS
2.1. TRATAMIENTO DE LOS RESULTADOS
Los datos relativos a cada animal se presentarán en forma de tabla. La unidad experimental es el animal. Se determinará en cada animal la cantidad de células analizadas, el número de aberraciones por célula y el porcentaje de células que presentan aberraciones cromosómicas estructurales. Se hará una relación de los distintos tipos, el número y la frecuencia de dichas aberraciones en los lotes tratados y los controles. Los gaps se registrarán por separado y se recogerán en el informe, pero por lo general no se incluirán en la frecuencia total de aberraciones. Si no se observan diferencias en la respuesta de un sexo y otro, los datos relativos a ambos sexos pueden considerarse conjuntamente en el análisis estadístico.
2.2. EVALUACIÓN E INTERPRETACIÓN DE LOS RESULTADOS
Son varios los criterios para determinar si un resultado es positivo (aumento de la proporción de células con aberraciones cromosómicas relacionado con la dosis, claro aumento del número de células que presentan aberraciones en un lote al que se ha administrado una sola dosis y en el que se ha realizado una única toma de muestras, etc.). Debe considerarse en primer lugar la importancia biológica de los resultados. Pueden emplearse métodos estadísticos como apoyo para evaluar los resultados del ensayo (6). El hecho de que los datos estadísticos sean significativos no ha de ser el único factor para determinar que una respuesta es positiva. Los resultados dudosos deberán aclararse mediante más ensayos en los que convendría modificar las condiciones experimentales.
El aumento de la poliploidía puede indicar que la sustancia de ensayo es capaz de producir aberraciones cromosómicas numéricas. El aumento de la endorreduplicación puede indicar que la sustancia de ensayo es capaz de inhibir la progresión del ciclo celular (7) (8).
No se considerarán mutágenas en el ensayo en cuestión las sustancias que den lugar a resultados que no se ajusten a los criterios anteriores.
Si bien en la mayoría de los experimentos los resultados serán claramente positivos o negativos, en algunos casos el conjunto de datos no permitirá emitir un juicio definitivo sobre la actividad de la sustancia de ensayo. Con independencia del número de veces que se repita el experimento, los resultados pueden seguir siendo ambiguos o dudosos.
Un resultado positivo en el ensayo de aberraciones cromosómicas in vivo indica que la sustancia de ensayo provoca aberraciones cromosómicas en la médula ósea de la especie estudiada. Un resultado negativo indica que, en las condiciones del ensayo, la sustancia de ensayo no provoca aberraciones cromosómicas en la médula ósea de la especie estudiada.
Deberá estudiarse la probabilidad de que la sustancia de ensayo o sus metabolitos pasen al torrente circulatorio o lleguen específicamente al tejido diana (toxicidad sistémica, etc.).
3. INFORME
3.1. INFORME DEL ENSAYO
El informe del ensayo incluirá la información siguiente:
|
|
Disolvente o vehículo:
|
|
|
Animales de ensayo:
|
|
|
Condiciones del ensayo:
|
|
|
Resultados:
|
|
|
Discusión de los resultados. |
|
|
Conclusiones. |
4. REFERENCIAS
|
(1) |
Adler, 1. D. (1984), Cytogcnetic Tests in Mammals, in: Mutagenicity Testing: a Practical Approach, S. Venitt and J. M. Parry (eds.). IRL Press, Oxford, Washington D. C, pp. 275–306. |
|
(2) |
Presten, R.)., Dean, B, J., Galloway, S., Holden, H., McFee, A. F. and Shelby, M. (1987), Mammalian In Vivo Cytogenetic Assays: Analysis of Chromosome Aberrations in Bone Marrow Cells, Mutation Res., 189, 157–165. |
|
(3) |
Richold, M., Chandly, A., Ashby, J., Gatehouse D. G., Bootman, J. and Henderson, L. (1990), In Vivo Cytogcnetic Assays, in: D. J. Kirkland, (ed.), Basic Mutagenicity Tests, UKEMS Recommended Procedures. UKEMS Sub-Committee on Guidelines for Mutagenicity Testing Report. Pan (revised, Cambridge University Press, Cambridge, New Cork, Port Chester, Melbourne, Sydney, pp. 115-141. |
|
(4) |
Tice, R. R. Hayashi, M., MacGregor, J. T., Anderson, D., Blakey, D. H., Holden, H. E., Kirch-Volders, M., Oleson Jr., F. B., Paccierotti, F., Preston R. J., Romagna F., Shimada, H., Sutou, S. and Vannier, B. (1994), Report from the Working Group on the In Vivo Mammalian Bone Marrow Chromosomal Aberration Test, Mutation Res., 312, pp. 305-312. |
|
(5) |
Fielder, R. J., Alleen, J. A., Boobis, A. R., Botham, P. A., Doe, J., Esdaile, D. J., Gatehouse, D. G., Hodson-Walker, G., Morton, D. B., Kirkland, D. J. and Richold, M. (1992), Report of British Toxicology Society/UK, Environmental Mutagen Society Working Group: Dose setting in In Vivo Mutagenicity Assays, Muta-genesis, 7, pp. 31 3-319. |
|
(6) |
Lovell, D. P., Anderson, D., Albanese, R., Amphlett, G. E., Clare, G., Ferguson, R., Richold, M., Papworth, D. G. and Savage, J. R. K. (1989), Statistical Analysis of In Vivo Cytogenetic Assays, in: UKEMS Sub-Committee on Guidelínes for Mutagenicity Testing. Repon Pan (III. Statistical Evahation of Mutagenicity Test Data, D. J. Kirkland, (ed.) Cambridge University Press, Cambridge, pp. 184-232. |
|
(7) |
Locke-Huhle, C. (1983). Endoreduplication in Chinese hamster cells during alpha-radiation-induced G2 arrest, Mutation Res. 119, pp. 403-413. |
|
(8) |
Huang, Y., Change, C. and Trosko, J. E. (1983), Aphidicolin-induced endoreduplication in Chinese hamster cells, Cancer Res., 43, pp. 1363-1364.» |
«B.12. MUTAGENICIDAD — ENSAYO DE MICRONÚCLEOS EN ERITROCITOS DE MAMÍFERO IN VIVO
1. MÉTODO
El presente método reproduce las directrices del documento OCDE TG 474 sobre el ensayo de micronúcleos en eritrocitos de mamífero (1997).
1.1. INTRODUCCIÓN
El ensayo in vivo de micronúcleos en mamíferos se emplea para detectar lesiones provocadas por la sustancia de ensayo en los cromosomas o el aparato mitótico de eritroblastos mediante el análisis de eritrocitos tomados de la médula ósea y/o la sangre periférica de animales (por lo general, roedores).
El ensayo de micronúcleos tiene por objeto determinar las sustancias que ocasionan lesiones citogenéticas que dan lugar a la formación de micronúcleos con fragmentos de cromosomas o cromosomas enteros retardados.
Cuando los eritroblastos de la médula ósea se transforman en eritrocitos policromáticos, el núcleo principal es expulsado y el micronúcleo, caso de haberse formado, puede permanecer en el citoplasma, que de lo contrario quedaría anucleado. Resulta más fácil visualizar los micronúcleos en estas células, pues carecen de núcleo principal. El aumento de la frecuencia de micronúcleos en los eritrocitos policromáticos de animales tratados es indicativo de la existencia de lesiones cromosómicas inducidas.
En este ensayo se emplea habitualmente médula ósea de roedores, pues es en ese tejido donde se forman los eritrocitos policromáticos. La detección de eritrocitos inmaduros (policromáticos) micronucleados en la sangre periférica también puede llevarse a cabo en otras especies en las que se haya demostrado que el bazo no es capaz de eliminar los eritrocitos micronucleados o que son suficientemente sensibles para detectar agentes que provocan aberraciones cromosómicas numéricas o estructurales. Entre los diversos criterios para distinguir los micronúcleos figura la determinación de la presencia o la ausencia de centrómero o de ADN centromérico en los mismos. El parámetro principal es la frecuencia de eritrocitos inmaduros (policromáticos) micronucleados. Si los animales se tratan de forma continua durante cuatro semanas o más, también puede emplearse como parámetro en el ensayo el número de eritrocitos maduros (normocromáticos) con micronúcleos en la sangre periférica respecto a un número determinado de eritrocitos maduros.
El presente ensayo in vivo de micronúcleos en mamíferos está especialmente indicado para evaluar el riesgo mutagénico, ya que permite tomar en consideración factores del metabolismo in vivo, de la farmacocinética y de los procesos de reparación del ADN, si bien dichos factores pueden variar según la especie, el tejido y el aspecto genético considerado. Los ensayos in vivo también resultan útiles para ahondar en el estudio de los efectos mutagénicos detectados en ensayos in vitro.
Si hay pruebas de que la sustancia de ensayo o un metabolito reactivo no llegan al tejido diana, no procede realizar el presente ensayo.
Véase asimismo la introducción general de la parte B.
1.2. DEFINICIONES
Centrómero (cinetocoro): región (o regiones) del cromosoma que se asocia a las fibras del huso acromático durante la división celular para permitir el movimiento ordenado de los cromosomas hijos hacia los polos de las células hijas.
Micronúcleo: núcleo pequeño, adicional al núcleo celular principal y separado de él, y producido durante la telofase de la mitosis o la meiosis por fragmentos cromosómicos o cromosomas enteros retardados.
Eritrocito normocromático: eritrocito maduro que carece de ribosomas y que se distingue del eritrocito policromático (inmaduro) mediante tinción selectiva de los ribosomas.
Eritrocito policromático: eritrocito inmaduro, en una fase intermedia de transformación, que aún tiene ribosomas y puede, por tanto, distinguirse del eritrocito normocromático (maduro) mediante tinción selectiva de dichos orgánulos.
1.3. PRINCIPIO DEL MÉTODO
Se exponen los animales a la sustancia de ensayo por una vía adecuada. Si se utiliza médula ósea, se sacrifican los animales a intervalos apropiados tras el tratamiento, se extrae la médula ósea, se preparan los portaobjetos y se tiñen (1) (2) (3) (4) (5) (6) (7). Si se emplea sangre periférica, se extrae a intervalos apropiados tras el tratamiento, se preparan frotis y se tiñen (4) (8) (9) (10). En los estudios con sangre periférica las células deben recolectarse lo antes posible después de la última exposición. Se analizan las preparaciones para detectar la presencia de micronúcleos.
1.4. DESCRIPCIÓN DEL MÉTODO
1.4.1. Preparación
1.4.1.1. Selección de la especie animal
Si se emplea médula ósea, se recomienda el uso de ratones o ratas, pero también pueden utilizarse otras especies de mamíferos adecuadas. Si se emplea sangre periférica, se recomienda el uso de ratones. Ahora bien, puede utilizarse cualquier otro mamífero adecuado, siempre que se trate de una especie en la que el bazo no elimine los eritrocitos micronucleados o en la que se haya demostrado una sensibilidad adecuada para detectar agentes que provocan aberraciones cromosómicas numéricas o estructurales. Deben usarse animales jóvenes y sanos de cepas utilizadas habitualmente en laboratorio. La variación del peso de los animales al empezar el estudio ha de ser mínima y no debe exceder de ± 20 % del peso medio de cada sexo.
1.4.1.2. Alojamiento y alimentación de los animales
Se aplicarán las condiciones generales recogidas en la introducción general de la parte B. La humedad debe ser del 50 al 60 %.
1.4.1.3. Preparación de los animales
Se reparten al azar animales adultos jóvenes y sanos entre los lotes tratados y los lotes control. Se identifica a los animales de forma unívoca y se los acostumbra a las condiciones de laboratorio durante al menos cinco días. Las jaulas se colocan de manera que los posibles efectos debidos a la posición de las mismas sean mínimos.
1.4.1.4. Preparación de las dosis
Las sustancias de ensayo sólidas deben disolverse o suspenderse en disolventes o vehículos adecuados y, si es preciso, diluirse antes de su administración a los animales. Las sustancias de ensayo líquidas pueden administrarse directamente o diluirse antes de la administración. Han de emplearse soluciones de la sustancia de ensayo recién preparadas, salvo que los datos relativos a la estabilidad demuestren que es posible conservarlas.
1.4.2. Condiciones del ensayo
1.4.2.1. Disolvente o vehículo
El disolvente o vehículo no deberá producir efectos tóxicos a las dosis empleadas. Es preciso cerciorarse de que el disolvente o vehículo no produce reacciones químicas con la sustancia de ensayo. Si se emplean disolventes o vehículos poco conocidos, debe disponerse de información de referencia que avale su compatibilidad. Siempre que sea posible, se recomienda considerar en primer lugar la utilización de un disolvente o vehículo acuoso.
1.4.2.2. Controles
Deberán realizarse controles positivos y negativos (disolvente o vehículo) en paralelo en ambos sexos y en cada ensayo. Salvo por lo que respecta a la exposición a la sustancia de ensayo, los animales de los lotes de control deben tratarse exactamente igual que los de los lotes tratados.
En los controles positivos deberían formarse micronúcleos in vivo con grados de exposición en los que quepa esperar un aumento detectable respecto a la frecuencia espontánea. Las dosis del control positivo deben ser tales que los efectos sean claros, pero no revelen inmediatamente al lector la identidad de los portaobjetos codificados. La sustancia del control positivo podrá administrarse por una vía distinta de la empleada para la sustancia de ensayo y la toma de muestras podrá realizarse una sola vez. Podrá considerarse la utilización en dichos controles de sustancias de clases químicas afines, cuando sea posible. A continuación figuran algunos ejemplos de sustancias para los controles positivos:
|
Sustancia |
N° CAS |
N° Einecs |
|
Metanosulfonato de etilo |
62-50-0 |
200-536-7 |
|
N-etilo-N-nitrosourea |
759-73-9 |
212-072-2 |
|
Mitomicina C |
50-07-7 |
200-008-6 |
|
Cidofosfamida Monohidrato de ciclofosfamida |
50-18-0 6055-19-2 |
200-015-4 |
|
Trietilenomelamina |
51-18-3 |
200-083-5 |
Salvo que haya datos anteriores sobre controles que pongan de manifiesto una variabilidad entre animales y una frecuencia de células con micronúdeos aceptables, se realizarán, para cada período de muestreo, controles negativos únicamente con el disolvente o vehículo, y se tratarán, por lo demás, de igual manera que los lotes tratados. Si se hace una sola toma de muestras en los controles negativos, el momento más oportuno para hacerla es tras el primer período de muestreo. Se realizarán asimismo controles sin tratar, salvo que exista información anterior o publicada sobre controles que demuestre que el disolvente o vehículo elegido no induce efectos nocivos ni mutágenos.
Si se emplea sangre periférica, puede aceptarse como control negativo en paralelo una muestra previa al tratamiento, pero únicamente en los estudios cortos (por ejemplo, de una a tres administraciones), si los resultados se encuentran en el intervalo previsto según controles anteriores.
1.5. PROCEDIMIENTO
1.5.1. Número y sexo de los animales
Cada lote tratado y cada control ha de estar compuesto al menos por cinco hembras y cinco machos analizables (11). Si en el momento en que se efectúe el estudio se dispone de datos sobre otros estudios realizados con la misma especie y la misma vía de exposición, que demuestren que no hay diferencia significativa entre sexos en cuanto a la toxicidad, será suficiente el ensayo en un solo sexo. En caso de que la exposición humana a las sustancias pueda ser específica de un sexo, como sucede con algunos productos farmacéuticos, el ensayo se realizará con animales del sexo correspondiente.
1.5.2. Pauta de tratamiento
No se recomienda ninguna pauta normalizada (una, dos o más administraciones a intervalos de 24 horas, etc.). Pueden aceptarse muestras tomadas en el marco de estudios de larga duración siempre que se haya demostrado que ese tipo de estudio proporciona resultados positivos o, si los resultados son negativos, que ha habido toxicidad o bien que se ha estado aplicando la dosis límite hasta el momento de tomar la muestra. También puede dividirse la dosis —por ejemplo, en dos veces al día separadas por algunas horas como máximo— con el fin de facilitar la administración de grandes cantidades.
El ensayo puede realizarse de dos maneras:
|
a) |
se administra la sustancia de ensayo a los animales en una sola vez. Se extraen muestras de la médula ósea al menos dos veces, la primera de ellas cuando hayan transcurrido más de 24 horas, pero menos de 48 horas desde el tratamiento. Debe respetarse un intervalo adecuado entre las tomas. Si se extraen muestras antes de que transcurran 24 horas desde el tratamiento, deberá justificarse. Las muestras de sangre periférica han de tomarse al menos en dos ocasiones, la primera de ellas cuando haya transcurrido un mínimo de 36 horas desde el tratamiento. Tras la primera toma, se respetan intervalos adecuados, sin sobrepasar las 72 horas. Si se observa una respuesta positiva en una toma de muestras, no es preciso realizar más; |
|
b) |
si se efectúan dos o más administraciones diarias (por ejemplo, dos o más administraciones a intervalos de 24 horas), ha de tomarse una muestra transcurridas de 18 a 24 horas desde la última administración si se trata de médula ósea, y de 36 a 48 horas desde la última administración en el caso de la sangre periférica (12). |
Si procede, pueden aplicarse otros tiempos de muestreo adicionales.
1.5.3. Dosis
Si se lleva a cabo un estudio previo de determinación de la gama de dosis por falta de datos adecuados, ha de hacerse en el mismo laboratorio, con la misma especie, cepa, sexo y protocolo de tratamiento que el estudio principal (13). Si se produce toxicidad, se administrarán tres dosis distintas para el primer período de muestreo. La gama de dosis deberá abarcar desde la toxicidad máxima hasta una toxicidad escasa o nula. En el último período de muestreo se administrará únicamente la dosis máxima. Se entenderá por «dosis máxima» la que produzca tales signos de toxicidad que, si se administrara una dosis superior según el mismo protocolo, resultaría probablemente letal. Las sustancias con actividades biológicas específicas a dosis bajas no tóxicas (como las hormonas y los mitógenos) pueden constituir excepciones en cuanto a los criterios de establecimiento de la dosis y han de evaluarse caso por caso. La dosis máxima también puede definirse como la dosis que produce algún signo de toxicidad en la médula ósea (por ejemplo, reducción de la proporción de eritrocitos inmaduros respecto al total de eritrocitos en la médula ósea o la sangre periférica).
1.5.4. Ensayo límite
Si en un ensayo realizado con una dosis de al menos 2 000 mg/kg de peso corporal administrada en una sola vez, o dos veces en el mismo día, no se observan efectos tóxicos y si, sobre la base de datos relativos a sustancias de estructura afín, no cabe esperar genotoxicidad, puede no ser necesario llevar a cabo un estudio completo con tres dosis diferentes. En el caso de estudios de mayor duración, la dosis límite será de 2 000 mg/kg de peso corporal/día cuando el tratamiento dure hasta 14 días y de 1 000 mg/kg de peso corporal/día cuando el tratamiento dure más de 14 días. Si se prevé la exposición humana a la sustancia, puede ser preciso administrar una dosis mayor en el ensayo límite.
1.5.5. Administración de las dosis
La sustancia de ensayo suele administrarse por sonda gástrica o con una cánula de intubación adecuada, o bien por inyección intraperítoneal. Pueden emplearse otras vías de administración, siempre que se justifiquen. El volumen máximo de líquido que puede administarse de una sola vez por sonda o inyección depende del tamaño del animal de ensayo, pero no excederá de 2 ml/100 g de peso corporal. Deberá justificarse la administración de volúmenes superiores a este. Salvo en el caso de sustancias irritantes o corrosivas, que por lo general producen efectos exacerbados con concentraciones mayores, deberá reducirse al mínimo la variabilidad del volumen de ensayo ajustando la concentración de manera que el volumen sea el mismo en todas las dosis.
1.5.6. Preparación de la médula ósea o la sangre periférica
Por lo general, la médula ósea se extrae del fémur o la tibia de los animales recién sacrificados. Las células suelen tomarse del fémur o la tibia, se preparan y se tiñen según métodos establecidos. La sangre periférica se extrae de la vena caudal o de otro vaso sanguíneo adecuado. Las células hemáticas se someten inmediatamente a una tinción supravital (8) (9) (10) o se extienden en frotis y luego se tiñen. Si se emplea un colorante específico del ADN [por ejemplo, naranja de acridina (14) o Hoechst 33258 más pironina-Y (15)] pueden evitarse algunos de los artefactos que aparecen cuando se utiliza un colorante que no es específico del ADN, pero no por ello han de descartarse los colorantes convencionales (coloración de Giemsa, etc.). También pueden utilizarse otros sistemas [por ejemplo, columnas de celulosa para eliminar las células nucleadas (16)], siempre que se tenga constancia de que resultan adecuados para la preparación de micronúcleos en laboratorio.
1.5.7. Análisis
Para cada animal, se determina la relación entre los eritrocitos inmaduros y el total de eritrocitos (inmaduros + maduros) en un recuento de al menos 200 eritrocitos si se trata de médula ósea y de 1 000 si se trata de sangre periférica (17). Antes de analizarlos al microscopio, se codifican independientemente todos los portaobjetos, incluidos los controles positivos y negativos. Se determinará la cantidad de eritrocitos inmaduros micronucleados en un mínimo de 2 000 eritrocitos inmaduros por animal. Puede obtenerse más información contando los eritrocitos maduros con micronúcleos. En el análisis de los portaobjetos la proporción de eritrocitos inmaduros respecto al total de eritrocitos no debe ser inferior al 20 % del valor del control. Si los animales han sido tratados de forma continua durante cuatro semanas o más, también puede determinarse la cantidad de micronúcleos en un mínimo de 2 000 eritrocitos maduros por animal. Pueden emplearse sistemas de análisis automatizados (análisis de la imagen o citometría de flujo continua de suspensiones celulares) en lugar del recuento manual, siempre que estén debidamente justificados y validados.
2. RESULTADOS
2.1. TRATAMIENTO DE LOS RESULTADOS
Los datos relativos a cada animal se presentarán en forma de tabla. La unidad experimental es el animal. Para cada animal analizado se relacionará por separado el número de eritrocitos inmaduros, el de eritrocitos inmaduros micronucleados y la proporción de eritrocitos inmaduros respecto al total de eritrocitos. Si los animales han sido tratados de forma continua durante cuatro semanas o más, se proporcionarán también los datos relativos a los eritrocitos maduros, caso de haberse recogido. Se facilitarán, para cada animal, la proporción de eritrocitos inmaduros respecto al total de los mismos y, si procede, el porcentaje de eritrocitos micronucleados. Si no se observan diferencias en la respuesta de un sexo y otro, los datos relativos a ambos sexos pueden considerarse conjuntamente en el análisis estadístico.
2.2. EVALUACIÓN E INTERPRETACIÓN DE LOS RESULTADOS
Son varios los criterios para determinar si un resultado es positivo (aumento del número de células micronu-cleadas relacionado con la dosis, claro aumento del número de dichas células en un lote al que se ha administrado una sola dosis y en el que se ha realizado una única toma de muestras, etc.). Debe considerarse en primer lugar la importancia biológica de los resultados. Pueden emplearse métodos estadísticos como apoyo para evaluar los resultados del ensayo (18) (19). El hecho de que los datos estadísticos sean significativos no ha de ser el único factor para determinar que una respuesta es positiva. Los resultados dudosos deberán aclararse mediante más ensayos en los que convendría modificar las condiciones experimentales.
No se considerarán mutágenas en el ensayo en cuestión las sustancias que den lugar a resultados que no se ajusten a los criterios anteriores.
Si bien en la mayoría de los experimentos los resultados serán claramente positivos o negativos, en algunos casos el conjunto de datos no permitirá emitir un juicio definitivo sobre la actividad de la sustancia de ensayo. Con independencia del número de veces que se repita el experimento, los resultados pueden seguir siendo ambiguos o dudosos.
Un resultado positivo en el ensayo de micronúcleos indica que la sustancia de ensayo induce la formación de micronúcleos, que son consecuencia de una lesión de los cromosomas o del aparato mitótico de los eritro-blastos de la especie estudiada. Un resultado negativo indica que, en las condiciones del ensayo, la sustancia de ensayo no induce la formación de micronúdeos en los eritrocitos inmaduros de la especie estudiada.
Deberá estudiarse la probabilidad de que la sustancia de ensayo o sus metabolitos pasen al torrente circulatorio o lleguen específicamente al tejido diana (toxicidad sistémica, etc.).
3. INFORME
INFORME DEL ENSAYO
El informe del ensayo incluirá la información siguiente:
|
|
Disolvente o vehículo:
|
|
|
Animales de ensayo:
|
|
|
Condiciones del ensayo:
|
|
|
Resultados:
|
|
|
— Discusión de los resultados. |
|
|
— Conclusiones. |
4. REFERENCIAS
|
(1) |
Heddle, J. A. (1973), A Rapid In Vivo Test for Chromosomal Damage, Mutation Res., 18, pp. 187-190. |
|
(2) |
Schmid, W. (1975), The Micronucleus Test, Mutation Res., 31, pp. 9-15, |
|
(3) |
Heddle, J. A., Salamone, M. F., Hite, M., Kirkhart, B., Mavournin, K., MacGregor, J. G. and Newell, G. W. (1983), The Induction of Micronuclei as a Measure of Genotoxicity, Mutation Res. 123, pp. 61-118. |
|
(4) |
Mavournin, K. H., Blakey, D. H., Cimino, M. C., Salamone, M. F. and Heddle, J. A. (1990), The In Vivo Micronucleus Assay in Mammalian Bone Marrow and Peripheral Blood. A report of the U.S. Environmental Protection Agency Gene-Tox Program, Mutation Res., 239, pp. 29-80. |
|
(5) |
MacGregor, J. T., Schlegel, R., Choy, W. N., and Wehr, C. M. (1983). Micronuclei in CircuLating Erythrocytes: A Rapid Screen for Chromosomal Damage During Routine Toxicity Testing in Mice, in: Developments in Science and Practice of Toxicology, ed. A. W. Hayes, R. C. Schnell and T. S. Miya, Elsevier, Amsterdam, pp., 555-558. |
|
(6) |
MacGregor, J. T., Heddle, J. A., Hite, M., Margolin, G, H., Ramel, C. Salamone, M. R. Tice, R. R. and Wild, D. (1987), Guidelines for the Conduct of Micronucleus Assays in Mammalian Bone Marrow Erythrocytes, Mutation Res., 189, pp. 103-112. |
|
(7) |
MacGregor, J. T., Wehr, C. M., Henika, P. R., and Shelby, M. E. (1990), The in vivo Erythrocyte Micronucleus Test: Measurement at Steady State Increases Assay Efficiency and Permits Integration with Toxicity Studies. Fund. Appl. Toxicol. 14, pp. 513-522. |
|
(8) |
Hayashi, M., Morita, T., Kodama, Y., Sofuni, T. and Ishidate, M. Jr. (1990), The Micronucleus Assay with Mouse Peripheral Blood Reticulocytes Using Acridine Orange-Coated Slides, Mutation Res., 245, pp. 245-249. |
|
(9) |
The Collaborative Study Group for the Micronucleus Test (1992). Micronucleus Test with Mouse Peripheral Blood Erythrocytes by Acridine Orange Supravital Staining: The Summary Repon of the 5th Collaborative Study by CSGMT/JKMMS, MMS, Mutation Res., 278, pp. 83-98. |
|
(10) |
The Collaborative Study Group for the Micronucleus Test (CSGMT/JEMMS, MMS: The Mammalian Mutagenesis Study Group of the Environmental Mutagen Society of Japan) (1995). Protocol recommended for the short-term mouse peripheral blood micronucleus test, Mutagenesis, 10, pp. 153-159. |
|
(11) |
Hayashi, M., Tice, R. R., MacGregor, J. T., Anderson, D., Blackey, D. H., Kirsch-Volders, M., Oleson, Jr. F. B., Pacchicrotti, F., Romagna, F., Shimada, H., Sutou, S. and Vannier, B. (1994), In Vivo, Rodent Erythrocyte Micronucleus Assay, Mutation Res., 312, pp. 293-304. |
|
(12) |
Higashikuni, N. and Sutou, S. (1995). An optimal, generalised sampling time of 30±6 h after double dosing in the mouse peripheral blood micronucleus test. Mutagenesis, 10, pp. 313-319. |
|
(13) |
Fielder, R. J., Alien, J. A., Boobis, A. R., Botham, P. A., Doe, J., Esdaile, D. J., Gatehouse, D. G., Hodson-Walker, C., Morton, D. B., Kirkland, D. J. and Rochold, M. (1992), Report of British Toxicology Society/UK Environmental Mutagen Society Working Group: Dose Setting in In Vivo Mutagenicity Assays. Mutagenesis, 7, pp. 313-319. |
|
(14) |
Hayashi, M., Sofuni, T. and Ishidate, M. Jr. (1983), An Application of Acridine Orange Fluorescent Staining to the Micronucleus Test, Mutation Res., 120, pp. 241-247. |
|
(15) |
MacGregor, J. T., Wehr, C. M. and Langlois, R. G. (1983), A Simple Fluorescent Staining Procedure for Micronuclei and RNA in Erythrocytes Using Hoechst 33258 and Pyronin Y. Mutation Res., 120, pp. 269-275. |
|
(16) |
Romagna, F. and Staniforth, C. D. (1989), The automated bone marrow micronucleus test, Mututton Res., 213, pp. 91-104. |
|
(17) |
Gollapudi, B. and McFadden, L. G. (1995), Sample size for the estimation of polychromatic to normochromatic erythrocyte ratio in the bone marrow micronucleus test, Mutation Res., 347, pp. 97-99. |
|
(18) |
Richold, M., Ashby, J., Bootman, J., Chandley, A., Gatehouse, D. G. and Henderson, L (1990), In Vivo Cytogenotics Assay, in: D. J. Kirkland (ed.), Basic Mutagenicity tests. UKEMS Recommended Procedures. UKEMS Sub-Committee on Guidelines for Mutagenícity Testing. Report, Part 1, revised, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 115-141. |
|
(19) |
Lovell, D. P., Anderson, D., Albanese, R., Amphlett, G. E., Clare, G., Ferguson, R., Richold, M., Papworth, D. G. and Savage, J. R. K. (1989), Statistical Analysis of In Vivo Cytogenetic Assays, in: D. J. Kirkland (ed.), Statistical Evalualion of Mutagenicity Test Data. UKEMS Sub-Commiltec on Guidelines for Mutagenicity Testing. Report, Part III, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 184-232.» |
«B.13/14. MUTAGENICIDAD — ENSAYO DE MUTACIÓN INVERSA EN BACTERIAS
1. MÉTODO
El presente método reproduce las directrices del documento OCDE TG 471 sobre el ensayo de mutación inversa en bacterias (1997).
1.1. INTRODUCCIÓN
En el ensayo de mutación inversa en bacterias se emplean cepas de Salmonella typhimurium y Escherichia coli auxotróficas respecto a un aminoácido para detectar mutaciones puntuales que impliquen sustitución, adición o deleción de uno o varios pares de bases del ADN (1) (2) (3). El ensayo se fundamenta en la detección de mutaciones que revierten mutaciones presentes en las cepas experimentales y restablecen la capacidad funcional de las bacterias de sintetizar un aminoácido esencial. Las bacterias revertientes se detectan por su capacidad de crecer en ausencia del aminoácido que la cepa experimental parental no es capaz de sintetizar.
Las mutaciones puntuales ocasionan numerosas enfermedades genéticas humanas. Además de ello, son muchas las pruebas de que dichas mutaciones en oncogenes y genes supresores de tumores de las células somáticas intervienen en la formación de tumores en los seres humanos y los animales de experimentación. El ensayo de mutación inversa en bacterias es rápido, poco costoso y relativamente sencillo de realizar. Muchas de las cepas experimentales presentan algunas características que las hacen más sensibles para la detección de mutaciones: secuencias de ADN sensibles en los sitios de reversión, mayor permeabilidad celular a las moléculas grandes y eliminación de sistemas de reparación del ADN o intensificación de los procesos de reparación del ADN propensos a errores. La especificidad de las cepas experimentales puede proporcionar información útil sobre los tipos de mutaciones inducidas por sustancias genotóxicas. Existe una importante base de datos relativa a una gran variedad de estructuras que recoge los resultados de ensayos de mutación inversa en bacterias. Asimismo, se han desarrollado metodologías bien consolidadas para analizar productos químicos, incluidos los compuestos volátiles, con diferentes propiedades fisicoquímicas.
Véase asimismo la introducción general de la parte B.
1.2. DEFINICIONES
Ensayo de mutación inversa en Salmonella typhimurium o Escherichia coli: ensayo que permite detectar una mutación en una cepa auxótrofa para un aminoácido (histidina y triptófano, respectivamente), que la transforma en una cepa independiente del aporte externo de dicho aminoácido.
Mutágenos que provocan la sustitución de un par de bases: agentes que provocan el cambio de una base en el ADN. En un ensayo de reversión el cambio puede producirse en el sitio de la mutación original o en un segundo sitio del genoma bacteriano.
Mutágenos que desplazan el marco de lectura: agentes que provocan la adición o deleción de uno o más pares de bases en el ADN, lo que modifica el marco de lectura en el ARN.
1.3. CONSIDERACIONES INICIALES
En el ensayo de mutación inversa en bacterias se utilizan células procariotas, que difieren de las células de mamífero en aspectos como la absorción, el metabolismo, la estructura cromosómica y los procesos de reparación del ADN. En los ensayos in vitro suele ser necesario emplear un sistema de activación metabólica exó-gena, si bien tales sistemas no pueden reproducir todas las condiciones que se dan in vivo en los mamíferos. Por tanto, el ensayo no proporciona información directa sobre la potencia mutagénica y carcinogénica de una sustancia determinada en mamíferos.
El ensayo de mutación inversa en bacterias se emplea habitualmente para hacer una selección inicial de la actividad genotóxica y, en particular, de la actividad inductora de mutaciones puntuales. Una importante base de datos ha puesto de manifiesto que muchas sustancias químicas que dan un resultado positivo en este ensayo también presentan actividad mutagénica en otros ensayos. También hay agentes mutagénicos que no son detectados por este ensayo. Esas deficiencias pueden deberse a la naturaleza específica del efecto detectado, a diferencias de la activación metabólica o de la biodisponibilidad. Por otra parte, los factores que incrementan la sensibilidad del ensayo de mutación inversa en bacterias pueden llevar a sobrestimar la actividad mutagénica.
El ensayo de mutación inversa en bacterias puede no ser apropiado para evaluar determinadas clases de sustancias químicas como los compuestos muy bactericidas (por ejemplo, algunos antibióticos) y aquellos de los que se cree (o se sabe) que interfieren de forma específica en el sistema de replicación celular de los mamíferos (por ejemplo, algunos inhibidores de la topoisomerasa y algunos análogos de los nucleósidos). En esos casos están más indicados los ensayos de mutaciones en mamíferos.
Pese a que muchos de los compuestos que dan un resultado positivo en este ensayo son carcinógenos para los mamíferos, la correlación no es absoluta, sino que depende de la clase química. Además de ello, hay carcinógenos que el ensayo no detecta, pues actúan por mecanismos que no son genotóxicos o que no existen en las células bacterianas.
1.4. PRINCIPIO DEL MÉTODO
Se exponen las suspensiones de células bacterianas a la sustancia de ensayo en presencia y en ausencia de un sistema de activación metabólica exógena. En el método de incorporación en placa, las suspensiones se mezclan con agar de recubrimiento y se vierten inmediatamente sobre una placa en medio mínimo. En el método de preincubación, la mezcla de tratamiento se incuba y luego se mezcla con agar de recubrimiento antes de verterla sobre una placa en medio mínimo. En ambos casos, tras dos o tres días de incubación, se cuentan las colonias revertientes y se compara su cantidad con la de colonias revertientes espontáneas en las placas de control con disolvente.
Entre los diversos procedimientos descritos para realizar el ensayo de mutación inversa en bacterias los métodos siguientes se utilizan comúnmente: incorporación en placa (1) (2) (3) (4), preincubación (2) (3) (5) (6) (7) (8), fluctuación (9) (10) y suspensión (11). También se han descrito modificaciones para los ensayos con gases y vapores (12).
Los procedimientos descritos en el método corresponden principalmente a la incorporación en placa y a la preincubación. Ambos son aceptables para realizar experimentos con y sin activación metabólica. El método de preincubación resulta más eficaz para detectar determinadas sustancias que pertenecen a clases químicas que incluyen nitrosaminas alifáticas de cadena corta, metales divalentes, aldehídos, azocolorantes y diazocom-puestos, alcaloides de la pirolizidina, compuestos alílicos y nitrocompuestos (3). También se reconoce que algunas clases de mutágenos no siempre se detectan con procedimientos ordinarios como los métodos de incorporación sobre placa y de preincubación. Esos casos deben considerarse «especiales» y se recomienda seriamente utilizar otros procedimientos para detectarlos. Se han determinado los «casos especiales» siguientes (junto con ejemplos de procedimientos de detección eficaces): azocolorantes y diazocompuestos (3) (5) (6) (13), gases y sustancias químicas volátiles (12) (14) (15) (16) y glicósidos (17) (18). Cualquier desviación respecto al procedimiento ordinario ha de estar científicamente justificada.
1.5. DESCRIPCIÓN DEL MÉTODO
1.5.1. Preparación
1.5.1.1. Bacterias
Se dejan crecer cultivos bacterianos recientes hasta el final de la fase exponencial o hasta el principio de la fase estacionaria (unas 109 células por ml). No deben emplearse cultivos que se encuentren al final de la fase estacionaria. Es fundamental que los cultivos utilizados para el experimento contengan un título elevado de bacterias viables. El título puede demostrarse sobre la base de datos de controles anteriores sobre curvas de crecimiento o bien determinando en cada ensayo el número de células viables mediante un experimento de cultivo en placas.
La temperatura de incubación recomendada es de 37 oC.
Se emplearán al menos cinco cepas bacterianas, de las cuales cuatro serán de S. typhimurium (TA1535; TA 1537, TA97a o TA97; TA98 y TA100), de fiabilidad demostrada y cuya respuesta sea reproducible entre laboratorios. Esas cuatro cepas de S. typhimurium poseen pares de bases GC en el sitio de reversión primaria y se sabe que no son capaces de detectar determinados mutágenos oxidantes, agentes de entrecruzamiento e hidracinas. Dichas sustancias son detectadas por las cepas E. coli WP2 y S. typhimurium TA102 (19), que poseen un par de bases AT en el sitio de reversión primaria. Así pues, se recomienda la combinación de cepas siguiente.
|
— |
S. typhimurium TA1535, y |
|
— |
S. typhimurium TA1537 y TA97 o TA97a, y |
|
— |
S. typhimurium TA98, y |
|
— |
S. typhimurium TA100, y |
|
— |
E. coli WP2 uvrA o E. coli WP2 uvrA (pKM101) o S. typhimurium TA102. |
Para detectar mutágenos de entrecruzamiento puede ser mejor incluir TA102 o añadir una cepa de E. coli eficaz en la reparación de ADN [por ejemplo, E. coli WP2 o E. coli WP2 (pKM101)].
Se seguirán procedimientos reconocidos de preparación de cultivos madre, comprobación de marcadores y conservación. Debe demostrarse en cada preparación de cultivo madre congelado que es necesario aportar el aminoácido en cuestión para que haya crecimiento (histidina en las cepas de S. typhimurium y triptófano en las de E. coli). Se controlarán de igual manera otras características fenotípicas, en concreto la presencia o ausencia de factores R, si procede [resistencia a la ampicilina en las cepas TA98, TA100 y TA97a o TA97, WP2 uvrA y WP2 uvrA (pKM101), y a la ampicilina + tetraciclina en la cepa TA102] y la presencia de mutaciones características (mutación rfa en S. typhimurium mediante la sensibilidad al violeta cristal, y mutación uvrA en E. coli o uvrB en S. typhimurium, mediante la sensibilidad a la luz ultravioleta) (2) (3). Las cepas deben producir un número de colonias revertientes espontáneas por placa en las gamas de frecuencia esperadas sobre la base de datos de controles anteriores del laboratorio y preferiblemente en la gama recogida en las publicaciones.
1.5.1.2. Medio
Se emplearán un agar mínimo adecuado (por ejemplo, con medio mínimo E de Vogel-Bonner y glucosa) y un agar de recubrimiento con histidina y biotina o triptófano para permitir pocas divisiones celulares (1) (2) (9).
1.5.1.3. Activación metabólica
Las bacterias deben exponerse a la sustancia de ensayo en presencia y en ausencia de un sistema adecuado de activación metabólica. El sistema más comúnmente empleado es una fracción postmitocondrial (S9) a la que se añaden cofactores y que se obtiene de hígados de roedores tratados con inductores enzimáticos como el Aroclor 1254 (1) (2) o una mezcla de fenobarbital y β-naftoflavona (18) (20) (21). La fracción postmitocondrial suele emplearse en concentraciones que varían del 5 al 30 % v/v en la mezcla S9. La elección y la condición de un sistema de activación metabólica puede depender de la clase de sustancia estudiada. En algunos casos puede ser oportuno utilizar varias concentraciones distintas de fracción postmitocondrial. En el caso de los azocolorantes y los diazocompuestos puede estar más indicado utilizar un sistema reductor de activación metabólica (6) (1 3).
1.5.1.4. Preparación de la sustancia de ensayo
Las sustancias de ensayo sólidas deben disolverse o suspenderse en disolventes o vehículos adecuados y, si es preciso, diluirse antes de tratar las bacterias. Las sustancias de ensayo líquidas pueden añadirse directamente al sistema de ensayo y/o diluirse antes del tratamiento. Han de emplearse soluciones recién preparadas, salvo que los datos relativos a la estabilidad demuestren que es posible conservarlas.
Es preciso cerciorarse de que el disolvente o vehículo no produce reacciones químicas con la sustancia de ensayo y es compatible con la supervivencia de las bacterias y la actividad de la S9 (22). Si se emplean disolventes o vehículos poco conocidos, debe disponerse de información que avale dicha compatibilidad. Siempre que sea posible, se recomienda considerar en primer lugar la utilización de un disolvente o un vehículo acuoso. Cuando se estudien sustancias inestables en presencia de agua, deberán emplearse disolventes orgánicos que no contengan agua.
1.5.2. Condiciones de ensayo
1.5.2.1. Cepas de ensayo (véase el punto 1.5.1.1)
1.5.2.2. Concentraciones de exposición
Para determinar la mayor concentración de la sustancia de ensayo deben considerarse, entre otros criterios, la citotoxicidad y la solubilidad en la mezcla de tratamiento final.
Puede ser útil determinar la toxicidad y la insolubilidad en un experimento preliminar. La citotoxicidad puede manifestarse por una reducción del número de colonias revertientes, la disminución o la aclaración de la confluencia del césped bacteriano, o la tasa de supervivencia de los cultivos tratados. La citotoxicidad de una sustancia puede verse alterada en presencia de sistemas de activación metabólica. Ha de valorarse la insolubilidad en forma de precipitación en la mezcla final en las condiciones reales de ensayo, visible por simple observación.
Si se trata de sustancias de ensayo solubles y no citotóxicas, la concentración máxima recomendada es de 5 mg/placa o 5 μl/placa. En el caso de sustancias no citotóxicas que no sean solubles a dicha concentración, una o varias de las concentraciones empleadas resultarán insolubles en la mezcla de tratamiento final. Las sustancias de ensayo que ya sean citotóxicas por debajo de 5 mg/placa o 5 μl/placa deberán ensayarse a concentraciones que llegarán como máximo hasta la concentración citotóxica. El precipitado no ha de interferir en la evaluación.
Se emplearán al menos cinco concentraciones analizables de la sustancia de ensayo con intervalos semilogarít-micos aproximadamente (√10) para el experimento inicial. Si se está estudiando la relación respuesta-concentración, puede estar indicado emplear intervalor menores. Para evaluar sustancias que contengan cantidades considerables de impurezas potencialmente mutagénicas podrá considerarse el uso de concentraciones superiores a 5 mg/placa o 5 μl/placa.
1.5.2.3. Controles positivos y negativos
En todos los experimentos deberán realizarse en paralelo controles positivos y negativos (disolvente o vehículo) específicos de la cepa utilizada, con y sin activación metabólica. Se seleccionarán las concentraciones del control positivo que demuestren ser eficaces en cada experimento.
En los experimentos con sistema de activación metabólica, la sustancia o sustancias de referencia del control positivo se elegirán con arreglo al tipo de cepa bacteriana empleada.
A continuación figuran algunos ejemplos de sustancias adecuadas para los controles positivos en ensayos con activación metabólica:
|
N° CAS |
N° Einecs |
Sustancia |
|
781-43-1 |
212-308-4 |
9,10-Dimetilantraceno |
|
57-97-6 |
200-359-5 |
7,12-Dimetilbenzo[a]aniraceno |
|
50-32-8 |
200-028-5 |
Benzo[a]pireno |
|
613-13-8 |
210-330-9 |
2-Aminoantraceno |
|
50-18-0 |
|
Ciclofosfamida |
|
6055-19-2 |
200-015-4 |
Monohidrato de ciclofosfamida |
La sustancia siguiente es apropiada para el control positivo del método de activación metabólica reductora:
|
Sustancia |
N° CAS |
N° Einecs |
|
Rojo Congo |
573-58-0 |
209-358-4 |
El 2-aminoantraceno no ha de emplearse como único indicador de la eficacia de la mezcla S9. Caso de emplearlo, todos los lotes de S9 han de caracterizarse asimismo con un mutágeno que requiera activación metabólica mediante enzimas microsomales, corno el benzo[a]pireno o el dimetilbenzoantraceno.
A continuación figuran algunos ejemplos de sustancias para los controles positivos específicas de determinadas cepas para ensayos sin sistema de activación metabólica exógena:
|
N° CAS |
N° Einecs |
Sustancia |
Cepa |
|
26628-22-8 |
247-852-1 |
Azida de sodio |
TA 1535 yTA 100 |
|
607-57-8 |
210-138-5 |
2-Nitrofluoreno |
TA 98 |
|
90-45-9 |
201-995-6 |
9-Aminoacridina |
TA 1537, TA 97 y TA 97a |
|
17070-45-0 |
241-129-4 |
ICR 191 |
TA 1537, TA 97 y TA 97a |
|
80-15-9 |
201-254-7 |
Hidroperóxido de cumeno |
TA 102 |
|
50-07-7 |
200-008-6 |
Mitomicina C |
WP2uvrA y TA 102 |
|
70-25-7 |
200-730-1 |
N-etil-N-nitro-N-nitrosoguanidina |
WP2, WP2uvrA y WP2uvrA(pKM101) |
|
56-57-5 |
200-281-1 |
4-Nitroquinolina-l-óxido |
WP2, WP2uvrA y WP2uvrA(pKM101) |
|
3688-53-7 |
|
Furilfuramida (AF2) |
Cepas que contengan plásmidos |
Pueden emplearse otras sustancias adecuadas para los controles positivos. Deberá considerarse la utilización en dichos controles de sustancias de clases químicas afines, cuando sea posible.
Se realizarán controles negativos sin sustancia de ensayo, únicamente con el disolvente o vehículo, y se tratarán de igual manera que los lotes tratados. Se prepararán asimismo controles sin tratar, salvo que exista información anterior sobre controles que demuestre que el disolvente elegido no induce efectos nocivos ni mutá-genos.
1.5.3. Procedimiento
En el método de incorporación sobre placa (1) (2) (3) (4), sin activación metabólica, suele mezclarse 0,05 ml o 0,1 ml de las soluciones de ensayo, 0,1 ml de cultivo bacteriano fresco (aproximadamente 108 células viables) y 0,5 ml de solución tampón estéril con 2,0 ml de agar de recubrimiento. En el ensayo con activación metabólica suele añadirse 0,5 ml de mezcla de activación metabólica que contenga una cantidad adecuada de fracción postmitocondrial (del 5 al 30 % v/v en dicha mezcla) al agar de recubrimiento (2,0 ml), las bacterias y la sustancia o solución de ensayo. Se mezcla el contenido de cada tubo y se vierte sobre una placa de agar mínimo. Se deja solidificar el agar de recubrimiento antes de incubar.
En el método de preincubación (2) (3) (5) (6) se incuba la sustancia o solución de ensayo con la cepa experimental (aproximadamente 108 células viables) y la solución tampón de ensayo o el sistema de activación metabólica (0,5 ml), por lo general durante 20 minutos o más a 30-37 oC. Después se añade el agar de recubrimiento y se vierte sobre una placa de agar mínimo. Habitualmente se mezcla 0,05 a 0,1 ml de sustancia o solución de ensayo, 0,1 ml de cultivo bacteriano y 0,5 ml de mezcla S9 o solución tampón estéril con 2,0 ml de agar de recubrimiento. Se airean los tubos durante la preincubación con un agitador.
Se prepararán tres placas por cada dosis para estimar adecuadamente la variación. Pueden prepararse solo dos placas por dosis si se justifica científicamente. La pérdida ocasional de una placa no invalida necesariamente el ensayo.
Las sustancias gaseosas o volátiles han de someterse a ensayo con métodos adecuados (recipientes de cultivo herméticos, etc.) (12) (14) (15) (16).
1.5.4. Incubación
Se incuban todas las placas de un mismo ensayo entre 48 y 72 horas a 37 oC. Al término de la incubación, se cuenta el número de colonias revertientes por placa.
2. RESULTADOS
2.1. TRATAMIENTO DE LOS RESULTADOS
Se facilitará el número de colonias revertientes por placa, así como el número de colonias revertientes en las placas del control negativo (control con disolvente y control sin tratar, en su caso) y del positivo. Debe proporcionarse el recuento por placa, la media de colonias revertientes por placa y la desviación estándar correspondientes a la sustancia de ensayo y a los controles positivos y negativos (sin tratar y/o con disolvente).
No será preciso comprobar los resultados positivos claros, si bien los resultados dudosos deberán aclararse mediante más ensayos en los que convendría modificar las condiciones experimentales. Los resultados negativos se confirmarán caso por caso. Los casos en que dicha confirmación no se considere necesaria deberán justificarse. En los experimentos ulteriores se considerará la posibilidad de modificar los parámetros del estudio para ampliar la gama de condiciones analizadas. Entre los parámetros modificables se encuentra la separación entre concentraciones, el método de tratamiento (incorporación sobre placa o prcincubación líquida) y las condiciones de activación metabólica.
2.2. EVALUACIÓN E INTERPRETACIÓN DE LOS RESULTADOS
Son varios los criterios para determinar si un resultado es positivo (aumento relacionado con la concentración en la gama utilizada y/o aumento rcproducible con una o varias concentraciones en el número de colonias revertientes por placa al menos en una cepa, con o sin sistema de activación metabólica, etc.) (23). Debe considerarse en primer lugar la importancia biológica de los resultados. Pueden emplearse métodos estadísticos como apoyo para evaluar los resultados del ensayo (24). No obstante, el hecho de que los datos estadísticos sean significativos no ha de ser el único factor para determinar que una respuesta es positiva.
No se considerarán mutágenas en el ensayo las sustancias que den lugar a resultados que no se ajusten a los criterios anteriores.
Si bien en la mayoría de los experimentos los resultados serán claramente positivos o negativos, en algunos casos el conjunto de datos no permitirá emitir un juicio definitivo sobre la actividad de la sustancia de ensayo. Con independencia del número de veces que se repita el experimento, los resultados pueden seguir siendo ambiguos o dudosos.
Un resultado positivo en el ensayo de mutación inversa en bacterias indica que la sustancia de ensayo induce mutaciones puntuales por sustitución de bases o desplazamientos del marco de lectura en el genoma de Sal-monella typhimurium y/o Escherichia coli. Un resultado negativo indica que, en las condiciones del ensayo, la sustancia de ensayo no resulta mutagénica en la especie estudiada.
3. INFORME
INFORME DEL ENSAYO
El informe del ensayo incluirá la información siguiente:
|
|
Disolvente o vehículo:
|
|
|
Cepas:
|
|
|
Condiciones del ensayo:
|
|
|
Resultados:
|
|
|
Discusión de los resultados. |
|
|
Conclusiones. |
4. REFERENCIAS
|
(1) |
Ames, B. N., McCann, J. and Yamasaki E. (1975), Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian-Microsome Mutagenicity Test, Mutation Res., 31, pp. 347-364. |
|
(2) |
Maron, D. M. and Ames, B. N. (1983), Revised Methods for the Salmonella Mutagenicity Test, Mutation Res., 113, pp. 173-215. |
|
(3) |
Gatehouse, D., Haworth, S., Cebula, T., Gocke, E., Kier, L., Matsushima, T., Melcion, C., Nohmi, T., Venitt, S. and Zeiger, E. (1994), Recommendations for the Performance of Bacterial Mutation Assays, Mutation Res., 312, pp. 217-233. |
|
(4) |
Kier, L. D., Brusick, D. J., Auletta, A. E., Von Halle, E. S., Brown, M. M., Simmon, V., F., Dunkel, V., McCann, J., Mortelmans, K., Prival, M., Rao, T. K. and Ray V. (1986), The Salmonella typhimurium/Mammalian Microsomal Assay: A Report of the U.S. Environmental Protection Agency Gene-Tox Program, Mutation Res., 168, pp. 69-240. |
|
(5) |
Yahagi, T., Degawa, M., Seino, Y.Y., Matsushima, T., Nagao, M., Sugimura, T. and Hashimoto, Y. (1975), Mutagenicity of Carcinogen Azo Dyes and their Derivatives, Cancer Letters, 1, pp. 91-96. |
|
(6) |
Matsushima, M., Sugimura, T., Nagao, M., Yahagi, T., Shirai, A. and Sawamura, M. (1980), Factors Modulating Mutagenicity Microbial Tests, in: Short-term Test Systems for Detecting Carcinogens, ed. Norpoth K. H. and Garner, R. C Springer, Berlin-Heidelberg-New York, pp. 273-285. |
|
(7) |
Gatehouse, D. G., Rowland, I. R., Wilcox, P., Callender, R. D. and Foster R. (1980), Bacterial Mutation Assays, in: Basic Mutagenicity Tests: UKEMS Pan 1 Revised, ed. D. J. Kirkland, Cambridge University Press, pp. 13-61. |
|
(8) |
Aeschacher, H. U., Wolleb, U. and Porchet, L. (1987), Liquid Preincubation Mutagenicity Test for Foods, J. Food Safety, S, pp, 167-177. |
|
(9) |
Green, M. H. L, Muriel, W. J. and Bridges, B. A. (1976), Use of a simplified fluctuation test to detect low levels of mutagens, Mutations Res., 38, pp. 33-42. |
|
(10) |
Hubbard, S. A., Green, M. H. L, Gatehouse, D. and Bridges, J. W. (1984), The Fluctuation Test in Bacteria, in: Handbook of Mutagenicity Test Procedures, 2nd Edition, ed. Kilbey, B. J., Legator, M., Nichols, W. and Ramel, C., Elsevier, Amsterdam-New York-Oxford, pp. 141-161. |
|
(11) |
Thompson, E. D. and Melampy, P. J. (1981), An Examination of the Quantitative Suspension Assay for Mutagenesis with Strains of Salmonella typhimurium, Environmental Mutagenesis, 3, pp. 453-465. |
|
(12) |
Araki, A., Noguchi, T., Kato, F. and Matsushima, T. (1994), Improved Method for Mutagenicity Testing of Gaseous Compounds by Using a Gas Sampling Bag, Mutation Res., 307, pp. 335-344. |
|
(13) |
Prival, M. J., Bell, S. J., Mitchell, V. D., Reipert, M. D, and Vaughan, V. L. (1984), Mutagenicity of Benzidine and Benzidine-Congener Dyes and Selected Monoazo Dyes in a Modified Salmonella Assay, Mutation Res., 136, pp. 33-47. |
|
(14) |
Zeiger, E., Anderson B. E., Haworth, S., Lawlor, T. and Mortelmans, K. (1992), Salmonella Mutagenicity Tests. V. Results from the Testing of 311 Chemicals, Environ. Mol. Mulagen., 19, pp. 2-141. |
|
(15) |
Simmon, V., Kauhanen K. and Tardiff, R. G. (1977), Mutagenic Activity of Chemicals Identified in Drinking Water, in Progress in Genetic Toxicology, D. Scott, B. Bridges and F. Sobels (eds.) Elsevier, Amsterdam, pp. 249-258. |
|
(16) |
Hughes, T. J., Simmons, D. M., Monteith, I. G. and Claxton, L. D. (1987), Vaporisation Technique to Measure Mutagenic Activity of Volatile Organic Chemicals in the Ames/Salmonella Assay, Environmental Mutagenesis, 9, pp. 421-441. |
|
(17) |
Matsushima, T., Matsumoto, A., Shirai, M., Sawamura, M. and Sugimura T. (1979), Mutagenicity of the Naturally Occurring Carcinogen Cycasin and Synthetic Methylazoxy Methane Conjugales in Salomonella typhimurium, Cancer Res., 39, pp, 3780-3782, |
|
(18) |
Tamura, G., Gold, C, Ferro-Luzzi, A. and Ames, B. N. (1980), Fecalase: A Model for Activation of Dietary Glycosides to Mutagens by Intestinal Flora, Proc. Natl. Acad. Sci. USA, 77, pp. 4961-4965. |
|
(19) |
Wilcox, P., Naidoo, A., Wedd, D. J. and Gatehouse, D. G. (1990), Comparison of Salmonella typhimurium TA 102 with Escherichia coli WP2 Tester strains, Mulagenesis, 5, pp. 285-291. |
|
(20) |
Matsushima, T., Sawamura, M., Hara, K. and Sugimura T. (1976), A Safe Substitute for Polychlorinated Biphenyls as an Inducer or Metabolic Activation Systems, in: In vitro Metabolic Activation in Mutagenesis Testing, eds, F. J. de Serres et al. Elsevier, North Holland, pp. 85-88. |
|
(21) |
Elliot, B. M., Combes, R. D., Elcombe, C. R., Tatehouse, D. G., Gibson, G, G., Mackay, J. M. and Wolf, R. C. (1992), Alternatives to Aroclor 1254-induced S9 in in vitro Genotoxicity Assays, Mutagenesis, 7, pp. 175-177. |
|
(22) |
Maron, D., Katzenellenbogen, J. and Ames, B. N. (1981), Compatibility of Organic Solvents with the Salmonella/Microsome Test, Mulalion Res., 88, pp. 343-350. |
|
(23) |
Claxton, L. D., Allen J., Auletta, A., Mortelmans, K., Nestmann, E. and Zeiger, E. (1987), Guide for the Salmonella typhimurium/Mammalian Microsome Tests for Bacterial Mutagenicity, Mutation Res., 189, pp. 83-91. |
|
(24) |
Mahon, G. A. T., Green, M. H. L., Middleton, B., Mitchell, I., Robinson, W. D. and Tweats, D. J. (1989), Analysis of Data from Microbial Colony Assays, in: UKEMS Sub-Committee on Guidelines for Mulagenicity Testing, Part II. Slatistical Evaluation of Mutagenicity Test Data, ed. Kirkland, D. J., Cambridge University Press, pp. 28-65.» |
B.15 ENSAYOS DE MUTAGÉNESIS Y DETECCIÓN DE CARCINOGÉNESIS — MUTACIÓN GÉNICA — SACCHAROMYCES CEREVISIAE
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIONES
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
Diversas cepas haploides y diploides de la levadura Saccharomyces cerevisiae pueden utilizarse para determinar la producción de mutaciones génicas por agentes químicos con y sin activación metabólica.
Es posible determinar la mutación directa en cepas haploides, en especial mediante la apreciación de la conversión de mutantes rojos consumidores de adenina (ade-1, ade-2) en una forma blanca que la precisa en cantidad doble, así como por sistemas selectivos, como la inducción de resistencia a la canavanina y la cicloheximida.
El método de mutación inversa, el más utilizado, comprende la utilización de la cepa haploide XV 185-14C, portadora de las mutaciones sin sentido ocres ade2-l, arg4-17, lys 1-1 y trp 5-48, reversibles mediante mutágenos causantes de sustituciones de bases que induzcan mutaciones en un punto específico o mutaciones supresoras ocres. La cepa XV 185-14C también lleva el marcador his 1-7, una mutación sin sentido invertida principalmente por mutaciones de segundo punto, y el marcador hom 3-10, invertido por mutágenos que desplazan el marco de lectura del código genético (frameshift mutagens).
De las cepas diploides de S. cerevisiae, la única que se utiliza ampliamente es D7, homocigota para ilv 1-92.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
Preparativos
Se prepararán soluciones de las sustancias químicas estudiadas y las de control inmediatamente antes de la prueba con ayuda de un vehículo apropiado. Cuando se trate de compuestos orgánicos no hidrosolubles, los disolventes orgánicos, como etanol, acetona o dimetilsulfóxido (DMSO), deberán emplearse en una concentración no superior al 2 % v/v. La concentración final del vehículo no debe influir de modo notable en la viabilidad de las células y en las características de crecimiento.
Activación metabólica
Las células han de exponerse a las sustancias analizadas en presencia de un sistema de activación metabólica exógena apropiado.
El método de uso más frecuente consiste en añadir la fracción posmitocondrial preparada a partir de hígados de roedores tratados previamente con inductores enzimáticos, a la que se añaden cofactores. También puede considerarse oportuno para la activación metabólica el empleo de especies, tejidos, fracciones posmitocondriales o métodos distintos.
Condiciones del ensayo
Cepas experimentales
La cepa haploide XV 185-14C y la cepa diploide D7 son las más utilizadas en estudios de mutación génica, pero también pueden ser apropiadas otras cepas.
Medios
Se utilizan medios de cultivo apropiados para la determinación del número de supervivientes y mutantes.
Uso de testigos negativos y positivos
Se utilizarán testigos positivos no tratados y tratados con disolventes de modo simultáneo. Es preciso utilizar sustancias químicas de control positivas para cada finalidad de mutación específica.
Concentración de exposición
Se utilizarán, al menos, cinco concentraciones debidamente escalonadas de la sustancia estudiada. En caso de sustancias tóxicas, la concentración máxima probada no reducirá el índice de supervivencias a menos del 5-10 %. Las sustancias relativamente insolubles en agua se estudiarán hasta su límite de solubilidad mediante métodos apropiados. En lo que respecta a las sustancias atóxicas claramente hidrosolubles, la concentración más alta se determinará en cada caso.
Condiciones de incubación
Las placas se incuban en la oscuridad, a 28-30 oC, durante 4-7 días.
Frecuencia de mutación espontánea
Se utilizarán subcultivos cuyas frecuencias de mutaciones espontáneas estén dentro de los límites normales admitidos.
Número de repeticiones
Se utilizarán al menos tres placas por concentración, a fin de determinar los prototrofos producidos por mutación génica, y para observar la viabilidad de las células. Si en los experimentos se emplearan marcadores, como hom 3-10, con tasa de mutaciones baja, puede aumentarse el número de placas utilizadas para lograr datos estadísticamente pertinentes.
Procedimiento
Las cepas de S. cerevisiae suelen tratarse en el curso de una prueba en medio líquido en la que se utilizan células en fase estacionaria o de crecimiento. Las experiencias iniciales deberán realizarse sobre células en crecimiento. Se expone a 1-5 × 107 células/ml a la sustancia objeto de estudio durante un período de hasta 18 horas a 28-37 oC, con agitación de la mezcla; durante el tratamiento, se añade una cantidad adecuada de un sistema de activación metabólica. Si la primera experiencia da resultados negativos, se procederá a una segunda, en esta ocasión con células en fase estacionaria; si los resultados de la primera fueran positivos, se confirmarán en una experiencia independiente. Al final del tratamiento, las células se centrifugan, lavan y siembran en un medio de cultivo apropiado. Tras la incubación, se examinan las placas para determinar la supervivencia y la inducción de mutaciones génicas.
2. RESULTADOS
Los resultados se presentarán en forma de tablas en las que aparezcan el número de colonias contadas, el de mutantes, el índice de supervivencia y la frecuencia de mutantes. Todos los resultados se confirmarán en un experimento independiente. Los resultados se evaluarán por métodos estadísticos apropiados.
3. INFORME
3.1. DATOS DEL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
cepa utilizada, |
|
— |
condiciones del ensayo: células en fase estacionaria o de crecimiento, composición de los medios, temperatura y duración de la incubación, sistema de activación metabólica, |
|
— |
condiciones de tratamiento: niveles de exposición, procedimiento y duración del tratamiento, temperatura del tratamiento, controles positivos y negativos, |
|
— |
número de colonias contadas, número de mutantes, supervivencia y frecuencia de mutantes, relación dosis-respuesta si es posible, evaluación estadística de los datos, |
|
— |
comentario de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.16. RECOMBINACIÓN MITÓTICA — SACCHAROMYCES CEREVISIAE
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIONES
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
En Saccharomyces cerevisiae, es posible apreciar la recombinación mitótica entre los genes (o, más generalmente, entre un gen y su centrómero) y en el interior de ellos. En el primer caso, se habla de crossing-over, generador de intercambios recíprocos, en tanto que, en el segundo los intercambios, casi nunca son recíprocos, y se habla de conversión génica. El crossing-over se determina generalmente por la producción de colonias o sectores recesivos homocigotos a partir de una cepa heterocigota, mientras que la conversión génica lo es mediante la producción de inversores prototróficos en una cepa auxotrofa heteroalélica portadora de dos alelos defectuosos distintos del mismo gen. Las cepas de uso más frecuente para descubrir una conversión génica mitótica son D4 (heteroalélica para ade 2 y trp 5), D7 (heteroalélica para trp 5), BZ34 (heteroalélica para arg 4) y JD1 (heteroalélica para his 4 y trp 5). El crossing-over mitótico productor de sectores homocigotos de color rojo y rosa se determina en D5 o en D7 (que también sirve para analizar la conversión génica mitótica y la mutación inversa para ilv 1-92), cepas ambas heteroalélicas complementarias de los alelos ade 2.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
Preparativos
Se preparan soluciones de las sustancias químicas estudiadas, y de los compuestos de control y de referencia, inmediatemente antes del ensayo, con ayuda de un vehículo apropiado. Cuando se trate de compuestos orgánicos no hidrosolubles, los disolventes orgánicos como etanol, acetona o dimetilsulfóxido (DMSO), deberán emplearse en una concentración no superior al 2 % v/v. La concentración final del vehículo no debe influir de modo notable en la viabilidad de las células ni en las características de crecimiento.
Activación metabólica
Las células han de exponerse a las sustancias analizadas en presencia y ausencia de un sistema de activación metabólica exógena apropiado. El método de uso más frecuente consiste en añadir a la fracción posmitocondrial preparada a partir de hígados de roedores tratados previamente con inductores enzimáticos, a la que se añaden cofactores. También puede considerarse oportuno para la activación metabólica el empleo de especies, tejidos, fracciones posmitocondriales o métodos distintos.
Condiciones del ensay
Cepas experimentales
Las cepas que se utilizan con más frecuencia son las diploides D4, D5, D7 y JD1, pero también pueden considerarse apropiadas otras.
Medios
Se utilizan medios de cultivo apropiados para la determinación de la supervivencia y de la frecuencia de recombinación mitótica.
Uso de testigos negativos y positivos
Se utilizarán testigos positivos no tratados y tratados con disolvente de modo simultáneo. Es preciso utilizar sustancias químicas de control positivas apropiadas para cada finalidad de recombinación específica.
Concentraciones de exposición
Se utilizarán, al menos, cinco concentraciones debidamente escalonadas de la sustancia estudiada. Entre los factores que deben tenerse en cuenta figuran la citotoxicidad y la solubilidad. La concentración mínima no debe influir en modo alguno en la viabilidad celular. En casos de sustancias tóxicas, la concentración máxima probada no reducirá el índice de supervivencia a menos del 5-10 %. Las sustancias relativamente insolubles en agua se estudiarán hasta su límite de solubilidad mediante métodos apropiados. En lo que respecta a las sustancias atóxicas claramente hidrosolubles, la concentración más alta se determinará en cada caso.
Las células pueden exponerse a las sustancias estudiadas en la fase estacionaria o durante el crecimiento hasta completar períodos de 18 horas, como máximo. No obstante, si se utilizan períodos de tratamiento largos, hay que examinar al microscopio los cultivos por si aparecieran esporas, ya que su presencia invalidaría la prueba.
Condiciones de incubación
Las placas se incuban en la oscuridad, a 28-30 oC, durante cuatro o siete días. Las placas que se utilicen para la determinación de los sectores homocigotos rojo y rosa producidos por crossing-over mitótico se conservarán en frigorífico a ± 4 oC durante uno o dos días más antes de la evaluación, a fin de que pueda intensificarse la pigmentación de las colonias de interés.
Frecuencia de recombinaciones mitóticas espontáneas
Se utilizarán subcultivos cuyas frecuencias de recombinaciones mitóticas espontáneas estén dentro de los límites normales admitidos.
Número de repeticiones
Se sembrarán al menos tres placas por concentración para determinar tanto la viabilidad como el número de prototrofos producidos por conversión génica mitótica. En caso de determinación de homocigosis recesiva producida por crossing-over mitótico, se aumentará el número de placas para obtener un número adecuado de colonias.
Procedimiento
Las cepas de S. cerevisiae suelen tratarse en el curso de una prueba en medio líquido en la que se utilizan células en fase estacionaria o de crecimiento. Las experiencias iniciales deberán realizarse sobre células en crecimiento. Se expone 1-5 × 107 células/ml a la sustancia objeto de estudio durante un período de hasta 18 horas a 28-37 oC, con agitación de la mezcla; durante el tratamiento, se añade, si procede, una cantidad adecuada de un sistema de activación metabólica.
Al final del tratamiento, las células se centrifugan, lavan y siembran en un medio de cultivo apropiado. Tras la incubación se examinan las placas para determinar la supervivencia y la inducción de recombinación mitótica.
Si la primera experiencia da resultados negativos, se procederá a una segunda, en esta ocasión con células en fase estacionaria; si los resultados de la primera fueran positivos, se confirmarán en una experiencia independiente.
2. RESULTADOS
Los resultados se presentarán en forma de tablas en las que aparezcan el número de colonias contadas, el de recombinantes, el índice de supervivencia y la frecuencia de recombinantes.
Los resultados se confirmarán en un experimento independiente.
Los resultados se evaluarán por métodos estadísticos apropiados.
3. INFORME
3.1. DATOS DEL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
cepa utilizada, |
|
— |
condiciones del ensayo: células en fase estacionaria o de crecimiento, composición de los medios, temperatura y duración de la incubación, sistema de activación metabólica, |
|
— |
condiciones del tratamiento: niveles de exposición, procedimiento y duración del tratamiento, temperatura del tratamiento; controles positivos y negativos, |
|
— |
número de colonias contadas, número de recombinantes, supervivencia y frecuencia de recombinación, relación dosis-respuesta si es posible, evaluación estadística de los datos, |
|
— |
comentario de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.17. «MUTAGENICIDAD — ENSAYO DE MUTACIÓN GÉNICA DE CÉLULAS DE MAMÍFERO IN VITRO
1. MÉTODO
El presente método reproduce las directrices del documento OCDE TG 476 sobre el ensayo de mutación génica de células de mamífero in vitro (1997).
1.1. INTRODUCCIÓN
El ensayo de mutación génica de células de mamífero in vitro puede realizarse para detectar mutaciones géni-cas inducidas por sustancias químicas. Entre las líneas celulares adecuadas para el ensayo se encuentran las células de linfoma de ratón L5178Y, las líneas celulares CHO, CHO-AS52 y V79 del hámster chino y las células linfoblastoides humanas TK6 (1). En dichas líneas celulares, los parámetros genéticos más empleados revelan una mutación en los loci de la timidinquinasa (TK) y la hipoxantina guanina fosforibosiltransferasa (HPRT), y en un transgen de la xantina guanina fosforibosiltransferasa (XPRT). Los ensayos de mutación TK, HPRT y XPRT detectan diversos espectros de fenómenos genéticos. La localización autosómica de TK y XPRT permite poner de manifiesto fenómenos genéticos (por ejemplo, deleciones importantes) que no se detectan en el locus HPRT de los cromosomas X (2) (3) (4) (5) (6).
En este ensayo pueden emplearse cultivos de líneas celulares establecidas o estirpes celulares. Las células deben seleccionarse sobre la base de su capacidad de crecimiento en cultivo y la estabilidad de la frecuencia de mutaciones espontáneas.
En los ensayos in vitro suele ser necesario emplear un sistema de activación metabólica exógena, si bien este no puede reproducir todas las condiciones que se dan in vivo en los mamíferos. Deben evitarse las circunstancias que puedan conducir a resultados que no reflejen la mutagenicidad intrínseca. Los resultados positivos que no reflejan la mutagenicidad intrínseca pueden deberse a cambios en el pH, la osmolalidad o altos grados de citotoxicidad (7).
El ensayo se emplea para detectar las posibles sustancias mutágenas o carcinógenas para los mamíferos. Pese a que muchos de los compuestos que dan un resultado positivo en este ensayo con carcinógenos para los mamíferos, no hay una correlación absoluta entre los resultados del mismo y la carcinogenicidad. La correlación depende de la clase química. Además de ello, cada vez hay más pruebas de que existen carcinógenos que el ensayo no detecta, pues, al parecer, estos actúan por mecanismos no genotóxicos o mecanismos ausentes en las células bacterianas (6).
Véase asimismo la introducción general de la parte B.
1.2. DEFINICIONES
Mutación directa: mutación génica del tipo parental a la condición mutante, que da lugar a una alteración o pérdida de la actividad enzimática o la función de la proteína codificada.
Mutágenos que provocan la sustitución de un par de bases: sustancias que provocan la sustitución de uno o varios pares de bases en el ADN.
Mutágenos que desplazan el marco de lectura: sustancias que provocan la adición o deleción de uno o más pares de bases en la molécula de ADN.
Período de expresión fenotípica: tiempo que tardan en desaparecer los productos génicos inalterados de las células mutadas recientemente.
Frecuencia de mutantes: número de células mutantes dividido por el número de células viables.
Crecimiento total relativo: aumento del número de células en el tiempo respecto a una población celular de control; es el producto del crecimiento en suspensión respecto al control negativo por la eficacia de clonado respecto al control negativo.
Crecimiento en suspensión relativo: aumento del número de células durante el período de expresión respecto al control negativo.
Viabilidad: eficacia de clonado de las células tratadas en el momento de la siembra en condiciones selectivas tras el período de expresión.
Supervivencia: eficacia de clonado de las células tratadas cuando se siembran al final del período de tratamiento; suele expresarse respecto a la supervivencia de la población celular de control.
1.3. PRINCIPIO DEL MÉTODO
Las células deficientes en timidinquinasa (TK) a causa de la mutación TK+/- → TK-/- son resistentes a los efectos citotóxicos de la trifluorotimidina (TFT), análogo de la pirimidina. Las células capaces de producir timidinquinasa son sensibles a la TFT, que inhibe el metabolismo y detiene la división celular. Así pues, las células imitantes son capaces de proliferar en presencia de TFT, mientras que las células normales, que contienen timidinquinasa, no lo son. Del mismo modo, las células deficientes en HPRT o XPRT se seleccionan por su resistencia a la 6-tioguanina (TG) o la 8-azaguanina (AG). Si en los ensayos de mutación génica en células de mamífero se utiliza una sustancia de ensayo análoga a una base o un compuesto afín al agente selectivo, deben considerarse detenidamente sus propiedades, por ejemplo, cuando se sospeche que la sustancia de ensayo presenta toxicidad selectiva en las células mutantes y no mutantes. Así pues, habrá que confirmar la eficacia del sistema o agente de selección cuando se estudien sustancias de estructura afín a la del agente selectivo (8).
Se exponen las células en suspensión o el cultivo en monocapa a la sustancia de ensayo, con y sin activación metabólica, durante un período apropiado. Se realiza un subcultivo para determinar la citotoxicidad y dejar que se exprese el fenotipo antes de la selección de los mutantes (9) (10) (11) (12) (13). La citotoxicidad suele determinarse midiendo la eficacia relativa de clonado (supervivencia) o el crecimiento total relativo de los cultivos tras el período de tratamiento. Los cultivos tratados se mantienen en un medio de crecimiento durante un período de tiempo suficiente y específico para cada locus y tipo celular seleccionado, de manera que la expresión fenotípica de las mutaciones inducidas sea casi óptima. La frecuencia de mutantes se determina sembrando un número conocido de células en un medio con agente selectivo y en otro medio sin dicho agente para determinar la eficacia de clonado (viabilidad). Tras un período de incubación adecuado, se cuentan las colonias. Se compara el número de colonias mutantes en el medio selectivo y en el no selectivo para hallar la frecuencia de mutantes.
1.4. DESCRIPCIÓN DEL MÉTODO
1.4.1. Preparación
1.4.1.1. Células
Entre los diversos tipos de células apropiados para este ensayo se encuentran las de los subclones de L5178Y, CHO, CHO-AS52, V79 y TK6. Deben emplearse tipos de células que presenten una sensibilidad probada a los mutágenos químicos, una elevada eficacia de clonado y una frecuencia estable de mutaciones espontáneas. Se comprobará la ausencia de contaminación por micoplasma. Se desecharán las células contaminadas.
El ensayo debe estar diseñado de manera que su sensibilidad y potencia estén predeterminadas. El número de células, cultivos y concentraciones de la sustancia de ensayo ha de reflejar dichos parámetros previamente definidos (14). En cada fase del ensayo, el número mínimo de células viables que sobrevivan al tratamiento deberá estar basado en la frecuencia de mutación espontánea. Por lo general, se emplea una cantidad de células al menos igual a diez veces el inverso de dicha frecuencia. Se recomienda, no obstante, utilizar un mínimo de 106 células. Para cerciorarse de que el ensayo proporciona resultados coherentes deberá disponerse de datos de controles históricos adecuados sobre el sistema celular utilizado.
1.4.1.2. Medios y condiciones de cultivo
Deben utilizarse medios de cultivo y condiciones de incubación apropiados (recipientes de cultivo, temperatura, concentración de CO2 y humedad). Los medios han de elegirse con arreglo a los sistemas selectivos y los tipos celulares empleados en el ensayo. Es especialmente importante aplicar condiciones de cultivo que favorezcan un crecimiento celular óptimo durante el período de expresión y la capacidad de las células, mutantes y no mutantes, de formar colonias.
1.4.1.3. Preparación de los cultivos
Se multiplican células de cultivos madre, se siembran en el medio de cultivo y se incuban a 37 oC. Antes de usar los cultivos en el ensayo, puede ser necesario lavarlos de las células mutantes que contengan.
1.4.1.4. Activación metabólica
Las células deben exponerse a la sustancia de ensayo en presencia y en ausencia de un sistema adecuado de activación metabólica. El sistema más comúnmente empleado es una fracción postmitocondrial (S9) a la que se añaden cofactores y que se obtiene de hígados de roedores tratados con inductores enzimáticos como el Aroclor 1254 (15) (16) (17) (19) o una mezcla de fenobarbital y β-naftoflavona (19) (20).
La fracción postmitocondrial suele emplearse en concentraciones que varían del 1 al 10 % v/v en el medio de ensayo final. La elección y condición de un sistema de activación metabólica puede depender de la clase química de la sustancia estudiada. En algunos casos puede ser oportuno utilizar varias concentraciones distintas de fracción postmitocondrial.
Son varias las técnicas —como la producción mediante ingeniería genética de líneas celulares que expresen enzimas activadoras específicas— que pueden proporcionar el potencial de activación endógena. La elección de las líneas celulares debe justificarse científicamente (por ejemplo, por la importancia de la isoenzima del citocromo P450 para el metabolismo de la sustancia de ensayo, etc.).
1.4.1.5. Preparación de la sustancia de ensayo
Las sustancias de ensayo sólidas deben disolverse o suspenderse en disolventes o vehículos adecuados y, si es preciso, diluirse antes de tratar las células. Las sustancias de ensayo líquidas pueden añadirse directamente al sistema de ensayo y/o diluirse antes del tratamiento. Han de emplearse soluciones de la sustancia de ensayo recién preparadas, salvo que los datos relativos a la estabilidad demuestren que es posible conservarlas.
1.4.2. Condiciones del ensayo
1.4.2.1. Disolvente o vehículo
Es preciso cerciorarse de que el disolvente o vehículo no produce reacciones químicas con la sustancia de ensayo y es compatible con la supervivencia de las células y la actividad de S9. Si se emplean disolventes o vehículos poco conocidos, debe disponerse de información que avale dicha compatibilidad. Siempre que sea posible, se recomienda considerar en primer lugar la utilización de un disolvente o vehículo acuoso. Cuando se estudien sustancias inestables en presencia de agua, deberán emplearse disolventes orgánicos que no contengan agua. Esta podrá eliminarse añadiendo un tamiz molecular.
1.4.2.2. Concentraciones de exposición
Para determinar la concentración máxima de exposición deben considerarse al menos los siguientes criterios: citotoxicidad, solubilidad en el sistema de ensayo y variaciones del pH o la osmolalidad.
La citotoxicidad se determinará con y sin activación metabólica en el experimento principal mediante un indicador adecuado de la integridad y el crecimiento celular, como la eficacia de clonado relativa (supervivencia) o el crecimiento total relativo. Puede ser útil determinar la citotoxicidad y la solubilidad en un experimento preliminar.
Se emplearán al menos cuatro concentraciones analizables. Si se produce citotoxicidad, la gama de concentraciones deberá abarcar desde la toxicidad máxima hasta una toxicidad escasa o nula, lo cual suele significar que la separación entre concentraciones corresponde a un factor comprendido entre 1 y √10. En el caso de sustancias escasamente citotóxicas, la concentración máxima de ensayo será la menor de estas tres: 5 mg/ml, 5 μl/ml o 0,01 M.
Las sutancias escasamente solubles deben ensayarse hasta concentraciones iguales o superiores al límite de solubilidad en las condiciones de cultivo. Ha de ponerse de manifiesto la insolubilidad en el medio de tratamiento final al que se exponen las células. Puede ser útil determinar la solubilidad al principio y al final de tratamiento, pues esta puede variar durante la exposición en el sistema de ensayo debido a la presencia de células, S9, suero, etc. La insolubilidad puede detectarse por simple observación. El precipitado no debe interferir en la evaluación del resultado.
1.4.2.3. Controles
En todos los experimentos deberán realizarse controles positivos y negativos (disolvente o vehículo) en paralelo, con y sin activación metabólica. Cuando se aplique esta, la sustancia del control positivo ha de requerir activación para provocar una respuesta mutagénica.
A continuación figuran algunos ejemplos de sustancias para el control positivo:
|
Activación metabólica |
Locus |
Sustancia |
N° CAS |
N° EINECS |
|
Ausencia de activación metabólica exógena |
HPRT |
Metanosulfonato de etilo |
62-50-0 |
200-536-7 |
|
Etil-nitrosourea |
759-73-9 |
212-072-2 |
||
|
TK (colonias grandes y pequeñas) |
Metanosulfonato de metilo |
66-27-3 |
200-625-0 |
|
|
XPRT |
Metanosulfonato de etilo |
62-50-0 |
200-536-7 |
|
|
Etil-nitrosourea |
759-73-9 |
212-072-2 |
||
|
Presencia de activación metabólica exógena |
HPRT |
3-metilcolantreno |
56-49-5 |
200-276-4 |
|
N-nitrosodimetilamina |
62-75-9 |
200-549-8 |
||
|
7,12-Dimetilbenzoantraceno |
57-97-6 |
200-359-5 |
||
|
TK (colonias grandes y pequeñas) |
Ciclofosfamida |
50-18-0 |
200-015-4 |
|
|
Monohidrato de ciclofosfamida |
6055-19-2 |
|
||
|
Benzo[a]pireno |
50-32-8 |
200-028-5 |
||
|
3-Metilcolantreno |
56-49-5 |
200-276-5 |
||
|
XPRT |
N-nitrosodimetilamina (para grandes cantidades de S9) |
62-75-9 |
200-549-8 |
|
|
Benzo[a]pireno |
50-32-8 |
200-028-5 |
Pueden utilizarse otras sustancias de referencia adecuadas para el control positivo, por ejemplo, la 5-bromo 2'-desoxiuridina (n° CAS 59-14-3, n° EINECS 200-415-9), si el laboratorio dispone de una base de datos al respecto. Deberá considerarse la utilización en dicho control de sustancias de clases químicas afines, cuando sea posible.
Se realizarán controles negativos únicamente con el disolvente o vehículo en el medio de tratamiento, y se tratarán de igual manera que los lotes tratados. Se prepararán asimismo controles sin tratar, salvo que exista información de controles históricos que demuestre que el disolvente elegido no induce efectos nocivos ni mutágenos.
1.4.3. Procedimiento
1.4.3.1. Tratamiento con la sustancia de ensayo
Las células en crecimiento se tratan con la sustancia de ensayo, en presencia y en ausencia de un sistema de activación metabólica. La duración de la exposición ha de ser suficiente (de 3 a 6 horas suelen bastar) y puede ampliarse hasta uno o varios ciclos celulares.
Pueden realizarse uno o dos cultivos con cada concentración de la sustancia de ensayo. En el caso de realizarse uno solo, se aumentará el número de concentraciones con el fin de disponer de suficientes cultivos para el análisis (por ejemplo, un mínimo de ocho concentraciones analizables). Deben realizarse dos cultivos para el control negativo (disolvente).
Las sustancias gaseosas o volátiles han de evaluarse con métodos adecuados, tales como recipientes de cultivo herméticos, etc. (21) (22).
1.4.3.2. Determinación de la supervivencia, la viabilidad y la frecuencia de mutantes
Al término del período de exposición se lavan las células y se cultivan para determinar la supervivencia y dejar que se exprese el fenotipo mutante. La evaluación de la citotoxicidad suele empezarse después del período de tratamiento, determinando la eficacia de clonado relativa (supervivencia) o el crecimiento total relativo de los cultivos.
Cada locus necesita un tiempo mínimo determinado para que la expresión fenotípica de las mutaciones inducidas recientemente sea casi óptima (un mínimo de seis a ocho días en el caso de HPRT y XPRT y de dos días en el de TK). Las células se cultivan con y sin agente o agentes selectivos para determinar el número de mutantes y la eficacia de clonado, respectivamente. La viabilidad, que sirve para calcular la frecuencia de mutantes, empieza a determinarse al final del período de expresión colocando los cultivos en un medio no selectivo.
Si la sustancia de ensayo da un resultado positivo en el ensayo con L5178Y TK+/-, deben medirse las colonias al menos en uno de los cultivos de ensayo (el de la concentración que dé resultados más positivos) y en los controles positivos y negativos. Si la sustancia de ensayo da un resultado negativo en el ensayo con L5178Y TK+/-, deben medirse las colonias en los controles positivos y negativos. También pueden medirse las colonias en los estudios realizados con TK6TK+/-.
2. RESULTADOS
2.1. TRATAMIENTO DE LOS RESULTADOS
Los datos deben incluir la determinación de la citotoxicidad y la viabilidad, los recuentos de colonias y la frecuencia de mutantes en los cultivos tratados y en los controles. Si el resultado del ensayo con L5178Y TK+/-es positivo, las colonias se cuentan distinguiendo las grandes de las pequeñas al menos en un cultivo tratado (con la concentración de sustancia de ensayo que dé los resultados más positivos) y en los controles positivos y negativos. Debe estudiarse detenidamente la naturaleza molecular y citogenética tanto de las colonias grandes como de las pequeñas (23) (24). En el ensayo TK+/-, las colonias se cuentan distinguiendo las de crecimiento normal (grandes) de las de crecimiento lento (pequeñas) (25). Las células mutantes que han sufrido las lesiones genéticas más importantes requieren un tiempo de duplicación mayor, de manera que forman colonias pequeñas. Dichas lesiones suelen variar desde la pérdida de un gen entero hasta las aberraciones cromo-sómicas visibles en el cariotipo. La inducción de pequeñas colonias mutantes se ha asociado a sustancias que provocan aberraciones cromosómicas importantes (26). Las células mutantes menos afectadas crecen a una velocidad similar a la de las células parentales y forman colonias grandes.
Debe proporcionarse el índice de supervivencia (eficacia de clonado relativa) o el crecimiento total relativo. La frecuencia de mutantes se expresa como el número de células mutantes respecto al número de células supervivientes.
Se proporcionarán los datos relativos a cada cultivo y se resumirán en forma de tabla.
No será preciso comprobar los resultados positivos claros, si bien los resultados dudosos deberán aclararse mediante más ensayos en los que convendría modificar las condiciones experimentales. Es preciso confirmar los resultados negativos caso por caso. Los casos en que dicha confirmación no se considere necesaria deberán justificarse debidamente. En los experimentos complementarios de aquellos que hayan dado resultados dudosos o negativos, se considerará la posibilidad de modificar los parámetros del estudio para ampliar la gama de condiciones analizadas. Entre los parámetros modificables se encuentran la separación entre concentraciones y las condiciones de activación metabólica.
2.2. EVALUACIÓN E INTERPRETACIÓN DE LOS RESULTADOS
Son varios los criterios para determinar si un resultado es positivo (aumento relacionado con la concentración o aumento reproducible de la frecuencia de mutantes, etc.). Debe considerarse en primer lugar la importancia bilógica de los resultados. Pueden emplearse métodos estadísticos como apoyo para evaluar los resultados del ensayo. El hecho de que los datos estadísticos sean significativos no ha de ser el único factor para determinar que una respuesta es positiva.
No se considerarán mutágenas en el sistema en cuestión las sutancias que den lugar a resultados que no se ajusten a los criterios anteriores.
Si bien en la mayoría de los experimentos los resultados eran claramente positivos o negativos, en algunos casos el conjunto de datos no permitirá emitir un juicio definitivo sobre la actividad de la sustancia de ensayo. Con independencia del número de veces que se repita el experimento, los resultados pueden seguir siendo ambiguos o dudosos.
Un resultado positivo en el ensayo de mutación génica de células de mamífero in vitro indica que la sustancia de ensayo provoca mutaciones génicas en los cultivos de células de mamífero utilizadas. El elemento más significativo es la relación dosis-respuesta positiva y reproducible. Un resultado negativo indica que, en las condiciones del ensayo, la sustancia de ensayo no provoca mutaciones génicas en los cultivos de células de mamífero utilizadas.
3. INFORME
INFORME DEL ENSAYO
El informe del ensayo incluirá la información siguiente:
|
|
Disolvente o vehículo:
|
|
|
Células:
|
|
|
Condiciones del ensayo:
|
|
|
Resultados:
|
|
|
Discusión de los resultados. |
|
|
Conclusiones. |
4. REFERENCIAS
|
(1) |
Moore, M. M., DeMarini, D. M., DeSerres, F. J. and Tindall, K. R. (eds.) (1987), Banbury Report 28: Mam-malian Cell Mutagenesis, Cold Spring Harbor Laboratory, New York. |
|
(2) |
Chu, E, H. Y. and Mailing, H. V. (1968), Mammalian Cell Genetics. II. Chemical Induction of Specific Locus Mutations in Chinese Hamster Cells In Vitro, Proc. Natl. Acad. Sci. USA, 61, pp. 1306-1312. |
|
(3) |
Liber, H. L. and Thilly, W. G. (1982), Mutation Assay at the Thymidine Kinase Locus in Diploid Human Lymphoblasts, Mulation Res., 94, pp. 467-485. |
|
(4) |
Moore, M. M., Harington-Brock, K., Doerr, C. L. and Dearfield, K. L. (1989), Differential Mutant Quanti-tation at the Mouse Lymphoma TK and CHO HGPRT Loct, Mulagenesis, 4, pp. 394-403. |
|
(5) |
Aaron, C. S. and Stankowski, Jr. L. F. (1989), Comparison of the AS52/XPRT and the CHO/HPRT Assays: Evaluation of Six Drug Candidates, Mutation Res., 223, pp. 121-128. |
|
(6) |
Aaron, C. S., Bolcsfoldi, G., Glatt, H. R., Moore, M., Nishi, Y., Stankowski, Jr. L. F., Theiss, J. and Thompson, E. (1994), Mammalian Cell Gene Mutation Assays Working Group Report. Report of the International Workshop on Standardisation of Genotoxicity Test Procedures. Mutation Res., 312, pp. 235-239. |
|
(7) |
Scott. D., Galloway, S. M., Marshall, R. R., Ishidate, M., Brusick, D., Ashby, J. and Myhr, B. C. (1991), Genotoxicity Under Extreme Culture Conditions. A report from ICPEMC Task Group 9, Mutation Res., 257, pp. 147-204. |
|
(8) |
Clive, D., McCuen, R., Spector, J. F. S., Piper, C. and Mavournin, K. H. (1983), Specific Gene Mutations in L5178Y Cells in Culture. A Report of the U. S. Environmental Protection Agency Gene-Tox Program, Mutation Res., 115, pp. 225-251. |
|
(9) |
Li, A. P., Gupta, R. S., Heflich, R. H. and Wasson, J. S. (1988), A Review and Analysis of the Chinese Hamster Ovary/Hypoxanthine Guanine Phosphoribosyl Transferase System to Determine the Mutagenicity of Chemical Agents: A Report of Phase III of the U. S. Environment Protection Agency Gene-Tox Program, Mutation Res., 196, pp. 17-36. |
|
(10) |
Li, A, P., Carver, J. H., Choy, W. N., Hsie, A. W., Gupta, R. S., Loveday, K. S., O'Neill, J. R, Riddle, J. C, Stankowski, L. F. Jr. and Yang, L. L. (1987), A Guide for the Performance of the Chinese Hamster Ovary Cell/Hypoxanthine-Cuanine Phosphoribosyl Transferase Gene Mutation Assay, Mutation Res., 189, pp. 135-141. |
|
(11) |
Liber, H. L., Yandell, D. W. and Little, J. B. (1989), A Comparison of Mutation Induction at the TK and HPRT Loci in Human Lymphoblastoid Cells: Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK Locus, Mutation Res., 216, pp. 9-17. |
|
(12) |
Stankowski. L. F. Jr., Tindall. K. R. and Hsie, A. W. (1986), Quantitative and Molecular Analyses of Ethyl Methanosulphonate — and ICR 191-lnduced Molecular Analyses of Ethyl Methanosulphonate — and ICR 191-induced Mutation in AS52 Cells, Mutation Res., 160, pp. 133.147. |
|
(13) |
Turner, N. T., Batson, A. G. and Clive, D. (1984), Procedures for the L5178Y/TK+/- — TK+/- Mouse Lymphoma Cell Mutagenicity Assay, in: Kilbey, B. J. et al. (eds.) Handbook of Mutagenichy Test Procedures, Elsevier Science Publishers, New York, pp. 239-268. |
|
(14) |
Arlett, C. F., Smith, D. M., Clarke, G. M., Creen, M. H. L., Cole, J., McGregor, D. B. and Asquith, J. C. (1989), Mammalian Cell Gene Mutation Assays Based upon Colony Formation, in: Statistical Evaluation of Mutagenicity Test Data. Kirkland D. J., ed., Cambridge University Press, pp. 66-101. |
|
(15) |
Abbondandolo, A., Bonatti, S., Corti, C, Fiorio, R., Loprieno, N. and Mazzaccaro, A. (1977), Induction of 6-Thioguanine-Resistant Mutants in V79 Chinese Hamster Cells by Mouse-Liver Microsome-Activated Dimethylnitrosamine, Mutation Res., 46, pp. 365-373. |
|
(16) |
Ames, B. N., McCann, J. and Yamasaki, E. (1975), Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian-Microsome Mutagenicity Test, Mutation Res., 31, pp. 347-364. |
|
(17) |
Clive, D., Johnson, K. O., Spector, J. R S., Batson, A. G. and Brown M. M. M. (1979), Validation and Characterisation of the L5178Y/TK+/- Mouse Lymphoma Mutagen Assay System, Mutat Res., 59, pp. 61-108. |
|
(18) |
Maron, D. M. and Ames, B. N. (1983), Revised Methods for the Salmonella Mutagenicity Test, Mutation Res., 113, pp. 173-215. |
|
(19) |
Elliott, B. M., Combes, R. D., Elcome, C. R., Gatehouse, D. G., Gibson, G. G., Mackay, J. M. and Wolf, R. C. (1992), Alternatives to Aroclor 1254-lnduced S9 in In Vitro Genotoxicity Assays, Mutagenesis, 7, pp. 175-177. |
|
(20) |
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976), A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems, in In Vitro Melabolic Activation in Mutagenesis Testing, de Serres, F. J., Fouts, J. R., Bend, J. R. and Philpot. R. M. (eds.) Elsevier, North-Holland, pp. 85-88. |
|
(21) |
Krahn, D. F., Barsky, F. C. and McCooey, K. T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids, In: Tice, R. R., Costa, D. L, Schaich, K. M. (eds.), Genotoxic Effects of Airborne Agenta, New York, Plenum, pp. 91-103. |
|
(22) |
Zamora, P. O., Benson, J. M., Li, A. P. and Brooks, A. L. (1983), Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay, Environmental Mutagenesis, 5, pp. 795-801. |
|
(23) |
Applegate, M. L., Moore, M. M., Broder, C. B., Burrell, A. and Hozier, J. C. (1990), Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells, Proc. Natl. Acad. Sci, USA, 87, pp. 51-55. |
|
(24) |
Moore, M. M., Clive, D., Hozier, J. C. Howard, B. L., Batson, A. G., Turner, N. T. and Sawyer, J. (1985), Analysis of Trifluorothymidine-Resistant (TFT) and Mutants of L51 78Y/TK+/- Mouse Lymphoma Cells, Mutation Res. 151, pp. 161-174. |
|
(25) |
Yandell, D. W., Dryja, T. P. and Little, J. B. (1990), Molecular Genetic Analysis of Recessive Mutations at a Heterozygous Autosomal Locus in Human Cells, Mutation Res., 229, pp. 89-102. |
|
(26) |
Moore, M. M. and Doerr, C. L. (1990), Comparison of Chromosome Aberration Frequency and Small Colony TK-Deficient Mutant Frequency in L5178Y/TK+/- — 3.7.2C Mouse Lymphoma Cells, Mutagenesis 5, pp. 609-614.» |
B.18. LESIÓN Y REPARACIÓN DE DNA — SÍNTESIS DE DNA NO PROGRAMADA — CÉLULAS DE MAMÍFEROS IN VITRO
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIÓN
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
El ensayo de síntesis no programada de DNA (UDS) mide la síntesis de reparación del DNA tras escisión y eliminación de un fragmento de DNA que contiene la región lesionada por agentes químicos y físicos. Se basa en la incorporación de timidina tritiada (3H-TdR) al DNA de células de mamífero que no se encuentren en la fase S del ciclo celular. Es posible determinar la captación de 3H-TdR mediante el examen del DNA procedente de células tratadas por autorradiografía o recuento en centelleo líquido (LSC): Las células de mamífero en cultivo, salvo si se utilizan hepatocitos primarios de rata, se tratan con la sustancia objeto de estudio con un sistema de activación metabólica exógeno y sin él. También es posible determinar la UDS por métodos in vivo.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
Preparativos
Las sustancias químicas analizadas y las de control o referencia se prepararán en un medio de crecimiento, o se disolverán o suspenderán en vehículos apropiados, para diluirse luego aún más en un medio de cultivo antes de utilizarse en el ensayo. La concentración final del vehículo no debe influir en la viabilidad celular.
Pueden utilizarse para este ensayo cultivos primarios de hepatocitos de rata, linfocitos humanos o líneas celulares establecidas (por ejemplo, fibroblastos humanos diploides).
Las células se expondrán a la sustancia química estudiada en presencia y ausencia de un sistema de activación metabólica apropiado.
Condiciones del ensayo
Número de cultivos
Son necesarios, para cada punto experimental, al menos dos cultivos celulares para la autorradiografía y seis (o menos, si está justificado científicamente) para el recuento en centelleo líquido.
Uso de testigos negativos y positivos
Se incluirán en cada experimento controles positivos y negativos (no tratados, con vehículo o ambos) simultáneos, con activación metabólica y sin ella.
Son ejemplos de controles positivos para la prueba con hepatocitos de ratas el 7,12-dimetilbenzantraceno (7,12-DMBA) o el 2-acetilaminfluoreno (2-AAF). En el caso de líneas celulares establecidas, el 4-NQO (4-nitriquinolina-N-óxido) es un ejemplo de control positivo para los ensayos por autorradiografía y LSC realizados sin activación metabólica; cuando se emplean sistemas de activación metabólica, un ejemplo de compuesto de control positivo sería la N-dimetilnitrosamina.
Concentraciones de exposición
Se utilizará una gama de concentraciones de la sustancia estudiada que permita determinar la respuesta de modo óptimo. La concentración máxima debe producir ciertos efectos citotóxicos. Los compuestos relativamente insolubles en agua se estudiarán hasta el límite de solubilidad. En lo que respecta a las sustancias atóxicas claramente hidrosolubles, la concentración más alta se determinará en cada caso.
Células
Para mantener los cultivos, se utilizarán medios de crecimiento, concentraciones de CO2 y temperatura y humedad apropiados. Las líneas celulares establecidas se examinarán periódicamente para comprobar si existe contaminación por mycoptasma.
Activación metabólica
En los cultivos primarios de hepatocitos no se utiliza ningún sistema de activación metabólica. Por su parte, las líneas celulares establecidas y los linfocitos se exponen a la sustancia estudiada en presencia y ausencia de un sistema de activación metabólica apropiado.
Procedimiento
Preparación de los cultivos
Líneas celulares establecidas, derivadas de cultivos madre (por ejemplo, por tripainización o agitación) se siembran con la densidad adecuada en recipientes de cultivo y se incuban a 37 oC.
Se establecen cultivos a corto plazo de hepatocitos de rata permitiendo a hepatocitos recién disociados en un medio apropiado fijarse a la superficie de crecimiento.
Los cultivos de linfocitos humanos se establecen por medio de técnicas apropiadas.
Tratamiento de los cultivos con la sustancia estudiada
Hepatocitos primarios de rata
Los hepatocitos de ratas recién aisladas se tratan con la sustancia estudiada, en un medio que contenga 3H-TdR, durante un período de tiempo apropiado. Al final de este, se eliminará el medio, y se aclararán, fijarán y secarán las células. Las preparaciones se sumergen en una emulsión autorradiográfica (también cabe utilizar película fotográfica), se exponen, se revelan, se tiñen y se cuentan.
Líneas celulares establecidas y linfocitos
Técnicas autorradiográficas: Los cultivos celulares se exponen a la sustancia estudiada durante períodos de tiempo adecuados, y se tratan después con 3H-TdR. El plazo de exposición dependerá de la naturaleza de la sustancia, de la actividad del sistema metabólico y del tipo de células. Para apreciar el pico de UDS, se añadirá 3H-TdR al mismo tiempo que la sustancia analizada o en los pocos minutos posteriores a su exposición. La elección de una u otra de estas técnicas vendrá determinada por posibles interacciones entre la sustancia y la 3H-TdR. Para distinguir esta última se utiliza, por ejemplo, un medio deficitario en arginina, un contenido de suero bajo o la adición de hidroxiurea al medio de cultivo.
Determinaciones de la UDS por LSC: Antes de proceder al tratamiento con la sustancia objeto de estudio, se bloqueará la entrada de las células en la fase S del modo antes indicado; a continuación, se expone a las células a la sustancia tal como se describe para la autorradiografía. Al final del período de incubación, se extrae el DNA de las células y se determinan el contenido total de DNA y el grado de incorporación de 3H-TdR.
Hay que señalar que cuando se utilicen linfocitos humanos en las técnicas expuestas no es necesario suprimir la replicación semiconservadora de DNA en cultivos no estimulados.
Análisis
Determinaciones autorradiográficas
Para determinar la UDS en células en cultivo, no se cuentan los núcleos en fase S. Se contarán por lo menos 50 células por concentración. Las preparaciones recibirán un código antes del recuento. Se contarán en cada una de ellas varios campos, elegidos al azar y lo bastante alejados entre sí. Se determinará la cantidad de 3H-TdR incorporada al citoplasma mediante el recuento de tres superficies del tamaño del núcleo en el citoplasma de cada célula contada.
Determinación por LSC
En las determinaciones de la UDS por LSC debe utilizarse un número adecuado de cultivos para cada concentración y en los controles.
Todos los resultados se confirmarán en un experimento independiente.
2. RESULTADOS
Los resultados se presentarán en forma de tablas.
2.1. DETERMINACIONES AUTORRADIOGRÁFICAS
Se registrarán por separado la cantidad de 3H-TdR incorporada al citoplasma y el número de granos encontrados sobre el núcleo celular.
Pueden utilizarse la media, la mediana y el modo para describir la distribución de la cantidad de 3H-TdR incorporada en el citoplasma, así como el número de granos por núcleo.
2.2. DETERMINACIONES POR LSC
Para las determinaciones por LSC, se indicará la incorporación de 3H-TdR en forma de dpm/μg de DNA. Puede utilizarse la media de dmp/μg de DNA, con su desviación estándar, para describir la distribución de la incorporación.
Los datos se evaluarán por métodos estadísticos apropiados.
3. INFORME
3.1. DATOS EL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
células utilizadas, densidad y número de pases en el momento del tratamiento, número de cultivos celulares, |
|
— |
métodos utilizados para el mantenimiento de los cultivos celulares, incluidos medio, temperatura y concentración de CO2, |
|
— |
sustancia estudiada, vehículo, concentraciones y justificación de la elección de las concentraciones empleadas en el ensayo, |
|
— |
detalles sobre los sistemas de activación metabólica, |
|
— |
programa de tratamiento, |
|
— |
controles positivos y negativos, |
|
— |
técnica autorradiográfica utilizada, |
|
— |
métodos empleados para bloquear la entrada de las células en la fase S, |
|
— |
técnicas utilizadas para extraer el DNA y determinar la cantidad total de DNA en la determinación por LSC, |
|
— |
relación dosis-respuesta, si es posible, |
|
— |
evaluación estadística, |
|
— |
comentario de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.19. ENSAYO IN VITRO DE INTERCAMBIO DE CROMÁTIDAS HERMANAS
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIÓN
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
El ensayo de intercambio de cromátidas hermanas (SCE) es una prueba a corto plazo encaminada a descubrir intercambios recíprocos de DNA entre 2 cromátidas hermanas de un cromosoma de desdoblamiento. Los SCE representan el intercambio recíproco de productos de replicación del DNA en loci aparentemente homólogos. Es probable que el proceso de intercambio comprenda la rotura y la reunión del DNA, aunque se sabe poco sobre su base molecular. Para descubrir los SCE, es necesario poder marcar de forma diferente las cromátidas hermanas, cosa que se consigue mediante la incorporación de bromodesoxiuridina (BrdU) al DNA cromosómico durante dos ciclos celulares.
Se exponen células de mamífero in vitro a la sustancia objeto de estudio en presencia y ausencia de un sistema de activación metabólica exógena de mamífero, si procede, y se cultivan durante dos ciclos de replicación en un medio que contenga BrdU. Después del tratamiento con un inhibidor del huso (por ejemplo, colchicina) para acumular las células en fase de mitosis de tipo metafásico (c-metafase), se recolectan las células y se realizan preparaciones de cromosomas.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
Preparativos
|
— |
Pueden utilizarse en el ensayo cultivos primarios (linfocitos humanos) o líneas celulares establecidas (por ejemplo, células ováricas de hámster chino). Las líneas celulares se controlarán en busca de una posible contaminación por mycoplasma. |
|
— |
Se utilizarán medios de cultivo y condiciones de incubación (por ejemplo, temperatura, recipientes de cultivo, concentración de CO2 y humedad) adecuados. |
|
— |
Las sustancias estudiadas pueden prepararse en medios de cultivo, o disolverse o suspenderse en vehículos apropiados, antes del tratamiento de las células. La concentración final de un vehículo en el sistema de cultivo no debe influir de modo notable en la viabilidad ni en el índice de crecimiento de las células, y los efectos sobre la frecuencia de SCE se controlarán con ayuda de un control del disolvente. |
|
— |
Las células se expondrán a la sustancia estudiada en presencia y ausencia de un sistema de activación metabólica exógeno de mamífero. Por otro lado, cuando se utilicen tipos de células con actividad metabólica intrínseca, el grado y la naturaleza de la actividad deben ser apropiados para la clase química objeto de estudio. |
Condiciones del ensayo
Número de cultivos
Para cada ensayo, se utilizarán dos cultivos separados, por lo menos.
Uso de controles positivos y negativos
Deben incluirse en cada experimento controles positivos en los que se utilicen una sustancia con acción directa y otra que precisa activación metabólica; también ha de utilizarse un control para el vehículo.
A continuación facilitamos ejemplos de sustancias que pueden utilizarse como controles positivos:
|
— |
compuesto de acción directa: etilmetanosulfonato, |
|
— |
compuesto de acción indirecta: ciclofosfamida. |
En caso oportuno, puede incluirse un control positivo complementario de la misma clase química que la sustancia estudiada.
Concentraciones de exposición
Se utilizarán al menos tres concentraciones de la sustancia objeto de estudio, escalonadas debidamente. La concentración máxima producirá un efecto tóxico significativo, pero no impedirá una replicación celular adecuada. Las sustancias relativamente insolubles en agua se estudiarán hasta el límite de solubilidad por métodos apropiados. En lo que respecta a las sustancias atóxicas claramente hidrosolubles, la concentración más alta se determinará en cada caso.
Procedimiento
Preparación de los cultivos
Líneas celulares establecidas, derivadas de cultivos madre (por ejemplo, por tripsinización o agitación), se siembran con la densidad adecuada en recipientes de cultivo y se incuban a 37 oC. En los cultivos de una sola capa celular, el número de células por recipiente de cultivo debe ajustarse de modo que los cultivos no confluyan en más del 50 % en el momento de la recogida. También es posible utilizar las células en forma de un cultivo en suspensión. Los cultivos de linfocitos humanos se preparan a partir de sangre heparinizada por medio de técnicas apropiadas, y se incuban a 37 oC.
Tratamiento
Se expone a células en fase de crecimiento exponencial a la sustancia analizada durante un período de tiempo apropiado; en la mayoría de los casos pueden bastar una o dos horas, pero en determinadas circunstancias puede prolongarse el período de tratamiento hasta abarcar dos ciclos celulares completos. Las células sin actividad metabólica intrínseca deben exponerse a la sustancia estudiada en presencia y ausencia de un sistema de activación metabólica apropiado. Al final del período de exposición, se lavan las células hasta eliminar la sustancia estudiada y se cultivan en presencia de BrdU durante dos ciclos de replicación. Otro método consiste en exponer simultáneamente las células a la sustancia y BrdU durante dos ciclos celulares completos.
Los cultivos de linfocitos humanos se tratan mientras se encuentran en un estado semisincrónico.
Las células se examinan en su segunda división tras el tratamiento, con lo que se garantiza la exposición de las fases más sensibles del ciclo celular a la sustancia estudiada. Todos los cultivos a los que se añada BrdU se manipularán en la oscuridad o con iluminación débil de lámparas incandescentes hasta la recolección de las células, a fin de reducir la fotolisis del DNA que contenga BrdU.
Recolección de las células
Los cultivos celulares se tratan con un inhibidor del huso (por ejemplo, colchicina) de 1 a 4 horas antes de la recolección. Cada cultivo se recolecta y trata por separado para la preparación de los cromosomas.
Preparación y teñido de los cromosomas
Las preparaciones de cromosomas se realizan por métodos citogenéticos estándar. Existen varias técnicas para teñirlos de forma que se aprecien los SCE (por ejemplo, el método de fluorescencia con Giemsa).
Análisis
El número de células analizadas dependerá de la frecuencia espontánea de control de SCE. Por lo general, se analizan al menos 25 metafases bien escalonadas por cultivo para contar los SCE. Las preparaciones reciben un código antes del análisis. En linfocitos humanos, solo se analizan las metafases que contienen 46 centrómeros. En las líneas celulares establecidas, solo se analizan las metafases que contienen ± 2 centrómeros del número modal. Se precisará si una inversión del marcado en el centrómero se considera SCE. Los resultados se confirmarán en un experimento independiente.
2. RESULTADOS
Los resultados se presentarán en forma de tablas. El número de SCE para cada metafase y el de SCE por cromosoma han de facilitarse por separado para todos los cultivos tratados y de control.
Los resultados se analizarán por métodos estadísticos adecuados.
3. INFORME
3.1. DATOS DEL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
células utilizadas, métodos de mantenimiento del cultivo celular |
|
— |
condiciones del ensayo: composición de los medios, concentración de CO2, concentración de la sustancia estudiada, vehículo empleado, temperatura de incubación, período de tratamiento, inhibidor del huso utilizado, su concentración y duración del tratamiento con él, tipo de sistema de activación de mamífero utilizado, controles positivos y negativos, |
|
— |
número de cultivos celulares por ensayo, |
|
— |
detalles de la técnica utilizada para la preparación de las extensiones, |
|
— |
número de metafases analizadas (datos facilitados por separado para cada cultivo), |
|
— |
número medio de SCE por célula y por cromosoma (datos facilitados por separado para cada cultivo), |
|
— |
criterio de recuento de los SCE, |
|
— |
justificación de la elección de las dosis, |
|
— |
relación dosis-respuesta, si es posible, |
|
— |
evaluación estadística, |
|
— |
comentario de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.20. ENSAYO DE LETALIDAD RECESIVA LIGADA AL SEXO EN DROSOPHILA MELANOGASTER
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIÓN
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
La prueba de letalidad recesiva ligada al sexo (SLRL) practicada con Drosophila melanogaster descubre la aparición de mutaciones (lo mismo mutaciones puntuales que pequeñas deleciones) en la línea germinal del insecto. Se trata de un análisis de mutación capaz de descubrir mutaciones en unos 800 loci de cromosoma X, lo que supone alrededor del 80 % del total de loci existentes. El cromosoma X supone aproximadamente una quinta parte del genoma haploide completo.
Las mutaciones del cromosoma X de Drosophila melanogaster se expresan fenotípicamente en los machos portadores del gen mutante. Cuando la mutación es letal en el estado hemicigoto, se deduce su presencia por la falta de uno de los grupos de descendencia masculina de los dos producidos normalmente por una hembra heterocigota. La prueba SLRL aprovecha estas circunstancias por medio de cromosomas marcados especialmente y con reordenaciones de su estructura.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
Preparativos
Animales de experimentación
Pueden utilizarse machos de un linaje de tipo salvaje bien definido, y hembras de linaje Muller-5. También cabe emplear otros linajes de hembras debidamente marcadas con cromosomas X invertidos de manera múltiple.
Sustancia objeto de estudio
Las sustancias estudiadas se disuelven en agua. Las que sean insolubles en agua pueden disolverse o suspenderse en vehículos apropiados (por ejemplo, una mezcla de etanol y Tween-60 u 80), para diluirse luego en agua o solución salina antes de la administración. Se evitará el uso de dimetilsulfóxido (DMSO) como vehículo.
Número de animales
Se determinarán previamente la sensibilidad y la potencia de la prueba. La frecuencia de mutantes espontáneos observada en el control apropiado influirá intensamente en el número de cromosomas tratados que deba analizarse.
Vía de administración
La exposición puede ser oral, por inyección o por exposición a gases o vapores. La sustancia objeto de estudio puede administrarse en una solución azucarada. Cuando sea necesario, pueden disolverse las sustancias en una solución de CINa al 0,7 % e inyectarlas así en el tórax o el abdomen.
Uso de controles negativos y positivos
Se incluirán controles negativos (vehículo) y positivos. No obstante, si el laboratorio dispone de datos de controles publicados adecuados, no son necesarios controles simultáneos.
Niveles de exposición
Se utilizarán tres niveles de exposición. Para una evaluación preliminar, puede utilizarse un solo nivel de exposición de la sustancia analizada, que corresponderá a la concentración máxima tolerada o a la que produzca ciertos signos de toxicidad. Cuando se trate de sustancias atóxicas, ha de utilizarse la exposición en la concentración máxima posible.
Procedimiento
Se tratan machos de fenotipo salvaje (de tres a cinco días de edad) con la sustancia objeto de estudio y se aparean individualmente con varias hembras vírgenes del linaje Muller-5, o de otro debidamente marcado (con cromosomas invertidos de manera múltiple). Las hembras se sustituyen por vírgenes nuevas cada dos o tres días para cubrir el ciclo germinal completo. Se examina luego la descendencia de estas hembras para apreciar los efectos letales correspondientes a los efectos sobre el esperma maduro, las espermátidas en estadios precoces, medios y tardíos, los espermatocitos y los espermatogonios en el momento del tratamiento.
Las hembras F1 heterocigotas procedentes de los cruces anteriores se aparean individualmente (es decir, una hembra por vial) con sus hermanos. En la generación F2, se examina cada cultivo para descubrir la ausencia de machos del tipo salvaje. Si un cultivo parece proceder de una hembra F1 portadora de un gen letal en el cromosoma X paterno (es decir, no se observa ningún macho portador del cromosoma tratado), se pondrán a prueba las hijas de esta hembra que presenten el mismo genotipo para comprobar si la letalidad se repite en la generación siguiente.
2. RESULTADOS
Los resultados se presentarán en forma de tablas en las que se consignen el número de cromosomas estudiados, el de machos infértiles y el de cromosomas letales en cada concentración de exposición y en cada período de apareamiento, para cada macho tratado. Se indicará el número de grupos de tamaños diferentes por macho. Los resultados se confirmarán en un experimento independiente.
Se utilizarán métodos estadísticos apropiados para evaluar el ensayo de letalidad recesiva ligada al sexo. El reagrupamiento de genes letales recesivos procedentes de un solo macho se tendrá en cuenta y se evaluará por un método estadístico adecuado.
3. INFORME
3.1. DATOS DEL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
linajes o cepas de Drosophila empleados, edad de los insectos, número de machos tratados, número de machos estériles, número de cultivos F2 establecidos, número de cultivos F2 sin progenie, número de cromosomas portadores de un gen letal encontrado en cada estadio de la célula germinal, |
|
— |
criterios adoptados para determinar el tamaño de los grupos tratados, |
|
— |
condiciones de la prueba: descripción detallada del programa de tratamiento y muestreo, niveles de exposición, datos de toxicidad, controles negativos (disolvente) y positivos si procede, |
|
— |
criterios de recuento de las mutaciones letales, |
|
— |
relación exposición/efecto, si es posible, |
|
— |
evaluación de los datos, |
|
— |
comentario de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.21. ENSAYO DE TRANSFORMACIÓN DE CÉLULAS DE MAMÍFERO IN VITRO
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIÓN
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
Es posible utilizar sistemas de cultivo de células de mamífero para descubrir modificaciones fenotípicas in vitro inducidas por sustancias químicas asociadas con transformación maligna in vivo. Entre las células de uso más frecuente figuran C3H10T1/2, 3T3, SHE, rata Fisher; las pruebas se basan en modificaciones de la morfología celular, la formación de focos o modificaciones vinculadas con el crecimiento o no en agar semisólido. Existen sistemas, empleados con menor frecuencia, que ponen de manifiesto otras alteraciones fisiológicas o morfológicas en las células tras exposición a sustancias químicas carcinogénicas. Ninguno de los criterios analizados en estas pruebas in vitro tiene una relación de mecanismo establecida con el cáncer. Algunos de los sistemas de prueba son capaces de descubrir promotores tumorales. Puede determinarse la citotoxicidad averiguando el efecto de la sustancia estudiada sobre la capacidad de formación de colonias (eficacia de clonado) o los índices de crecimiento de los cultivos. La determinación de la citotoxicidad tiene por objeto establecer si la exposición a la sustancia analizada ha sido toxicológicamente significativa, pero no permite calcular la frecuencia de transformaciones en todas las pruebas, dado que en algunas de estas pueden ser necesarias una incubación prolongada, una nueva siembra o ambas.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCION DEL MÉTODO
Preparativos
Células
Según la prueba de transformación que se realice, pueden utilizarse líneas celulares o células primarias diversas. El investigador se encargará de que las células que se empleen presenten la modificación fenotípica apropiada tras exposición a sustancias carcinogénicas conocidas, y de que la fiabilidad y la validez de la prueba efectuada en su laboratorio estén contrastadas por datos documentados.
Medio
Se utilizarán medios y condiciones experimentales apropiados para la prueba de transformación elegida.
Sustancia objeto de estudio
Las sustancias puestas a prueba pueden prepararse en medios de cultivo, disolverse o suspenderse en vehículos apropiados antes de tratar las células. La concentración final del vehículo en el medio de cultivo no influirá en la viabilidad de las células, el índice de crecimiento ni en la incidencia de transformación.
Actividad metabólica
Las células se expondrán a la sustancia estudiada en presencia y en ausencia de un sistema de activación metabólica de mamífero apropiado. Cuando se utilicen tipos celulares con actividad metabólica intrínseca, deberá saberse si la naturaleza de la actividad es apropiada para la clase química analizada.
Condiciones del ensayo
Uso de controles negativos y positivos
Se incluirán en cada experimento controles positivos a base de una sustancia de acción directa y otra que precise activación metabólica; también se utilizará un control negativo (vehículo).
A continuación se ofrecen ejemplos de sustancias que pueden emplearse como controles positivos:
|
— |
Compuestos de acción directa:
|
|
— |
Compuestos que precisan activación metabólica:
|
Cuando se juzgue oportuno, se incluirá un control positivo complementario perteneciente a la misma clase química que la sustancia estudiada.
Concentraciones de exposición
Se utilizarán varias concentraciones de la sustancia objeto de estudio, que producirán un efecto tóxico acorde con su potencia; así, la concentración máxima provocará una supervivencia escasa, mientras que la supervivencia con la concentración mínima será aproximadamente igual a la encontrada en el control negativo. Las sustancias relativamente insolubles en agua se estudiarán hasta el límite de solubilidad por medio de métodos apropiados. En lo que respecta a las sustancias atóxicas claramente hidrosolubles, la concentración más alta de la sustancia estudiada se determinará en cada caso.
Procedimiento
Las células deben exponerse durante un período de tiempo adecuado, que dependerá del sistema celular utilizado; por tanto, quizá sea necesario un nuevo tratamiento, acompañado de un cambio de medio (y, en caso preciso, de una nueva mezcla de activación metabólica), si la exposición es prolongada. Las células carentes de actividad metabólica intrínseca suficiente se expondrán a la sustancia estudiada en presencia y en ausencia de un sistema de activación metabólica apropiado. Al final del período de exposición, se lavan las células hasta eliminar la sustancia analizada y se cultivan en condiciones que permitan la aparición del fenotipo transformado en estudio; se determina también la incidencia de esta transformación. Todos los resultados se confirman en un experimento independiente.
2. RESULTADOS
Los resultados se presentarán en forma de tablas que recogerán datos diversos según el método que se utilice, como, por ejemplo, recuentos por placa, placas positivas o número de células transformadas. Cuando proceda, se expresará el índice de supervivencia en porcentaje de los índices de control y frecuencia de transformación a modo del número de células transformadas frente al de supervivientes. Los resultados se evaluarán por métodos estadísticos apropiados.
3. INFORME
3.1. DATOS DEL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
tipo de célula utilizado, número de cultivos celulares, métodos de mantenimiento de los cultivos celulares, |
|
— |
condiciones del ensayo; concentración de la sustancia estudiada, vehículo utilizado, período de incubación, duración y frecuencia del tratamiento, densidad celular durante el tratamiento, tipo de sistema de activación metabólica exógena utilizado, controles positivos y negativos, especificación del fenotipo estudiado, sistema selectivo empleado (si procede), justificación de la elección de las dosis, |
|
— |
método utilizado para contar las células viables y transformadas, |
|
— |
evaluación estadística, |
|
— |
discusión de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.22. ENSAYO DE LETALIDAD DOMINANTE EN ROEDORES
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIÓN
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
La letalidad dominante provoca la muerte del embrión o feto. Su inducción por exposición a una sustancia química indica que esta ha alterado el tejido embrionario de la especie estudiada. Se admite generalmente que la letalidad dominante se debe a una lesión cromosómica (anomalías estructurales y numéricas). Si los animales tratados son hembras, la muerte del embrión también puede deberse a efectos tóxicos.
En general, los machos se exponen a la sustancia objeto de estudio y se aparean con hembras vírgenes no tratadas. Es posible investigar por separado los distintos estadios de las células embrionarias gracias al empleo de intervalos de apareamiento sucesivos. Un número de implantes muertos por hembra, en el grupo tratado, superior al observado en el grupo de control, es reflejo de las pérdidas tras la implantación. Por otro lado, es posible calcular las pérdidas antes de ella mediante recuento de los cuerpos lúteos, o por comparación entre número total de implantes por hembra en los grupos tratados y los de control. El efecto total de letalidad dominante es igual a la suma de las pérdidas antes y después de la implantación. El cálculo de tal efecto se basa en la comparación del número de implantes vivos por hembra, en el grupo de prueba, en relación con el registrado para el grupo de control. Una reducción del número de implantes en determinados intervalos puede deberse a destrucción de células (es decir, de espermatocitos, espermatogonios o ambos).
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
Preparativos
Cuando sea posible, las sustancias objeto de estudio se disolverán o suspenderán en solución salina isotónica. Las sustancias químicas insolubles en agua pueden disolverse o suspenderse en vehículos apropiados. El vehículo utilizado no alterará el compuesto estudiado ni producirá efectos tóxicos. Deben emplearse preparaciones recientes de la sustancia objeto de estudio.
Condiciones del ensayo
Vía de administración
En general, la sustancia estudiada solo debe administrarse una vez, aunque cabe utilizar un programa de tratamiento reiterado si lo aconseja la información toxicológica disponible. Las vías de administración usuales son la intubación oral y la inyección intraperitoneal. Pueden ser apropiadas otras vías de administración.
Animales de experimentación
Se recomienda utilizar ratas o ratones, jóvenes y en plena madurez sexual, que se distribuirán al azar para formar grupos de tratamiento y de control.
Número y sexo
Se utilizará un número apropiado de machos tratados, teniendo en cuenta la frecuencia espontánea del carácter biológico evaluado según la cepa empleada. El número elegido se basará en la sensibilidad de detección y el grado de significación, determinados previamente. Por ejemplo, en un experimento tipo, el número de machos en cada grupo posológico deberá ser suficiente para obtener alrededor de 30 a 50 hembras grávidas por período de apareamiento.
Uso de controles negativos y positivos
Se incluirán simultáneamente en cada experimento controles positivos y negativos (vehículos). Cuando se disponga de resultados aceptables obtenidos con controles positivos en experimentos recientes en el mismo laboratorio, podrán emplearse en lugar de un control positivo simultáneo. En lo que respecta a las sustancias de control positivas, se utilizará una dosis baja apropiada (por ejemplo, MMS, intraperitonalmente, 10 mg/kg) para demostrar la sensibilidad de la prueba.
Dosis
Normalmente, se utilizan tres niveles posológicos. La dosis alta producirá signos de toxicidad o reducirá la fecundidad de los animales tratados. En determinados casos, puede bastar un solo nivel posológico alto.
Prueba de límite
Las sustancias atóxicas se estudian en concentración de 5 g/kg en caso de administración única, y de 1 g/kg/día en caso de administración reiterada.
Procedimiento
Hay varios esquemas de tratamiento posibles. El método más frecuente es el de la administración única, pero pueden utilizarse otros.
Cada macho se aparea de forma secuencial con 1 o 2 hembras vírgenes no tratadas, a intervalos de tiempo apropiados tras el tratamiento. Las hembras se mantendrán con los machos durante al menos un ciclo estral completo o hasta que se compruebe el apareamiento por la presencia de esperma en la vagina o por la existencia de tapón vaginal.
El número de apareamientos tras el tratamiento dependerá del programa de tratamiento, y garantizará la obtención de muestras de todos los estadios de células embrionarias tras el tratamiento.
Las hembras se sacrifican en la segunda mitad de la gestación para examinar el contenido de su útero y determinar el número de implantes vivos y muertos. Pueden inspeccionarse los ovarios para averiguar el número de cuerpos lúteos.
2. RESULTADOS
Los resultados se presentarán en forma de tablas en las que aparezcan el número de machos, el de hembras grávidas y el de hembras no grávidas. Se facilitarán por separado los resultados de cada apareamiento, incluida la identidad de cada macho y hembra. Se recogerán, para cada hembra, la semana de apareamiento y la dosis recibida por los machos, así como las frecuencias de implantes vivos y muertos.
El cálculo del efecto total de la letalidad dominante se basa en la comparación del número de implantes vivos por hembra en el grupo tratado y en el de control. Se analiza la relación entre implantes vivos y muertos en el grupo tratado, en comparación con la obtenida en el grupo de control, para deducir las pérdidas tras la implantación.
Si se registran los datos en forma de muertes precoces y tardías, se indicará claramente esta diferencia en las tablas. Si se calculan las pérdidas antes de la implantación, debe informarse de ellas. Tales pérdidas pueden calcularse a modo de diferencias entre el número de cuerpos lúteos y el de implantes, o de reducción del número medio de implantes por hembra en relación con los apareamientos de control.
Los resultados se evaluarán por métodos estadísticos apropiados.
3. INFORME
3.1. DATOS DEL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
especie, cepa, edad y pesos de los animales utilizados, número de animales de cada sexo en los grupos tratados y controles, |
|
— |
sustancia estudiada, vehículo, niveles posológicos ensayados y justificación de su elección, controles negativos y positivos, datos relativos a la toxicidad, |
|
— |
vía y programa de tratamiento, |
|
— |
programa de apareamiento, |
|
— |
método utilizado para comprobar el apareamiento, |
|
— |
momento del sacrificio, |
|
— |
criterios de recuento de los efectos de la letalidad dominante, |
|
— |
relación dosis-respuesta, si es posible, |
|
— |
evaluación estadística, |
|
— |
comentario de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.23. «ENSAYO DE ABERRACIONES CROMOSÓMICAS EN ESPERMATOGONIAS DE MAMÍFERO
1. MÉTODO
El presente método reproduce las directrices del documento OCDE TG 483 sobre el ensayo de aberraciones cromosómicas en espermatogonias de mamífero (1997).
1.1. INTRODUCCIÓN
El ensayo de aberraciones cromosómicas en espermatogonias de mamífero tiene por objeto detectar sustancias que provocan aberraciones cromosómicas estructurales en espermatogonias de mamífero (1) (2) (3) (4) (5). Las aberraciones estructurales pueden ser cromosómicas o cromatídicas. La mayoría de los mutágenos químicos dan lugar a aberraciones cromatídicas, aunque también pueden producirse aberraciones cromosómicas. El presente método no está pensado para detectar aberraciones numéricas ni se emplea habítualmente con ese fin. Las mutaciones cromosómicas y los efectos relacionados ocasionan numerosas enfermedades genéticas humanas.
El presente ensayo detecta alteraciones cromosómicas en las células germinales de las espermatogonias y debería, por tanto, permitir prever la inducción de mutaciones hereditarias en las células germinales.
El ensayo suele realizarse con roedores; es un ensayo citogenético in vivo que detecta aberraciones cromosómicas en las mitosis de las espermatogonias. El presente método no se refiere a otras células diana.
Para detectar aberraciones cromatídicas en las espermatogonias es preciso analizar la primera mitosis celular después del tratamiento, antes de que las lesiones desaparezcan en las divisiones celulares siguientes. Puede obtenerse información adicional del análisis de los cromosomas meióticos de células madre de espermatogonias tratadas, para detectar aberraciones cromosómicas de la diacinesis a la metafase I cuando las células tratadas se convierten en espermatocitos.
El presente ensayo in vivo está diseñado para determinar si los mutágenos de las células somáticas también son activos en las células germinales. Asimismo, está indicado para evaluar el riesgo mutagénico, ya que permite tomar en consideración factores del metabolismo in vivo, de la toxicocinética y de los procesos de reparación del ADN.
Los testículos contienen varias generaciones de espermatogonias, que presentan distinta sensibilidad al tratamiento químico. Por tanto, las aberraciones detectadas corresponden a una respuesta global de las poblaciones de espermatogonias tratadas, en las que predominan las células diferenciadas, que son las más numerosas. Según su localización en el testículo y debido a la barrera física y fisiológica de las células de Sertoli, así como a la barrera entre la sangre y el testículo, algunas generaciones de espermatogonias están expuestas a la circulación general y otras no.
Si hay pruebas de que la sustancia de ensayo o un metabolito reactivo no llegan al tejido diana, no procede realizar el presente ensayo.
Véase asimismo la introducción general de la parte B.
1.2. DEFINICIONES
Aberración cromatídica: lesión cromosómica estructural que consiste en la rotura de una cromátida o la rotura y reunión de cromátidas.
Aberración cromosómica: lesión cromosómica estructural que consiste en la rotura o en en la rotura y reunión de ambas cromátidas en el mismo sitio.
Gap: lesión acromática más estrecha que la cromátida, con alteración mínima de la alineación de las cromátidas.
Aberracción numérica: número de cromosomas distinto del número normal propio de los animales empleados.
Poliploidía: un múltiplo del número de dotación cromosómica haploide (n) superior al diploide (3n, 4n, etc.).
Aberración estructural: alteración de la estructura cromosómica (deleciones, intracambios o intercambios) detectable mediante examen microscópico de la metafase de la división celular.
1.3. PRINCIPIO DEL MÉTODO
Se exponen los animales a la sustancia de ensayo por una vía adecuada y se sacrifican a intervalos apropiados tras el tratamiento. Antes de sacrificarlos, se les administra una sustancia que detenga la división celular en la metafase (por ejemplo, colchicina o Colcemid®). A continuación, se realizan preparaciones de cromosomas de células germinales y se tiñen, tras lo cual se analizan las células en metafase para detectar aberraciones cromo-sómicas.
1.4. DESCRIPCIÓN DEL MÉTODO
1.4.1. Preparación
1.4.1.1. Selección de la especie animal
Si bien suelen emplearse hámsters chinos y ratones machos, pueden utilizarse machos de otra especie de mamíferos apropiada. Deben usarse animales adultos jóvenes y sanos de cepas utilizadas habitualmente en laboratorio. La variación del peso de los animales al empezar el estudio ha de ser mínima y no debe exceder de ± 20 % del peso medio.
1.4.1.2. Alojamiento y alimentación de los animales
Se aplicarán las condiciones generales recogidas en la Introducción general de la parte B. La humedad debe ser del 50 al 60 %.
1.4.1.3. Preparación de los animales
Se reparten al azar machos adultos jóvenes y sanos entre los lotes tratados y los lotes de control. Las jaulas se colocan de manera que los posibles efectos debidos a la posición de las mismas sean mínimos. Se identifica a los animales de forma unívoca y se los acostumbra a las condiciones de laboratorio durante al menos cinco días antes de empezar el estudio.
1.4.1.4. Preparación de las dosis
Las sustancias de ensayo sólidas deben disolverse o suspenderse en disolventes o vehículos adecuados y, si es preciso, diluirse antes de su administración a los animales. Las sustancias de ensayo líquidas pueden administrarse directamente o diluirse antes de la administración. Han de emplearse soluciones de la sustancia de ensayo recién preparadas, salvo que los datos relativos a la estabilidad demuestren que es posible conservarlas.
1.4.2. Condiciones del ensayo
1.4.2.1. Disolvente o vehículo
El disolvente o vehículo no deberá producir efectos tóxicos a las dosis empleadas. Es preciso cerciorarse de que el disolvente o vehículo no produce reacciones químicas con la sustancia de ensayo. Si se emplean disolventes o vehículos poco conocidos, debe disponerse de información que avale su compatibilidad. Siempre que sea posible, se recomienda considerar en primer lugar la utilización de un disolvente o vehículo acuoso.
1.4.2.2. Controles
Deberán realizarse controles positivos y negativos (disolvente o vehículo) en paralelo en cada ensayo. Salvo por lo que respecta a la exposición a la sustancia de ensayo, los animales de los lotes de control deben tratarse exactamente igual que los de los lotes tratados.
En los controles positivos deberían producirse aberraciones cromosómicas estructurales en las espermatogo-nias in vivo con grados de exposición en los que quepa esperar un aumento detectable respecto al nivel de fondo.
Las dosis del control positivo deben ser tales que los efectos sean claros, pero no revelen immediatamente al lector la identidad de los portaobjetos codificados. La sustancia empleada en el control positivo podrá administrarse por una vía distinta de la empleada para la sustancia de ensayo y la toma de muestras podrá realizarse una sola vez. Podrá considerarse la utilización en dichos controles de sustancias de clases químicas afines, cuando sea posible. A continuación figuran algunos ejemplos de sustancias para los controles positivos:
|
Sustancia |
N° CAS |
N° EINECS |
|
Ciclofosfamida Monohidrato de ciclofosfamida |
50-18-0 6055-19-2 |
200-015-4 |
|
Ciclohexilamina |
108-91-8 |
203-629-0 |
|
Mitomicina C |
50-07-7 |
200-008-6 |
|
Monómero de acrilamida |
79-06-1 |
201-173-7 |
|
Trietilenomelamina |
51-18-3 |
200-083-5 |
Salvo que haya datos anteriores sobre controles que pongan de manifiesto una variabilidad entre animales y una frecuencia de células con aberraciones cromosómicas aceptables, se realizarán, para cada período de muestreo, controles negativos únicamente con el disolvente o vehículo, y se tratarán, por lo demás, de igual manera que los lotes tratados. Se realizarán asimismo controles sin tratar, salvo que exista información anterior o publicada sobre controles que demuestre que el disolvente o vehículo elegido no induce efectos nocivos ni mutágenos.
1.5. PROCEDIMIENTO
1.5.1. Número de animales
Los lotes tratados y los controles han de estar compuestos al menos por cinco machos analizables cada uno.
1.5.2. Pauta de tratamiento
Las sustancias de ensayo se administrarán preferiblemente una o dos veces (uno o dos tratamientos), aunque también puede dividirse la dosis —por ejemplo, en dos veces al día separadas por algunas horas como máximo— con el fin de facilitar la administración de grandes cantidades. Si se siguen otras pautas debe justificarse científicamente.
En el lote al que se ha administrado la dosis máxima se toman dos muestras tras el tratamiento. Dado que la sustancia de ensayo puede afectar a la cinética del ciclo celular, suele tomarse la primera muestra hacia las 24 horas y la segunda, unas 48 horas después del tratamiento. En los demás lotes se toma una muestra transcurridas 24 horas o un período de 1,5 veces la duración del ciclo celular desde el tratamiento, salvo que se conozca otro período de muestreo más adecuado para detectar los efectos estudiados (6).
Pueden seguirse otros períodos de muestreo. Por ejemplo, en el caso de sustancias que puedan provocar la pérdida de cromosomas o tener efectos independientes de la fase S, pueden estar indicados períodos de muestreo más cortos (1).
Debe valorarse caso por caso la pertenencia de una pauta de administración repetida. Si se aplica una pauta de ese tipo, los animales deben sacrificarse 24 horas (1,5 veces la duración del ciclo celular) después de la última administración. Cuando proceda, pueden aplicarse períodos de muestreo complementarios.
Antes de sacrificar los animales, se les inyecta por vía intraperitoneal una dosis adecuada de una sustancia que detenga la división celular en la metafase (por ejemplo, Colcemid® o colchicina). Se extraen las muestras transcurrido un plazo oportuno, que en el caso de los ratones es de unas 3 a 5 horas y en el de los hámsters chinos, de unas 4 a 5 horas.
1.5.3. Dosis
Si se lleva a cabo un estudio previo de determinación de la gama de dosis por falta de datos adecuados, ha de hacerse en el mismo laboratorio, con la misma especie, cepa y protocolo de tratamiento que el estudio principal (7). Si se produce toxicidad, se administrarán tres dosis distintas para el primer período de muestreo. La gama de dosis deberá abarcar desde la toxicidad máxima hasta una toxicidad escasa o nula. En el último período de muestreo se administrará únicamente la dosis máxima. Se entenderá por «dosis máxima» la que produzca tales signos de toxicidad que, si se administrara una dosis superior según el mismo protocolo resultaría problamente letal.
Las sustancias con actividades biológicas específicas a dosis bajas no tóxicas (como las hormonas y los mitó-genos) pueden constituir excepciones en cuanto a los criterios de establecimiento de la dosis y han de evaluarse caso por caso. La dosis máxima también puede definirse como la dosis que produce algún signo de toxicidad en las espermatogonias (por ejemplo, disminución de la proporción entre el número de mitosis de las espermatogonias y el número de metafases I y II de la meiosis; la disminución no ha de ser superior al 50 %).
1.5.4. Ensayo límite
Si en un ensayo realizado con una dosis de al menos 2 000 mg/kg de peso corporal/día administrada en una sola vez, o dos veces en el mismo día, no se observan efectos tóxicos y si, sobre la base de datos relativos a sustancias de estructura afín, no cabe esperar genotoxicidad, puede no ser necesario llevar a cabo un estudio completo con tres dosis diferentes. Si se prevé la exposición humana a la sustancia, puede ser preciso administrar una dosis mayor en el ensayo límite.
1.5.5. Administración de las dosis
La sustancia de ensayo suele administrarse por sonda gástrica o con una cánula de intubación adecuada, o bien por inyección intraperitoneal. Pueden emplearse otras vías de administración, siempre que se justifiquen. El volumen máximo de líquido que puede administrarse de una sola vez por sonda o inyección depende del tamaño del animal de ensayo, pero no excederá de 2 ml/100 g de peso corporal. Deberá justificarse la administración de volúmenes superiores a este. Salvo en el caso de sustancias irritantes o corrosivas, que por lo general producen efectos exacerbados con concentraciones mayores, deberá reducirse al mínimo la variabilidad del volumen de ensayo ajustando la concentración de manera que el volumen sea el mismo en todas las dosis.
1.5.6. Preparación de los cromosomas
Immediatamente después del sacrificio, se preparan suspensiones con células de uno o ambos testículos, se tratan con una solución hipotónica y se fijan. A continuación, se distribuyen las células en portaobjetos y se tiñen.
1.5.7. Análisis
Deben analizarse al menos 100 metafases bien extendidas de cada animal (es decir, un mínimo de 500 meta-fases por lote). La cantidad puede ser menor si se observan numerosas aberraciones. Antes de analizarlos al microscopio, se asigna un código independiente a cada portaobjetos, incluidos los controles positivos y negativos. Dado que los métodos de fijación provocan con frecuencia la rotura de células en metafase y la pérdida de cromosomas, las células analizadas deben contener un número de centrómeros iqual a 2n ± 2.
2. RESULTADOS
2.1. TRATAMIENTO DE LOS RESULTADOS
Los datos relativos a cada animal se presentarán en forma de tabla. La unidad experimental es el animal. Se determinará en cada animal la cantidad de células con aberraciones cromosómicas estructurales y el número de aberraciones cromosómicas por célula. Se hará una relación de los distintos tipos, el número y la frecuencia de dichas aberraciones en los lotes tratados y los controles. Los gaps se registrarán por separado y se recogerán en el informe, pero por lo general no se incluirán en la frecuencia total de aberraciones.
Si se observan tanto mitosis como meiosis, se determinará la relación entre las mitosis de espermatogonias y la primera y segunda metafases meióticas en una muestra de 100 células en división por cada animal de los lotes tratados y de los controles negativos, con el fin de establecer si ha habido citotoxicidad. Si se observan mitosis únicamente, se determinará el índice mitótico al menos en 1 000 células por animal.
2.2. EVALUACIÓN E INTERPRETACIÓN DE LOS RESULTADOS
Son varios los criterios para determinar si un resultado es positivo (aumento de la proporción de células con aberraciones cromosómicas relacionado con la dosis, claro aumento del número de células que presentan aberraciones en un lote al que se ha administrado una sola dosis y en el que se ha realizado una única toma de muestras, etc.). Debe considerarse en primer lugar la importancia biológica de los resultados. Pueden emplearse métodos estadísticos como apoyo para evaluar los resultados del ensayo (8). El hecho de que los datos estadísticos sean significativos no ha de ser el único factor para determinar que una respuesta es positiva. Los resultados dudosos deberán aclararse mediante más ensayos en los que convendría modificar las condiciones experimentales.
No se considerarán mutágenas en el ensayo en cuestión las sustancias que den lugar a resultados que no se ajusten a los criterios anteriores.
Si bien en la mayoría de los experimentos los resultados serán claramente positivos o negativos, en algunos casos el conjunto de datos no permitirá emitir un juicio definitivo sobre la actividad de la sustancia de ensayo. Con independencia del número de veces que se repita el experimento, los resultados pueden seguir siendo ambiguos o dudosos.
Un resultado positivo en el ensayo de aberraciones cromosómicas en espermatogonias in vivo indica que la sustancia de ensayo provoca aberraciones cromosómicas estructurales en las células germinales de la especie estudiada. Un resultado negativo indica que, en las condiciones del ensayo, la sustancia de ensayo no provoca aberraciones cromosómicas en las células germinales de la especie estudiada.
Deberá estudiarse la probabilidad de que la sustancia de ensayo o sus metabolitos lleguen al tejido diana.
3. INFORME
INFORME DEL ENSAYO
El informe del ensayo incluirá la información siguiente:
|
|
Disolvente o vehículo:
|
|
|
Animales de ensayo:
|
|
|
Condiciones del ensayo:
|
|
|
Resultados:
|
|
|
Discusión de los resultados. |
|
|
Conclusiones. |
4. REFERENCIAS
|
(1) |
Adler, I. D. (1986), Clastogenic Potential in Mouse Spermatogonia of Chemical Mutagens Related to their Cell-Cycle Specifkations, in: Genetic Toxicology of Environmental Chemicals, Part B: Genetic Effects and Applied Mutagenesis, Ramel, C., Lambert B., and Magnusson, J. (eds.) Liss, New York, pp. 477-484. |
|
(2) |
Adler, I. D. (1984). Cytogenic tests in Mammals, in: Mutagenicity Testing: a Practical Approach, ed. S. Venitt and J. M. Parry, IRL Press, Oxford, Washington DC, pp. 27 5-306. |
|
(3) |
Evans, E. P., Breckon, G. and Ford, C. E. (1964), An Air-Drying Method for Meiotic Preparations from Mammalian Testes, Cytogenetics and Cell Genetics, 3, pp. 289-294. |
|
(4) |
Richold, M., Ashby, J., Chandley, A., Gatehouse, D., G., and Henderson L. (1990), ln Vivo Cytogenetic Assays, in: D. J. Kirkland (ed.), Basic Mutagenicity Tests, UKEMS Recommended Procedures. UKEMS Subcommittee on Guidelines for Mutagenicity Testing. Report. Part I revised, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 115-141. |
|
(5) |
Yamamoto, K. and Kikuchi, Y. (1978), A New Method for Preparation of Mammalian Spermatogonial Chromosomes, Mutation Res., 52, pp. 207-209. |
|
(6) |
Adler, I. D., Shelby, M. D., Bootman, J., Favor, J., Generoso, W., Pacchierotti, F., Shibuya, T. and Tanaka, N. (1994), International Workshop on Standardisation of Genotoxicity Test Procedures. Summary Report of the Working Group on Mammalian Germ Cell Tests, Mutation Res., 312, pp. 313-318. |
|
(7) |
Fielder, R. J., Allen, J. A., Boobis, A. R., Botham, P. A., Doe, J., Esdaile, D. J., Gatehouse, D. G., Hodson-Walker, G., Morton, D. B., Kirkland D. J. and Richold, M. (1992), Report of British Toxicology Society/UK Environmental Mutagen Society Working group: Dose setting in ln Vivo Mutagenicity Assays, Mutagenesis, 7, pp. 313-319. |
|
(8) |
Lovell, D. P., Anderson, D., Albanese, R., Amphlett, G. E., Clare, G., Ferguson, R., Richold, M., Papworth, D. G. and Savage, J. R. K. (1989), Statistical Analysis of ln Vivo Cytogenetic Assays, in: D. J. Kirkland (ed.), Slatistical Evaluation of Mutagenicity Test Data. UKEMS Sub-Committee on Guidelines for Mutagenicity Testing, report, Part III, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 184-232.» |
B.24. ENSAYO DE LA MANCHA EN EL RATÓN
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIÓN
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
Se trata de una prueba que se realiza in vivo en ratones, cuyos embriones en desarrollo se exponen a la sustancia estudiada. Las células efectoras de los embriones en desarrollo son los melanoblastos, y los genes efectores son los responsables de la pigmentación del pelo. Los embriones son heterocigotos para cierto número de estos genes. Una mutación en el alelo dominante de tal gen o su pérdida (a consecuencia de fenómenos genéticos diversos) en un melanoblasto provoca la expresión del fenotipo recesivo en sus células descendientes, con la aparición consiguiente de una mancha de otro color en el pelo del ratón resultante. Se cuenta el número de descendientes portadores de manchas (mutaciones), y se compara su frecuencia con la observada en la descendencia procedente del desarrollo de embriones tratados únicamente con el disolvente. La prueba de la mancha en el ratón descubre supuestas mutaciones somáticas en las células fetales.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
Preparativos
Cuando sea posible, las sustancias se disuelven u suspenden en solución salina isotónica. Las que sean insolubles en agua se disolverán o suspenderán en vehículos apropiados. El vehículo utilizado no debe alterar la sustancia estudiada ni provocar efectos tóxicos. Deben emplearse preparaciones recientes de la sustancia objeto de estudio.
Animales de experimentación
Se aparean animales de la cepa T (nonagouti, a/a; chinchilla, pink eye, cchp/cchp; brown, b/b; dilute, short ear, d se/d se; piebald spotting, s/s) con la cepa HT (pallid, nonagouti, brachypody, pa a bp/pa a bp; leaden fuzzy, in fz/ln fz; pearlpe/pe) o con la C57 BL (nonagouti, a/a). Pueden utilizarse otros cruces apropiados, como entre NMRI (nonagouti, a/a; albino, c/c) y DBA (nonagouti, a/a; brown, b/b; dilute d/d), siempre que produzcan descendencia nonagouti.
Número y sexo
Se tratan hembras grávidas suficientes para obtener un número de descendientes vivos apropiado para cada nivel posológico utilizado. El tamaño de la muestra dependerá del número de manchas observadas en los ratones tratados y de la importancia del número de datos de control. Solo se considerará aceptable un resultado negativo si se han examinado al menos 300 descendientes de hembras tratadas con la dosis máxima.
Uso de controles negativos y positivos
Ha de disponerse de datos de control simultáneos procedentes de ratones tratados solo con el vehículo (controles negativos). Pueden agruparse datos de control históricos obtenidos en el mismo laboratorio para mejorar la sensibilidad de la prueba, siempre que tales datos sean homogéneos. Si la sustancia analizada no resulta ser mutágena, deberá disponerse de datos de control positivos obtenidos en fecha reciente en el mismo laboratorio con una sustancia de potencial mutágeno conocido en esta prueba.
Vía de administración
Las vías de administración usuales son la intubación oral y la inyección intraperitoneal de las hembras grávidas. Cuando convenga, se recurrirá al tratamiento por inhalación u otras vías de administración.
Dosis
Se utilizan al menos dos niveles posológicos, incluido uno que origine signos de toxicidad o reduzca el tamaño de las camadas. En caso de sustancias atóxicas, la exposición se efectuará a la dosis máxima practicable.
Procedimiento
Normalmente, se administra un tratamiento único los días 8,9 o 10 de la gestación, considerando como día 1 aquel en que se observe por primera vez el tapón vaginal. Estos días corresponden, a 7,25, 8,25 y 9,25 días tras la concepción. Pueden efectuarse, pasados estos días, tratamientos sucesivos.
Análisis
Al cabo de tres o cuatro semanas del nacimiento, se asignan códigos a la descendencia y se comprueba si existen manchas. Se distinguen tres clases de manchas:
|
a) |
las manchas blancas a menos de 5 mm de la línea mesoventral, que se suponen consecuencia de muerte celular (WMVS), |
|
b) |
las manchas amarillas, de tipo agouti, asociadas con los pezones, los órganos sexuales, la garganta, las regiones axilar e inguinal y la parte central de la frente, que se suponen debidas a falta de diferenciación (MDS), y |
|
c) |
las manchas pigmentadas y blancas repartidas al azar por el pelaje, que se creen resultado de mutaciones somáticas (RS). |
Se cuentan las tres clases, pero solo la última, la RS, tiene significación genética. Los problemas que plantea la diferenciación de las clases MDS y RS pueden solucionarse examinando muestras de pelo con el microscopio de fluorescencia.
Se observarán las anomalías morfológicas macroscópicas evidentes de la descendencia.
2. RESULTADOS
Los datos se presentarán en forma del número total de crías examinadas y del número de las que presenten una o varias manchas supuestamente debidas a mutación somática. Se compararán por métodos apropiados los datos de los animales tratados y los controles negativos. Asimismo, se ofrecerán los resultados considerando la camada como unidad.
3. INFORME
3.1. DATOS DEL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
cepas utilizadas en el cruce, |
|
— |
número de hembras grávidas en los grupos experimental y de control, |
|
— |
tamaño medio de las camadas en los grupos experimentales y de control en el nacimiento y al destete, |
|
— |
dosis de la sustancia estudiada, |
|
— |
disolvente utilizado, |
|
— |
día de la gestación en que se administró el tratamiento, |
|
— |
vía de administración, |
|
— |
número total de descendientes examinados, y número con WMVS, MDS y RS en los grupos experimental y de control, |
|
— |
anomalías morfológicas macroscópicas, |
|
— |
relación dosis/respuesta de RS, si es posible, |
|
— |
evaluación estadística, |
|
— |
comentario de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.25. TRANSLOCACIÓN HEREDITARIA EN EL RATÓN
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIÓN
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
La prueba de translocación hereditaria en el ratón descubre alteraciones cromosómicas estructurales y numéricas en las células embrionarias de mamíferos, tal como se ponen de manifiesto en la descendencia de primera generación. Los tipos de alteraciones cromosómicas apreciados son translocaciones recíprocas y, si se incluye la descendencia femenina, pérdida del cromosoma X. Los portadores de translocaciones y las hembras XO muestran una fertilidad reducida que permite la selección de una descendencia F1 para practicar un análisis citogenético. Una esterilidad total es consecuencia de tipos de translocación determinados (autosoma X y tipo c-t). Las translocaciones se observan citogenéticamente en las células meióticas en diacinesis-metafase I de los individuos machos, ya sean machos F1 o hijos de hembras F1. Las hembras XO se identifican citogenéticamente por la presencia de solo 39 cromosomas en las mitosis de la médula ósea.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
Preparativos
Las sustancias objeto de estudio se disuelven en solución salina isotónica. Si son insolubles en agua, se disuelven o suspenden en vehículos apropiados. Se emplean soluciones recién preparadas de la sustancia estudiada. Si se utiliza un vehículo para facilitar la administración, no debe alterar el compuesto analizado ni provocar efectos tóxicos.
Vía de administración
Las vías de administración usuales son la intubación oral y la inyección intraperitoneal, aunque pueden ser apropiadas otras vías.
Animales de experimentación
Para facilitar la reproducción y la verificación citológica, estos experimentos se realizan con ratones; no es preciso que sean de una cepa determinada. Sin embargo, el tamaño medio de una camada de la cepa empleada deberá ser superior a 8, y relativamente constante.
Se utilizarán animales sanos y sexualmente maduros.
Número de animales
El número de animales necesario depende de la frecuencia de translocaciones espontáneas, así como del índice mínimo de inducción preciso para obtener un resultado positivo.
La prueba suele realizarse mediante análisis de la descendencia masculina F1. Al menos 500 descendientes machos F1 se analizan por grupo de dosis. Si se incluye la descendencia femenina F1 son necesarios 300 machos y 300 hembras.
Uso de controles negativos y positivos
Debe disponerse de datos sobre controles adecuados procedentes de pruebas realizadas simultáneamente y de controles históricos. Si existen resultados aceptables de controles positivos procedentes de experimentos efectuados en fecha reciente en el mismo laboratorio, pueden utilizarse en lugar de un control positivo simultáneo.
Dosis
Se prueba una dosis, por lo general la máxima asociada con la producción de efectos tóxicos mínimos pero que no altere el comportamiento reproductor ni la supervivencia. Para establecer una relación dosis/respuesta, son necesarias otras dos dosis. En caso de sustancias atóxicas, la exposición se efectuará a la dosis máxima posible.
Procedimiento
Tratamiento y apareamiento
Existen dos programas de tratamiento. De ellos, el más utilizado es la administración única de la sustancia estudiada. La sustancia también puede administrarse los 7 días de la semana durante 35 días. El número de apareamientos tras el tratamiento dependerá del programa de administración, y garantizará la obtención de muestras de todos los estadios de las células embrionarias tratadas. Al final del período de apareamiento, se coloca a las hembras en jaulas individuales. Cuando dan a luz, se registra la fecha, el tamaño de la camada y el sexo de los descendientes. Se desteta a toda la descendencia masculina y se descarta toda la femenina, salvo si se incluyen en el experimento.
Búsqueda de los heterocigotos de translocación
Se utiliza uno de los dos métodos posibles:
|
— |
estudio de la fertilidad de la descendencia F1, y verificación posterior de posibles portadores de translocaciones por análisis citogenéticos, |
|
— |
análisis citogenético de toda la progenie F1 masculina sin selección previa mediante estudio de fertilidad. |
|
a) |
Estudio de la fertilidad Es posible determinar la fertilidad reducida de un individuo F1 observando el tamaño de la camada, analizando el contenido uterino de su compañera femenina o por ambos medios. Es necesario fijar los criterios de determinación de la fertilidad normal y reducida de la cepa de ratón utilizada. Observación del tamaño de la camada: Los machos F1 sometidos a estudio se colocan en jaulas individuales con hembras procedentes del mismo experimento o de la colonia. Las jaulas se inspeccionan a diario a partir de los 18 días tras el apareamiento. Se registran, en el momento del nacimiento, el tamaño de la camada y el sexo de la descendencia F2, y se descartan luego las crías. Si se estudia la descendencia femenina F1, se conserva la descendencia F2 procedente de camadas pequeñas para un análisis en mayor profundidad. Las hembras portadoras de una translocación se someten a verificación por análisis citogenético de una translocación en cualquiera de sus descendientes machos. Las hembras XO se identifican por la modificación de sexos en su descendencia, que pasa de 1:1 a 1:2 machos/hembras. En un método secuencial, los machos F1 normales no se someten a nueva verificación si la primera camada F2 alcanza o supera un valor normal predeterminado; de lo contrario, se observan una segunda y una tercera camadas F2. Los animales F1 que no puedan clasificarse como normales tras observación de hasta un máximo de tres camadas F2 se someten a un nuevo control mediante análisis del contenido uterino de sus compañeras femeninas, o sufren directamente análisis citogenético. Análisis del contenido uterino: La reducción del tamaño de las camadas en los portadores de translocaciones se debe a la muerte de embriones, por lo que un número alto de implantes muertos indica la presencia de una translocación en el animal sometido a la prueba. Cada macho F1 estudiado se aparea con 2 o 3 hembras. Se comprueba si existe concepción mediante el examen matinal diario para descubrir la presencia de tapones vaginales. Las hembras se sacrifican 14 o 16 días después, y se registra el número de implantes vivos y muertos presentes en su útero. |
|
b) |
Análisis citogenético Se obtienen preparaciones de testículos por el método de secado con aire. Los portadores de translocaciones se identifican por la presencia de configuraciones multivalentes en diacinesis metafase I en los espermatocitos primarios. La observación de al menos dos células con una asociación multivalente constituye la prueba necesaria de que el animal estudiado es portador de una translocación. Si no se ha realizado selección mediante análisis de la fecundidad, se practica estudio citogenético en todos los machos F1. Deben analizarse al microscopio un mínimo de 25 células en diacinesis/metafase I por macho. En los machos F1 con testículos pequeños y que presenten una detención meiótica antes de la diacinesis, o en las hembras F1 que se sospeche sean XO, es necesario el examen de las metafases mitóticas en los espermatogonios o en la médula ósea. La presencia de un cromosoma desusadamente largo o corto en 10 células, demuestra la existencia de una translocación especial que origina la esterilidad del macho (tipo c-t). Algunas translocaciones X autosomas que provocan la esterilidad del macho solo pueden identificarse mediante un análisis de las bandas de cromosomas mitóticos. La presencia de 39 cromosomas en la totalidad de 10 micosis es demostrativa de un estado XO en una hembra. |
2. RESULTADOS
Los resultados se presentarán en forma de tablas.
Se registran, en el nacimiento y el destete para cada período de apareamiento, el tamaño medio de las camadas y la relación entre sexos.
En lo que respecta a la evaluación de la fertilidad de los animales F1, se expondrán el tamaño medio de las camadas procedentes de todos los apareamientos normales, así como el tamaño de cada una de las surgidas de animales F1 portadores de translocación. En cuanto al análisis del contenido uterino, se harán constar el número medio de implantes vivos y muertos producto de apareamientos normales, y el de implantes vivos y muertos de cada apareamiento de portadores de translocación.
Respecto al análisis citogenético de la diacinesis-metafase I, se enumerarán el número de tipos diferentes de configuraciones multivalentes y el número total de células en cada portador de translocación.
Se comunicarán el número total de apareamientos y la duración del período de apareamiento de los individuos estériles F1. Se facilitarán asimismo el peso de los testículos y detalles del análisis citogenético.
Para hembras XO, se dan resultados del tamaño medio de la camada, de la razón de sexos de la progenie F2 y del análisis citogenético.
Cuando se preseleccionan, por pruebas de fertilidad, posibles F1 portadores de translocación, las tablas deben incluir información relativa a cuántos de estos eran heterocigotos de translocación confirmados.
Se dan también datos de los experimentos de control negativo y control positivo.
3. INFORME
3.1. DATOS DE LA PRUEBA
El informe incluirá los datos siguientes:
|
— |
cepa de ratones, edad de los animales, pesos de los animales tratados, |
|
— |
número de animales reproductores de cada sexo en los grupos experimentales y de control, |
|
— |
condiciones de la prueba, descripción detallada del tratamiento, niveles de las dosis, disolventes, frecuencia de apareamientos, |
|
— |
número y sexo de los descendientes por cada hembra, número y sexo de los descendientes criados para análisis de translocación, |
|
— |
tiempos y criterios de análisis de translocación, |
|
— |
número y descripción detallada de los portadores de translocación, incluyendo datos de reproducción y de contenido uterino, cuando proceda, |
|
— |
procedimientos citogenéticos y detalles del análisis microscópico, preferiblemente con fotografías, |
|
— |
evaluación estadística, |
|
— |
discusión de resultados, |
|
— |
interpretación de resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.26. ENSAYO DE TOXICIDAD ORAL SUBCRÓNICA TOXICIDAD ORAL POR ADMINISTRACIÓN CONTINUADA (90 DÍAS) EN ROEDORES
1. MÉTODO
El presente método de ensayo de toxicidad oral subcrónica reproduce las directrices del documento OCDE TG 408 (1998).
1.1. INTRODUCCIÓN
Al determinar y evaluar las características tóxicas de una sustancia química, el estudio de la toxicidad oral subcrónica por administración continuada puede estar indicado cuando los ensayos de toxicidad aguda o por administración continuada durante 28 días hayan proporcionado información relativa a la toxicidad. El estudio de 90 días aporta información sobre los peligros que puede presentar para la salud una exposición continuada durante un período prolongado, que abarque la maduración posterior al destete y el crecimiento hasta la edad adulta. La información obtenida se refiere a los efectos tóxicos principales, los órganos diana y la posibilidad de acumulación, y puede proporcionar una estimación de la dosis de exposición sin efectos adversos observados, que puede emplearse para seleccionar las dosis de los estudios de toxicidad crónica y establecer los criterios de inocuidad de la exposición humana.
El presente método hace especial hincapié en los parámetros neurológicos y proporciona una indicación de los efectos sobre el sistema inmunitario y la reproducción. Cabe destacar la necesidad de realizar observaciones clínicas detenidas de los animales, a fin de obtener la máxima información posible. El método sirve para detectar sustancias químicas que pueden tener efectos tóxicos sobre el sistema nervioso, inmunitario o los órganos reproductores, lo cual puede justificar la realización de estudios más exhaustivos.
Véase también la introducción general de la Parte B.
1.2. DEFINICIONES
Dosis: cantidad de sustancia de ensayo administrada. Se expresa en peso (g, mg), en peso de sustancia de ensayo por unidad de peso del animal (por ejemplo, mg/kg) o en concentración constante en la dieta (ppm).
Posología: termino general que abarca la dosis administrada, su frecuencia y duración.
NOAEL: sigla inglesa referente a la dosis de exposición sin efectos adversos observados, es decir, la dosis más alta a la que no se observa ningún efecto adverso debido al tratamiento.
1.3. PRINCIPIO DEL MÉTODO DE ENSAYO
Se administra diariamente la sustancia de ensayo por vía oral en dosis graduadas a distintos lotes de animales de experimentación, a razón de una dosis por lote durante 90 días. A lo largo del período de administración, se observa atentamente a los animales por si aparecen signos de toxicidad. Se practica la autopsia a los animales que mueran o sean sacrificados durante el ensayo, así como a los que sobrevivan al final del mismo.
1.4. DESCRIPCIÓN DEL MÉTODO
1.4.1. Preparación de los animales
Deben emplearse animales sanos, que se hayan mantenido al menos 5 días en las condiciones de laboratorio para su aclimatación y que no hayan sido sometidos a experimentos previos. Se caracteriza la especie, cepa, procedencia, sexo, peso y/o edad de los animales de experimentación. Estos se reparten al azar entre los lotes tratados y los de control. Las jaulas se disponen de forma que se reduzcan al mínimo los posibles efectos debidos al enjaulamiento. Se asigna a cada animal un número de identificación distinto.
1.4.2. Preparación de las dosis
La sustancia de ensayo se administra por sonda o con el alimento o el agua de bebida. El método de administración oral depende del objetivo del estudio y de las propiedades fisicoquímicas de la sustancia.
En caso necesario, la sustancia de ensayo se disuelve o suspende en un vehículo adecuado. Se recomienda considerar en primer lugar, siempre que sea posible, el uso de una solución o suspensión acuosa, después el uso de una solución o emulsión oleosa (por ejemplo, en aceite de maíz) y, por último, la posible disolución en otros vehículos. Si se emplean vehículos distintos del agua, deben conocerse sus características toxicológicas. Debe determinarse la estabilidad de la sustancia de ensayo en las condiciones de administración.
1.4.3. Condiciones del ensayo
1.4.3.1. Animales de experimentación
La especie idónea es la rata, si bien pueden emplearse otras especies de roedores como el ratón. Hay que utilizar animales adultos jóvenes y sanos de una cepa de laboratorio habitual. Las hembras han de ser nulíparas y no grávidas. La administración ha de empezar lo antes posible tras el destete y, en cualquier caso, antes de que los animales tengan nueve semanas de edad. Al principio del experimento, la diferencia de peso entre los animales empleados ha de ser mínima y no superar el ± 20 % del peso medio de cada sexo. Cuando el ensayo se realice, como fase previa de un estudio de toxicidad crónica a largo plazo, deben utilizarse en ambos estudios, animales de la misma cepa y procedencia.
1.4.3.2. Número y sexo
Deben utilizarse por lo menos 20 animales (10 hembras y 10 machos) para cada dosis. Si se van a sacrificar animales durante el experimento, habrá que añadir el número de animales que se haya previsto sacrificar antes de acabar el estudio. Sobre la base de los conocimientos previos relativos a la sustancia química u otra sustancia muy próxima, puede tratarse un lote satélite de 10 animales (5 de cada sexo) en el control y con la dosis más elevada para observar la reversibilidad o persistencia de efectos tóxicos una vez finalizado el tratamiento. La duración del período de observación posterior al tratamiento ha de establecerse adecuadamente en función de los efectos observados.
1.4.3.3. Posología
Se emplean al menos tres dosis de ensayo y un lote de control, salvo si se lleva a cabo un ensayo límite (véase el punto 1.4.3.4). Para establecer las dosis pueden emplearse los resultados de los estudios de administración continuada o de determinación de dosis y debe tomarse en consideración toda la información toxicológica y toxicocinética disponible sobre la sustancia de ensayo o productos afines. A menos que las características fisicoquímicas o los efectos biológicos de la sustancia de ensayo impongan restricciones, la dosis superior debe seleccionarse con el propósito de inducir efectos tóxicos, pero sin llegar a provocar la muerte ni un sufrimiento intenso. Se selecciona una serie de dosis decrecientes para poner de manifiesto las respuestas en función de la dosis. La dosis mínima será la dosis sin efectos adversos observados (NOAEL). Los intervalos del doble al cuádruple suelen ser óptimos para establecer las dosis decrecientes y a menudo es preferible añadir un cuarto lote de ensayo en lugar de utilizar intervalos muy amplios (por ejemplo, con un factor superior a 6-10) entre dosis.
El lote de control no recibe la sustancia de ensayo, pero sí recibe el vehículo en caso de que este se utilice para la sustancia. A excepción de la administración de la sustancia de ensayo, los animales del lote de control deben tratarse de la misma manera que los de los lotes de ensayo. Si se emplea un vehículo, el lote de control ha de recibir el mayor volumen utilizado. Si la sustancia de ensayo se administra con los alimentos y provoca una disminución de la ingesta, puede ser útil utilizar un lote de control alimentado en paralelo para saber si la disminución se debe a las características organolépticas o a alteraciones toxicológicas del modelo de ensayo.
Debe prestarse atención a las siguientes características del vehículo u otros aditivos, según proceda: efectos sobre la absorción, distribución, metabolismo o retención de la sustancia de ensayo; efectos sobre las propiedades químicas de la sustancia de ensayo que puedan modificar su toxicidad y efectos sobre el consumo de alimentos y agua o sobre el estado nutricional de los animales.
1.4.3.4. Ensayo límite
Si en un ensayo con una sola dosis equivalente, al menos, a 1 000 mg/kg peso corporal/día y siguiendo el procedimiento descrito en el presente estudio, no se produce ningún efecto adverso observable y si, a la luz de los datos de sustancias estructuralmente afines, no cabe esperar efectos tóxicos, puede considerarse innecesario realizar un estudio completo con tres dosis. El ensayo límite es válido excepto cuando la exposición humana indique la necesidad de utilizar una dosis superior.
1.5. PROCEDIMIENTO
1.5.1. Administración de las dosis
Las dosis de ensayo se administran diariamente a los animales durante 90 días. Cualquier otra posología, por ejemplo, de 5 días por semana, debe justificarse. Si la sustancia de ensayo se administra por sonda, debe hacerse en una sola dosis y con una sonda gástrica o una cánula de intubación adecuada. El volumen máximo de líquido que puede administrarse de una sola vez depende del tamaño del animal y no debe superar 1 ml/100 g de peso corporal, salvo en el caso de las soluciones acuosas, en que puede llegarse a 2 ml/100 g de peso corporal. Excepto en el caso de sustancias irritantes o corrosivas que provoquen normalmente efectos exacerbados a concentraciones superiores, la variabilidad del volumen de ensayo debe reducirse al mínimo ajustando la concentración para que el volumen sea constante en todas las dosis.
En el caso de sustancias administradas con los alimentos o el agua de bebida, es importante cerciorarse de que las cantidades de sustancia de ensayo administradas no interfieren con la nutrición normal ni el equilibrio hídrico. Cuando la sustancia de ensayo se administre con los alimentos, puede utilizarse una concentración constante en la dicta (ppm) o bien una dosis constante en términos de peso corporal de los animales, pero debe indicarse qué método se ha elegido. Si la sustancia se administra por sonda, la dosis debe darse todos los días a la misma hora y ajustarse según sea necesario para mantener una dosis constante en términos de peso corporal del animal. Si se realiza un estudio de 90 días como fase previa de un estudio de toxicidad crónica a largo plazo, debe emplearse la misma dieta en ambos.
1.5.2. Observaciones
El período de observación debe durar al menos 90 días. Deben mantenerse animales en un lote satélite para las observaciones de seguimiento durante un período adecuado y sin tratamiento, a fin de detectar la persistencia o la desaparición de los efectos tóxicos.
Debe hacerse una observación clínica general al menos una vez al día, preferentemente a la misma hora y teniendo en cuenta el período más agudo de los efectos previstos tras la administración. Se registra el estado clínico de los animales. Se examinan todos los animales al menos dos veces al día, por lo general a primera y a última hora, para detectar signos de morbilidad y mortalidad.
Todos los animales deben someterse al menos a una observación clínica exhaustiva antes de la primera exposición (para poder realizar comparaciones en un mismo sujeto) y, después, a una por semana. Dichas observaciones han de efectuarse fuera de la jaula de alojamiento, de preferencia en un ambiente normal y siempre a la misma hora. Las observaciones se registran cuidadosamente, preferentemente mediante sistemas de puntuación definidos de forma explícita por el laboratorio de ensayo. Debe procurarse que las variaciones en las condiciones de observación sean mínimas. Los signos anotados deben incluir, sin ánimo de exhaustividad, los cambios de la piel, pelo, ojos, mucosas, presencia de secreciones y excreciones y actividad neurovegetativa (lagrimeo, piloerección, tamaño de la pupila, respiración anómala, etc.). Deben registrarse también los cambios en la marcha, postura y respuesta a la manipulación, así como la presencia de movimientos clónicos o tónicos, o estereotipados (por ejemplo, realización excesiva de movimientos de limpieza o recorridos circulares repetitivos) o comportamientos anómalos (automutilación, marcha hacia atrás, etc.) (1).
Debe realizarse una exploración oftalmológica con un oftalmoscopio o equipo similar adecuado antes de administrar la sustancia de ensayo y al término del estudio, a ser posible en todos los animales, pero al menos en el lote con la dosis más alta y en el lote de control. Si se observan cambios oculares en esos animales, deben examinarse todos los demás.
Hacia el final del período de exposición, pero en ningún caso antes de la undécima semana, debe evaluarse la reactividad sensorial frente a estímulos de distintos tipos (1) (por ejemplo, auditivos, visuales y propioceptivos) (2) (3) (4), la fuerza de prensión (5) y la actividad motriz (6). La bibliografía respectiva recoge más información sobre los métodos que pueden seguirse, si bien es posible emplear procedimientos distintos de los ahí descritos.
Las observaciones funcionales hacia el final del estudio pueden omitirse si se dispone de datos sobre dichas observaciones realizadas en otros estudios y las observaciones clínicas diarias no ponen de manifiesto alteraciones funcionales.
De forma excepcional, también pueden omitirse las observaciones funcionales en los lotes que muestren signos de toxicidad de otro tipo tales que interfieran significativamente con los resultados de las pruebas funcionales.
1.5.2.1. Peso corporal y consumo de alimentos y agua
Al menos una vez por semana deben pesarse todos los animales, medirse el consumo de alimentos y, si la sustancia de ensayo se administra con el agua de bebida, también el consumo de agua. Este último puede vigilarse, asimismo, en los estudios en que la sustancia de ensayo se administra con los alimentos o mediante sonda y la ingestión de agua pueda verse alterada.
1.5.2.2. Hematología y bioquímica clínica
Las muestras de sangre deben tomarse de un punto indicado y conservarse, si procede, en condiciones adecuadas. Al final del período de ensayo, las muestras se toman justo antes del sacrificio de los animales o como parte del método de sacrificio.
Deben practicarse los siguientes exámenes hematológicos al final del período de ensayo y con las muestras tomadas a lo largo del mismo: hematocrito, concentración de hemoglobina, recuento de eritrocitos, recuento de leucocitos y fórmula leucocitaria, recuento de plaquetas y medida del tiempo o capacidad de coagulación sanguínea.
Deben hacerse determinaciones bioquímicas para investigar efectos tóxicos importantes en los tejidos, y especialmente en el riñón y el hígado, con muestras sanguíneas obtenidas de todos los animales justo antes de su sacrificio o como parte del método de sacrificio (aparte de los moribundos o sacrificados a lo largo del ensayo). Al igual que los exámenes hematológicos, los análisis de bioquímica clínica pueden realizarse en muestras tomadas en el transcurso del ensayo. Se recomienda que los animales estén en ayunas desde el día anterior a la toma de muestras (3). Los parámetros medidos en el plasma y el suero deben incluir las concentraciones de sodio, potasio, glucosa, colesterol total, urea, nitrógeno residual en sangre, creatinina, proteínas totales y albúmina, y al menos dos enzimas indicadoras de los efectos hepatocelulares (alanina aminotrans-ferasa, aspartato aminotransferasa, fosfatasa alcalina, gamma-glutamil transpeptidasa o sorbitol deshidroge-nasa). La determinación de otras enzimas (de origen hepático o no) y de ácidos biliares puede proporcionar información útil en ciertas circunstancias.
Pueden realizarse, con carácter facultativo, las siguientes determinaciones en la última semana del estudio utilizando la recogida programada de orina: aspecto, volumen, osmolalidad o densidad, pH, proteínas, glucosa, sangre y células sanguíneas.
Además de ello, debe plantearse el estudio de los marcadores séricos de lesiones tisulares generales. Otras determinaciones que deben realizarse si se sabe o sospecha que las propiedades conocidas de la sustancia de ensayo pueden afectar a las funciones metabólicas correspondientes incluyen la concentración de calcio, fósforo, triglicéridos en ayunas, hormonas específicas, metahemoglobina y colinesterasa. Debe valorarse la necesidad de hacer estos análisis con las sustancias de determinadas clases o bien según cada caso.
Por lo general, debe aplicarse un enfoque flexible, en función de las especies y los efectos observados o esperados con una sustancia determinada.
Si los datos disponibles sobre antecedentes no son adecuados, debe plantearse la determinación de variables hematológicas y bioquímicas clínicas antes de iniciar la administración de la sustancia; por lo general, se desaconseja obtener dichos datos antes del tratamiento (7).
1.5.2.3. Autopsia macroscópica
Debe practicarse una autopsia macroscópica completa y detallada a todos los animales empleados en el estudio, que incluya un examen detenido de la superficie corporal externa, todos los orificios y las cavidades craneana, torácica y abdominal con su contenido. El hígado, los riñones, cápsulas suprarrenales, testículos, epidí-dimos, útero, ovarios, timo, bazo, cerebro y corazón de todos los animales (aparte de los moribundos y/o sacrificados a lo largo del ensayo) han de limpiarse de los tejidos adherentes, según convenga, y pesarse lo antes posible tras la disección para evitar su desecación.
Los tejidos que se enumeran a continuación deben conservarse en el medio de fijación más adecuado teniendo en cuenta tanto el tipo de tejido como el examen histopatológico a que vayan a someterse: todas las lesiones macroscópicas, encéfalo (zonas representativas, con inclusión del cerebro, cerebelo y protuberancia), médula espinal (cervical, torácica media y lumbar), hipófisis, tiroides, paratiroides, timo, esófago, glándulas salivares, estómago, intestino delgado y grueso (incluidas las placas de Peyer), hígado, páncreas, riñones, cápsulas suprarrenales, bazo, corazón, tráquea y pulmones (conservados mediante inflado con fijador, seguido de inmersión), aorta, gónadas, útero, órganos sexuales secundarios, glándula mamaria de las hembras, próstata, vejiga urinaria, vesícula biliar (ratón), ganglios linfáticos (preferentemente un ganglio relacionado con la vía de administración y otro distante de la misma para tener en cuenta los efectos sistémicos), nervios periféricos (ciático o tibial), preferentemente muy próximos al músculo, una sección de la médula ósea (y/o médula ósea aspirada y recién montada), piel y ojos (si se han observado cambios durante los exámenes oftalmológicos). Las observaciones clínicas y de otro tipo pueden indicar la necesidad de examinar otros tejidos. También deben conservarse todos los posibles órganos diana, según las propiedades conocidas de la sustancia de ensayo.
1.5.2.4. Examen histopatológico
Es preciso practicar un examen histopatológico completo de los órganos y tejidos conservados de todos los animales del lote expuesto a la dosis más elevada y del lote de control. En caso de que se hayan observado cambios relacionados con el tratamiento en el lote con la dosis más elevada, estos exámenes se ampliarán a animales de todos los demás lotes tratados.
Deben examinarse todas las lesiones macroscópicas.
Si se utiliza un lote satélite, debe hacerse un examen histopatológico de los órganos y tejidos que presenten efectos en los grupos tratados.
2. RESULTADOS E INFORME
2.1. RESULTADOS
Deben proporcionarse datos de cada animal. Además, deben resumirse todos los datos en un cuadro que recoja, para cada lote de ensayo, el número de animales al inicio del ensayo, el número de animales hallados muertos durante el mismo o sacrificados por razones compasivas, el momento de la muerte o sacrificio, el número de animales que presenten signos de toxicidad, una descripción de dichos signos (con inclusión del momento de su aparición, duración y gravedad), el número de animales que presenten lesiones, el tipo de lesiones y el porcentaje de animales afectado por cada tipo de lesión.
Siempre que sea posible, deben evaluarse los resultados numéricos mediante un método estadístico adecuado y comúnmente aceptado. La elección de los métodos estadísticos y de los datos que vayan a analizarse debe efectuarse en la fase de diseño del estudio.
2.2. INFORME DEL ENSAYO
El informe del ensayo debe incluir la información siguiente:
2.2.1. Sustancia de ensayo:
|
— |
naturaleza física, pureza y propiedades fisicoquímicas, |
|
— |
identificación química, |
|
— |
vehículo (si procede): justificación de la elección del vehículo, si es distinto del agua. |
2.2.2. Especie sometida a ensayo:
|
— |
especie y cepa empleada, |
|
— |
número, edad y sexo de los animales, |
|
— |
procedencia, condiciones de alojamiento, dieta, etc., |
|
— |
peso de cada animal al inicio del ensayo. |
2.2.3. Condiciones de ensayo:
|
— |
justificación de la elección de las dosis, |
|
— |
datos sobre la formulación de la sustancia de ensayo o su preparación con los alimentos, concentración obtenida, estabilidad y homogeneidad del preparado, |
|
— |
datos de la administración de la sustancia de ensayo, |
|
— |
dosis reales (mg/kg peso corporal/día) y factor de conversión de la concentración (ppm) de la sustancia de ensayo en los alimentos o en el agua de bebida a dosis reales, en su caso, |
|
— |
datos de la calidad de los alimentos y el agua. |
2.2.4. Resultados:
|
— |
peso corporal y cambios en el mismo, |
|
— |
consumo de alimentos y de agua, en su caso, |
|
— |
datos de reacciones tóxicas por sexo y dosis, incluidos los signos de toxicidad, |
|
— |
naturaleza, gravedad y duración de las observaciones clínicas (reversibles o no), |
|
— |
resultados del examen oftalmológico, |
|
— |
evaluación de la actividad sensorial, fuerza de prensión y actividad motriz (si procede), |
|
— |
pruebas hematológicas con los correspondientes valores de referencia, |
|
— |
pruebas de bioquímica clínica con los correspondientes valores de referencia, |
|
— |
peso corporal y peso de los órganos en el momento del sacrificio, y relación peso órgano/peso corporal, |
|
— |
hallazgos de la autopsia, |
|
— |
descripción pormenorizada de todas las observaciones histopatológicas, |
|
— |
datos sobre la absorción, si los hay, |
|
— |
tratamiento estadístico de los resultados, si procede. |
Discusión de los resultados.
Conclusiones.
3. REFERENCIAS
|
(1) |
IPCS (1986). Principies and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document No. 60. |
|
(2) |
Tupper, D.E., Wallace. R.B. (1980). Utility of the Neurologic Examination in Rats. Acta Neurobiol. Exp., 40, 999-1003. |
|
(3) |
Gad, S.C. (1982). A Neuromuscular Screen for Use in Industrial Toxicology. J. Toxicol. Environ. Health, 9. 691-704. |
|
(4) |
Moser, V.C., Mc Daniel, K.M., Phillips, .M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol., 108, 267-283. |
|
(5) |
Meyer O.A., Tilson H.A., Byrd W.C., Riley M.T. (1979). A Method for the Routine Assessment of Foreand Hind-limb grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233-236. |
|
(6) |
Crofton K.M., Howard J.L., Moser V.C., Gill M.W., Reiter L.W., Tilson H.A., MacPhail R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol., 13, 599-609. |
|
(7) |
Weingand K., Brown G., Hall R. et al. (1996). ”Harmonisation of Animal Clinic Pathology Testing in Toxicity and Safety Studies”, Fundam. & Appl. Toxicol., 29, 198-201. |
B.27. ENSAYO DE TOXICIDAD ORAL SUBCRÓNICA TOXICIDAD ORAL POR ADMINISTRACIÓN CONTINUADA (90 DÍAS) EN NO ROEDORES
1. MÉTODO
El presente método de ensayo de toxicidad oral subcrónica reproduce las directrices del documento OCDE TG 409 (1998).
1.1. INTRODUCCIÓN
Al determinar y evaluar las características tóxicas de una sustancia química, el estudio de la toxicidad oral subcrónica por administración continuada puede estar indicado cuando los ensayos de toxicidad aguda o por administración continuada durante 28 días hayan proporcionado información relativa a la toxicidad. El estudio de 90 días aporta información sobre los peligros que puede presentar para la salud una exposición continuada durante un período de crecimiento rápido hasta el principio de la edad adulta. La información obtenida se refiere a los efectos tóxicos principales, los órganos diana y la posibilidad de acumulación, y puede proporcionar una estimación de la dosis de exposición sin efectos adversos observados, que puede emplearse para seleccionar las dosis de los estudios de toxicidad crónica y establecer los criterios de inocuidad de la exposición humana.
El presente ensayo pone de manifiesto los efectos adversos, en animales no roedores, de la exposición a sustancias químicas y está indicado solo en los casos siguientes:
|
— |
cuando los efectos observados en otros estudios indiquen la necesidad de aclarar y precisar algunos aspectos en una especie no roedora, o |
|
— |
cuando los estudios toxicocinéticos indiquen que los animales de experimentación idóneos han de pertenecer a una especie determinada no roedora, o |
|
— |
cuando haya otros motivos concretos para emplear una especie no roedora. |
Véase también la introducción general de la Parte B.
1.2. DEFINICIONES
Dosis: cantidad de sustancia de ensayo administrada. Se expresa en peso (g, mg), en peso de sustancia de ensayo por unidad de peso del animal (por ejemplo, mg/kg) o en concentración constante en la dieta (ppm).
Posología: término general que abarca la dosis administrada, su frecuencia y duración.
NOAEL: sigla inglesa referente a la dosis de exposición sin efectos adversos observados, es decir, la dosis más alta a la que no se observa ningún efecto adverso debido al tratamiento.
1.3. PRINCIPIO DEL MÉTODO DE ENSAYO
Se administra diariamente la sustancia de ensayo por vía oral en dosis graduadas a distintos lotes de animales de experimentación, a razón de una dosis por lote durante 90 días. A lo largo del período de administración, se observa atentamente a los animales por si aparecen signos de toxicidad. Se practica la autopsia a los animales que mueran o sean sacrificados durante el ensayo, así como a los que sobrevivan al final del mismo.
1.4. DESCRIPCIÓN DEL MÉTODO
1.4.1. Selección de la especie animal
Suele emplearse el perro, preferiblemente de una raza definida. Con frecuencia se utiliza el sabueso. También pueden emplearse otras especies como el cerdo o el cerdo enano. No se recomiendan los primates; su uso ha de justificarse. Deben emplearse animales jóvenes y sanos y, en el caso del perro, la administración de sustancia de ensayo debe comenzar preferiblemente a la edad de 4-6 meses, pero nunca después de los 9 meses. Si el ensayo se realiza como fase previa de un estudio de toxicidad crónica a largo plazo, deben utilizarse en ambos estudios animales de la misma especie y raza.
1.4.2. Preparación de los animales
Deben emplearse animales jóvenes y sanos, que se hayan mantenido en las condiciones de laboratorio para su aclimatación y que no hayan sido sometidos a experimentos previos. La duración de la aclimatación dependerá de la especie de ensayo seleccionada y de su procedencia: se recomienda un mínimo de 5 días para los perros y los cerdos criados con ese propósito en un animalario del laboratorio, y de dos semanas si proceden de una fuente exterior. Se caracteriza la especie, cepa, procedencia, sexo, peso y/o edad de los animales de experimentación. Estos se reparten al azar entre los lotes tratados y los de control. Las jaulas se ponen de forma que se reduzcan al mínimo los posibles efectos debidos al enjaulamiento. Se asigna a cada animal un número de identificación distinto.
1.4.3. Preparación de las dosis
La sustancia de ensayo se administra con el alimento o el agua de bebida, por sonda o en cápsulas. El método de administración oral depende del objetivo del estudio y de las propiedades fisicoquímicas de la sustancia.
En caso necesario, la sustancia de ensayo se disuelve o suspende en un vehículo adecuado. Se recomienda considerar en primer lugar, siempre que sea posible, el uso de una solución o suspensión acuosa, después el uso de una solución o emulsión oleosa (por ejemplo, en aceite de maíz) y, por último, la posible disolución en otros vehículos. Si se emplean vehículos distintos del agua, deben conocerse sus características toxicoló-gicas. Debe determinarse la estabilidad de la sustancia de ensayo en las condiciones de administración.
1.5. PROCEDIMIENTO
1.5.1. Número y sexo de los animales
Deben utilizarse por lo menos 8 animales (4 hembras y 4 machos) para cada dosis. Si se van a sacrificar animales durante el experimento, habrá que añadir el número de animales que se haya previsto sacrificar antes de acabar el estudio. El número de animales vivos al término del estudio debe permitir una evaluación significativa de los efectos tóxicos. Sobre la base de los conocimientos previos relativos a la sustancia química u otra sustancia muy próxima, puede tratarse un lote satélite de 8 animales (4 de cada sexo) en el control y con la dosis más elevada para observar la reversibilidad o persistencia de efectos tóxicos una vez finalizado el tratamiento. La duración del período de observación posterior al tratamiento ha de establecerse adecuadamente en función de los efectos observados.
1.5.2. Posología
Se emplean al menos tres dosis de ensayo y un lote de control, salvo si se lleva a cabo un ensayo límite (véase el punto 1.5.3). Para establecer las dosis pueden emplearse los resultados de los estudios de administración continuada o de determinación de dosis y debe tomarse en consideración toda la información toxi-cológica y toxicocinética disponible sobre la sustancia de ensayo o productos afines. A menos que las características fisicoquímicas o los efectos biológicos de la sustancia de ensayo impongan restricciones, la dosis superior debe seleccionarse con el propósito de inducir efectos tóxicos, pero sin llegar a provocar la muerte ni un sufrimiento intenso. Se selecciona una serie de dosis decrecientes para poner de manifiesto las respuestas en función de la dosis. La dosis mínima será la dosis sin efectos adversos observados (NOAEL). Los intervalos del doble al cuádruple suelen ser óptimos para establecer las dosis decrecientes y a menudo es preferible añadir un cuarto lote de ensayo en lugar de utilizar intervalos muy amplios (por ejemplo, con un factor superior a 6-10) entre dosis.
El lote de control no recibe la sustancia de ensayo, pero sí recibe el vehículo en caso de que este se utilice para la sustancia. A excepción de la administración de la sustancia de ensayo, los animales del lote de control deben tratarse de la misma manera que los de los lotes de ensayo. Si se emplea un vehículo, el lote de control ha de recibir el mayor volumen utilizado. Si la sustancia de ensayo se administra con los alimentos y provoca una disminución de la ingesta, puede ser útil utilizar un lote de control alimentado en paralelo para saber si la disminución se debe a las características organolépticas o a alteraciones toxicológicas del modelo de ensayo.
Debe prestarse atención a las siguientes características del vehículo u otros aditivos, según proceda: efectos sobre la absorción, distribución, metabolismo o retención de la sustancia de ensayo, efectos sobre las propiedades químicas de la sustancia de ensayo que puedan modificar su toxicidad y efectos sobre el consumo de alimentos y agua o sobre el estado nutricional de los animales.
1.5.3. Ensayo límite
Si en un ensayo con una sola dosis equivalente, al menos, a 1 000 mg/kg peso corporal/día y siguiendo el procedimiento descrito en el presente estudio, no se produce ningún efecto adverso observable y si, a la luz de los datos de sustancias estructuralmente afines, no cabe esperar efectos tóxicos, puede considerarse innecesario realizar un estudio completo con tres dosis. El ensayo límite es válido excepto cuando la exposición humana indique la necesidad de utilizar una dosis superior.
1.5.4. Administración de las dosis
Las dosis de ensayo se administran diariamente a los animales durante 90 días. Cualquier otra posología, por ejemplo, de 5 días por semana, debe justificarse. Si la sustancia de ensayo se administra por sonda, debe hacerse en una sola dosis y con una sonda gástrica o una cánula de intubación adecuada. El volumen máximo de líquido que puede administrarse de una sola vez depende del tamaño del animal. Por lo general, el volumen ha de ser el mínimo posible. Excepto en el caso de sustancias irritantes o corrosivas que provoquen normalmente efectos exacerbados a concentraciones superiores, la variabilidad del volumen de ensayo debe reducirse al mínimo ajustando la concentración para que el volumen sea constante en todas las dosis.
En el caso de sustancias administradas con los alimentos o el agua de bebida, es importante cerciorarse de que las cantidades de sustancia de ensayo administradas no interfieren con la nutrición normal ni el equilibrio hídrico. Cuando la sustancia de ensayo se administre con los alimentos, puede utilizarse una concentración constante en la dieta (ppm) o bien una dosis constante en términos de peso corporal de los animales, pero debe indicarse qué método se ha elegido. Si la sustancia se administra por sonda o en cápsulas, la dosis debe darse todos los días a la misma hora y ajustarse según sea necesario para mantener una dosis constante en términos de peso corporal del animal. Si se realiza un estudio de 90 días como fase previa de un estudio de toxicidad crónica a largo plazo, debe emplearse la misma dieta en ambos.
1.5.5. Observaciones
El período de observación debe durar al menos 90 días. Deben mantenerse animales en un lote satélite para las observaciones de seguimiento durante un período adecuado y sin tratamiento, a fin de detectar la persistencia o la desaparición de los efectos tóxicos.
Debe hacerse una observación clínica general al menos una vez al día, preferentemente a la misma hora y teniendo en cuenta el período más agudo de los efectos previstos tras la administración. Se registra el estado clínico de los animales. Se examinan todos los animales al menos dos veces al día, por lo general a primera y a última hora, para detectar signos de morbilidad y mortalidad.
Todos los animales deben someterse al menos a una observación clínica exhaustiva antes de la primera exposición (para poder realizar comparaciones en un mismo sujeto) y, después, a una por semana. Dichas observaciones han de efectuarse, si es posible, fuera de la jaula de alojamiento en un ambiente normal y de preferencia siempre a la misma hora. Debe procurarse que las variaciones en las condiciones de observación sean mínimas. Los signos de toxicidad han de anotarse cuidadosamente, así como el momento de aparición, la gravedad y la duración. Las observaciones deben incluir, sin ánimo de exhaustividad, los cambios de la piel, pelo, ojos, mucosas, presencia de secreciones y excreciones y actividad neurovegetativa (lagrimeo, piloerección, tamaño de la pupila, respiración anómala, etc.). Deben registrarse también los cambios en la marcha, postura y respuesta a la manipulación, así como la presencia de movimientos clónicos o tónicos, o estereotipados (por ejemplo, realización excesiva de movimientos de limpieza o recorridos circulares repetitivos) o comportamientos anómalos.
Debe realizarse una exploración oftalmológica con un oftalmoscopio o equipo similar adecuado antes de administrar la sustancia de ensayo y al término del estudio, a ser posible, de todos los animales, pero al menos en el lote con la dosis más alta y en el lote de control. Si se observan en esos animales cambios oculares relacionados con el tratamiento, deben examinarse todos los demás.
1.5.5.1. Peso corporal y consumo de alimentos y agua
Al menos una vez por semana deben pesarse todos los animales, medirse el consumo de alimentos y, si la sustancia de ensayo se administra con el agua de bebida, también el consumo de agua. Este último puede vigilarse, asimismo, en los estudios en que la sustancia de ensayo se administra con los alimentos o mediante sonda y la ingestión de agua pueda verse alterada.
1.5.5.2. Hematología y bioquímica clínica
Las muestras de sangre deben tomarse de un punto indicado y conservarse, si procede, en condiciones adecuadas. Al final del período de ensayo, las muestras se toman justo antes del sacrificio de los animales o como parte del método de sacrificio.
Al principio del ensayo y, a continuación, bien una vez al mes bien a la mitad del período de ensayo, así como al término del mismo, debe practicarse un examen hematológico) que incluya hematocrito, concentración de hemoglobina, recuento de eritrocitos, recuento de leucocitos y fórmula leucocitaria, recuento de plaquetas y medida de la capacidad de coagulación como el tiempo de coagulación, de protrombina o de trom-boplastina.
Deben hacerse determinaciones bioquímicas para investigar efectos tóxicos importantes en los tejidos, y especialmente en el riñón y el hígado, con muestras sanguíneas obtenidas de todos los animales al principio del ensayo y, a continuación, bien una vez al mes bien a la mitad del período de ensayo, así como al término del mismo. Debe estudiarse el equilibrio electrolítico, el metabolismo glucídico y las funciones hepática y renal. La elección de determinados análisis dependerá de las observaciones sobre el modo de actuación de la sustancia de ensayo. Los animales deben estar en ayunas antes de las tomas de sangre, durante un período adecuado según la especie. Se propone determinar el calcio, fósforo, cloro, sodio, potasio, glucosa en ayunas, alanina aminotransferasa, aspartato aminotransferasa, ornitina descarboxilasa, gamma-gluta-mil transpeptidasa, nitrógeno residual, albúmina, creatinina en sangre, bilirrubina total y proteínas séricas totales.
Deben practicarse análisis de orina, como mínimo, al inicio, a la mitad y al final del ensayo, utilizando la recogida programada, con el fin de estudiar los parámetros siguientes: aspecto, volumen, osmolalidad o densidad, pH, proteínas, glucosa, sangre y células sanguíneas. Si es necesario, pueden analizarse otros parámetros adicionales para profundizar el estudio de los efectos observados.
Además de ello, debe plantearse el estudio de los marcadores de lesiones tisulares generales. Para realizar una evaluación toxicológica adecuada pueden ser necesarias otras determinaciones como el análisis de los lípidos, hormonas, equilibrio ácido-básico, metahemoglobina e inhibición de la colinesterasa. Si es preciso, pueden realizarse análisis de bioquímica clínica adicionales para profundizar el estudio de los efectos observados. Debe valorarse la necesidad de hacer estos análisis con las sustancias de determinadas clases o bien según cada caso.
Por lo general, debe aplicarse un enfoque flexible, en función de las especies y los efectos observados o esperados con una sustancia determinada.
1.5.5.3. Autopsia macroscópica
Debe practicarse una autopsia macroscópica completa y detallada a todos los animales empleados en el estudio, que incluya un examen detenido de la superficie corporal externa, todos los orificios y las cavidades craneana, torácica y abdominal con su contenido. El hígado y la vesícula biliar, los riñones, cápsulas suprarrenales, testículos, epidídimos, ovarios, útero, glándulas tiroides y paratiroides, timo, bazo, cerebro y corazón de todos los animales (aparte de los moribundos y/o sacrificados a lo largo del ensayo) han de limpiarse de los tejidos adherentes, según convenga, y pesarse lo antes posible tras la disección para evitar su desecación.
Los tejidos que se enumeran a continuación deben conservarse en el medio de fijación más adecuado teniendo en cuenta tanto el tipo de tejido como el examen histopatológico a que vayan a someterse: todas las lesiones macroscópicas, encéfalo (zonas representativas, con inclusión del cerebro, cerebelo y protuberancia), médula espinal (cervical, torácica media y lumbar), hipófisis, ojos, tiroides, paratiroides, timo, esófago, glándulas salivares, estómago, intestino delgado y grueso (incluidas las placas de Peyer), hígado, vesícula biliar, páncreas, riñones, cápsulas suprarrenales, bazo, corazón, tráquea y pulmones, aorta, gónadas, útero, órganos sexuales secundarios, glándula mamaria de las hembras, próstata, vejiga urinaria, ganglios linfáticos (de preferencia un ganglio relacionado con la vía de administración y otro distante de la misma para tener en cuenta los efectos sistémicos), nervios periféricos (ciático o tibial), preferentemente muy próximos al músculo, una sección de la médula ósea (y/o médula ósea aspirada y recién montada) y piel. Las observaciones clínicas y de otro tipo pueden indicar la necesidad de examinar otros tejidos. También deben conservarse todos los posibles órganos diana, según las propiedades conocidas de la sustancia de ensayo.
1.5.5.4. Examen histopatológico
Es preciso practicar un examen histopatológico completo de los órganos y tejidos conservados, al menos, de todos los animales del lote expuesto a la dosis más elevada y del lote de control. En caso de que se hayan observado cambios relacionados con el tratamiento en el lote con la dosis más elevada, este examen se ampliará a animales de todos los demás lotes tratados.
Deben examinarse todas las lesiones macroscópicas.
Si se utiliza un lote satélite, debe hacerse un examen histopatológico de los órganos y tejidos que presenten efectos en los grupos tratados.
2. RESULTADOS E INFORME
2.1. RESULTADOS
Deben proporcionarse datos de cada animal. Además, deben resumirse todos los datos en un cuadro que recoja, para cada lote de ensayo, el número de animales al inicio del ensayo, el número de animales hallados muertos durante el mismo o sacrificados por razones compasivas, el momento de la muerte o sacrificio, el número de animales que presenten signos de toxicidad, una descripción de dichos signos (con inclusión del momento de su aparición, duración y gravedad), el número de animales que presenten lesiones, el tipo de lesiones y el porcentaje de animales afectado por cada tipo de lesión.
Siempre que sea posible, deben evaluarse los resultados numéricos mediante un método estadístico adecuado y comúnmente aceptado. La elección de los métodos estadísticos y de los datos que vayan a analizarse debe efectuarse en la fase de diseño del estudio.
2.2. INFORME DEL ENSAYO
El informe del ensayo debe incluir la información siguiente:
2.2.1. Sustancia de ensayo:
|
— |
naturaleza física, pureza y propiedades fisicoquímicas, |
|
— |
identificación química, |
|
— |
vehículo (si procede): justificación de la elección del vehículo, si es distinto del agua. |
2.2.2. Especie sometida a ensayo:
|
— |
especie y cepa empleada, |
|
— |
número, edad y sexo de los animales, |
|
— |
procedencia, condiciones de alojamiento, dieta, etc., |
|
— |
peso de cada animal al inicio del ensayo. |
2.2.3. Condiciones de ensayo:
|
— |
justificación de la elección de las dosis, |
|
— |
datos sobre la formulación de la sustancia de ensayo o su preparación con los alimentos, concentración obtenida, estabilidad y homogeneidad del preparado, |
|
— |
datos de la administración de la sustancia de ensayo, |
|
— |
dosis reales (mg/kg peso corporal/día) y factor de conversión de la concentración (ppm) de la sustancia de ensayo en los alimentos o en el agua de bebida a dosis reales, en su caso, |
|
— |
datos de la calidad de los alimentos y el agua. |
2.2.4. Resultados:
|
— |
peso corporal y cambios en el mismo, |
|
— |
consumo de alimentos y de agua, en su caso, |
|
— |
datos de reacciones tóxicas por sexo y dosis, incluidos los signos de toxicidad, |
|
— |
naturaleza, gravedad y duración de las observaciones clínicas (reversibles o no), |
|
— |
examen oftalmológico, |
|
— |
pruebas hematológicas con los correspondientes valores de referencia, |
|
— |
pruebas de bioquímica clínica con los correspondientes valores de referencia, |
|
— |
peso corporal y peso de los órganos en el momento del sacrificio, y relación peso órgano/peso corporal, |
|
— |
hallazgos de la autopsia, |
|
— |
descripción pormenorizada de todas las observaciones histopatológicas, |
|
— |
datos sobre la absorción, si los hay, |
|
— |
tratamiento estadístico de los resultados, si procede. |
Discusión de los resultados.
Conclusiones.
B.28. TOXICIDAD DÉRMICA SUBCRÓNICA ENSAYO DE 90 DÍAS EN ROEDORES
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIONES
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
Se aplican a diario dosis crecientes de la sustancia estudiada en la piel de animales de experimentación integrados en varios grupos, a razón de una dosis por grupo durante un período de 90 días. Durante el período de aplicación se observa diariamente a los animales en busca de signos de toxicidad. Los animales que mueren durante la prueba se someten a autopsia, que se practica también a los supervivientes al concluir el estudio.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
Preparativos
Se mantiene a los animales en las condiciones de alojamiento y alimentación experimentales durante al menos 5 días previos a la prueba. Antes de comenzarla, se distribuye al azar a los animales, que serán jóvenes y sanos, para formar grupos de tratamiento y de control. Antes de proceder a la aplicación, se elimina el pelo de la región dorsal del tronco de los animales. Puede emplearse afeitado, pero deberá practicarse aproximadamente 24 horas antes de la prueba. Suelen ser necesarios cortes o afeitados reiterados del pelo, más o menos a intervalos semanales. Al practicarlos, hay que cuidar de no lesionar la piel. Deberá dejarse despejada, para la aplicación de la sustancia en estudio, una superficie no inferior al 10 % de la corporal. Para decidir la zona que debe despejarse y las dimensiones de la superficie a tratar, se tendrá en cuenta el peso del animal. Cuando se prueben sólidos, que pueden pulverizarse si se considera oportuno, se humedecerá lo suficiente la sustancia estudiada con agua o, en caso necesario, un vehículo apropiado para garantizar un buen contacto con la piel. Las sustancias líquidas se emplean por lo general sin diluir. Se practican aplicaciones diarias 5 o 7 días a la semana.
Condiciones del ensayo
Animales de experimentación
Pueden emplearse ratas, conejos o cobayas adultos. Es posible emplear otras especies, pero su uso exigiría justificación. Al principio de la prueba, los pesos no diferirán en un porcentaje superior al ± 20 % del peso medio. Cuando se realice una prueba dérmica subcrónica como preliminar a una de índole crónica, se utilizará en ambas la misma especie y cepa.
Número y sexo
Se utilizarán al menos 20 animales (10 hembras y 10 machos) con piel sana para cada dosis. Las hembras serán nulíparas y no estarán preñadas. Si está previsto el sacrificio de algunos animales durante la prueba, debe aumentarse su número en una cantidad igual a la de los que vayan a sacrificarse antes de terminar el estudio. Además, puede tratarse a un grupo satélite de 20 animales (10 de cada sexo) con la dosis más alta durante 90 días para observar en ellos la reversibilidad, la persistencia o la aparición tardía de efectos tóxicos durante 28 días tras el tratamiento.
Dosis
Se utilizarán al menos tres niveles posológicos y un control, o un control para el vehículo si se emplea este. El período de exposición no será inferior a 6 horas diarias. La aplicación de la sustancia de prueba debe hacerse a las mismas horas todos los días, y la cantidad del producto se ajustará a intervalos (semanales o quincenales) de forma que se mantenga una dosis constante en relación con el peso del animal. Salvo por la omisión de la sustancia estudiada, los animales del grupo de control deben recibir un trato idéntico al de los integrantes del grupo de prueba. Cuando se utilice un vehículo para facilitar la administración, el grupo de control lo recibirá de igual modo que los grupos tratados, y en cantidad igual a la administrada al grupo con dosis más alta. La dosis más alta debe originar efectos tóxicos, pero producir pocos fallecimientos, o ninguno. La dosis más baja no debe originar signo alguno de toxicidad. Cuando se disponga de una estimación válida de la exposición humana, la dosis mínima será superior a ese valor. Lo ideal sería que la dosis media produjera el efecto tóxico mínimo observable. Si se emplea más de una dosis intermedia, la diferencia entre dosis será suficiente para que los efectos tóxicos resultantes sean escalonados. En los grupos con dosis bajas e intermedias, así como en los controles, la mortalidad debe ser baja, a fin de permitir una evaluación significativa de los resultados.
Si la aplicación de la sustancia probada provoca una irritación cutánea grave, se reducirán las concentraciones, lo que quizá origine una reducción, o la ausencia, de otros efectos tóxicos con la dosis alta. Si la piel ha sufrido lesiones importantes, quizá sea necesario dar por concluido el estudio y emprender uno nuevo a concentraciones menores.
Prueba de límite
Si una experiencia previa realizada con una dosis de 1 000 mg/kg, o superior, en función de la posibilidad de exposición humana (siempre que esta se conozca) no ha provocado efecto tóxico alguno, cabe considerar inútil la continuación de la experiencia.
Período de observación
Es preciso observar a diario a los animales de experimentación en busca de signos de toxicidad. Hay que tomar nota del momento del fallecimiento y del de aparición y desaparición de signos de toxicidad.
Procedimiento
Los animales se alojarán en jaulas individuales. El régimen ideal comprende la administración, a los animales, de la sustancia ensayada los 7 días de la semana durante un período de 90 días.
Los animales de grupos satélites destinados a observaciones complementarias deben observarse 28 días más sin tratamiento para apreciar la recuperación de los efectos tóxicos, o su persistencia. El período de exposición diario será de 6 horas.
La sustancia estudiada se aplicará uniformemente sobre una zona que corresponda aproximadamente al 10 % de la superficie corporal total. En caso de sustancias muy tóxicas, la zona tratada puede ser inferior, pero se cubrirá con una capa lo más uniforme y delgada posible.
Durante la exposición, la sustancia estudiada se mantiene en contacto con la piel con ayuda de un apósito de gasa poroso y esparadrapo no irritante. Además, la zona tratada se cubrirá de forma que se mantengan colocados el apósito y la sustancia y se impida a los animales la ingestión de esta. Cabe utilizar dispositivos de sujeción para evitar la ingestión de la sustancia objeto de estudio, pero no se recomienda la inmovilización completa.
Al final del período de exposición, se eliminará la sustancia restante, siempre que sea posible, por medio de agua o algún otro método apropiado de limpieza de la piel.
Es preciso observar a diario a todos los animales y registrar los signos de toxicidad, incluidos el momento de comienzo, el grado y la duración. Entre las observaciones que deben practicarse sobre los animales enjaulados destacan las de modificaciones de la piel y el pelo, los ojos y las membranas mucosas, así como de los sistemas respiratorio, circulatorio, autonómico y nervioso central, la actividad somatomotriz y el comportamiento. Cada semana, se determinarán el consumo de alimento y el peso de los animales. Son necesarias observaciones periódicas de los animales para evitar en lo posible la pérdida de alguno por causa de canibalismo, autólisis de tejidos o enjaulamiento erróneo. Al final del período de prueba, se realiza la autopsia a todos los supervivientes de los grupos de tratamiento no satélites. Los animales moribundos se retirarán y someterán a autopsia.
En todos los animales, incluidos los controles, se practican habitualmente los exámenes siguientes:
|
a) |
Exploración oftalmológica con un oftalmoscopio o equipo apropiado equivalente, antes de administrar la sustancia estudiada y al final del estudio, preferiblemente en todos los animales, pero por lo menos en los grupos con dosis altas y de control. Si se apreciaran alteraciones oculares, se examinará a todos los animales. |
|
b) |
Pruebas hematológicas al final del período de estudio: hematocrito, concentración de hemoglobina, recuento eritrocitario, recuento y fórmula leucocitarios, y determinación de la coagulación por pruebas como tiempo de coagulación, tiempo de protrombina, tiempo de tromboplastina o recuento de plaquetas. |
|
c) |
Determinaciones bioquímicas clínicas en la sangre al final del período de prueba. Se consideran aspectos de interés en todos los estudios el balance electrolítico, el metabolismo de hidratos de carbono y las funciones hepáticas y renal. En la selección de las pruebas concretas influirán las observaciones sobre el modo de acción de la sustancia. Se sugiere la determinación de los parámetros siguientes: calcio, fósforo, cloruro, sodio, potasio, glucosa en ayunas (con período de ayuno apropiado a cada especie), transaminasas glutamicopirúvica (4) y gluamicooxaloacética (5) séricas, ornitindescarboxilasa, gammaglutamiltranspeptidasa, nitrógeno ureico, albúmina, creatinina plasmática, bilirrubina total y proteínas séricas totales. Entre otras determinaciones que pueden ser necesarias para una evaluación toxicológica adecuada figuran los análisis de lípidos, hormonas, equilibrio acidobásico, metahemoglobina y actividad de colinesterasa. Siempre que sea preciso, se emplearán otras pruebas bioquímicas clínicas para ampliar la investigación de los efectos observados. |
|
d) |
El análisis de orina no es necesario de forma sistemática, sino solo en caso de efectos tóxicos probables o manifiestos. |
Si los datos de referencia previos son insuficientes, habrá que considerar la posible determinación de los parámetros hematológicos y de bioquímica clínica antes de iniciar la administración.
Autopsia
Se practicará en todos los animales una autopsia completa que comprenda la inspección de la superficie externa del cuerpo, todos sus orificios y las cavidades craneal, torácica y abdominal y sus contenidos. Hígado, riñones, suprarrenales y testículos deben pesarse frescos lo antes posible tras la disección, a fin de evitar la desecación. Se conservarán en un medio adecuado para un posible examen histopatológico posterior los órganos y tejidos siguientes: todas las lesiones macroscópicas, el cerebro —incluidos cortes de bulbo/protuberancia—, corteza cerebelosa y encefálica, hipófisis, tiroides/paratiroides, tejido tímico, (tráquea), pulmones, corazón, aorta, (glándulas salivales), hígado, bazo, riñones, suprarrenales, páncreas, gónadas, útero, órganos genitales accesorios, vesícula biliar (si existe), esófago, estómago, duodeno, yeyuno, íleon, ciego, colon, recto, vejiga urinaria, ganglio linfático representativo, (glándula mamaria femenina), (musculatura del muslo), nervio periférico, (ojos), (esternón con médula ósea), (fémur, incluida superficie articular), (médula espinal a tres niveles: cervical, mesotorácico y lumbar) y (glándulas lagrimales exorbitarias). Los tejidos que aparecen entre paréntesis solo se examinarán si así lo aconsejan los signos de toxicidad o afectación del órgano efector.
Examen histopatológico
|
a) |
Se someterán a un examen histopatológico completo la piel normal y la tratada, y los órganos y tejidos de los animales de los grupos de control y los de dosis máxima. |
|
b) |
Se examinarán todas las lesiones macroscópicas. |
|
c) |
Se examinarán los órganos efectores de los animales de grupos con otras dosis. |
|
d) |
Si se utilizan ratas, se someterán los pulmones de los animales integrantes de los grupos con dosis mínimas e intermedias a examen histopatológico en busca de signos de infección, ya que esta medida permite una evaluación cómoda del estado de salud de los animales. Es posible que en los animales de estos grupos no sean necesarios por sistema exámenes histopatológicos complementarios, pero siempre se llevarán a cabo en los órganos de los del grupo con dosis máxima que presenten indicios de lesiones. |
|
e) |
Cuando se utilice un grupo satélite, se practicará un examen histopatológico de los tejidos y órganos que presenten signos de toxicidad en los grupos tratados. |
2. RESULTADOS
Los resultados se resumirán en forma de tabla, y mostrarán para cada grupo de prueba el número de animales al comienzo del ensayo, el de los que presenten lesiones y el porcentaje de animales con cada tipo de lesión. Los resultados se evaluarán por un método estadístico apropiado. Puede usarse cualquier método estadístico reconocido.
3. INFORME
3.1. DATOS DEL ENSAYO
El informe sobre el ensayo incluirá si fuera posible:
|
— |
especie, cepa, origen, condiciones ambientales, alimentación, |
|
— |
condiciones de la prueba, |
|
— |
dosis (incluido el vehículo, si procede) y concentraciones, |
|
— |
datos de respuesta tóxica en función del sexo y la dosis, |
|
— |
dosis carente de efectos, si es posible, |
|
— |
momento de la muerte durante el estudio, o indicación de que los animales sobrevivieron a la experiencia, |
|
— |
descripción de los efectos tóxicos o de otro tipo, |
|
— |
momento de observación de cada signo anómalo y evolución de este, |
|
— |
datos sobre alimentación y peso, |
|
— |
hallazgos oftalmológicos, |
|
— |
pruebas hematológicas practicadas y sus resultados completos, |
|
— |
pruebas de bioquímica clínica empleadas y sus resultados completos (incluidos los del análisis de orina, si procede), |
|
— |
hallazgos de autopsia, |
|
— |
descripción detallada de los hallazgos histopatológicos, |
|
— |
tratamiento estadístico de los resultados, si es posible, |
|
— |
comentario de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.29. TOXICIDAD SUBCRÓNICA POR INHALACIÓN ENSAYO DE 90 DÍAS EN ROEDORES
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIONES
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
Se expone a diario, durante un período de tiempo determinado, a varios grupos de animales de experimentación, a concentraciones diferentes de la sustancia objeto de estudio, una concentración por grupo, durante un plazo de 90 días. En los casos en que se emplea un vehículo como ayuda para lograr una concentración apropiada de la sustancia ensayada en la atmósfera, se utilizará un grupo de control del vehículo. Durante el período de administración, se observa diariamente a los animales en busca de signos de toxicidad. Los animales que mueran durante la prueba se someterán a autopsia, que se practicará también a los supervivientes al concluir el estudio.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
Preparativos
Se mantiene a los animales en las condiciones de alojamiento y alimentación experimentales durante al menos 5 días previos a la prueba. Antes de comenzarla, se distribuye al azar a los animales, jóvenes y sanos, para formar grupos de tratamiento y de control. En caso necesario, se añadirá a la sustancia estudiada un vehículo adecuado que ayude a lograr una concentración apropiada de la sustancia en la atmósfera. Si se utilizaran un vehículo u otros aditivos para facilitar la administración, deberán saberse exentos de efectos tóxicos. Si se juzga oportuno, pueden emplearse datos publicados.
Condiciones del ensayo
Animales de experimentación
Salvo indicación en contrario, la especie preferible es la rata. Se utilizarán animales jóvenes y sanos de cepas de laboratorio de uso habitual. Al principio del estudio, los pesos no diferirán en un porcentaje superior al ± 20 % del valor medio apropiado. Cuando se realice un estudio de inhalación subcrónico previo a otro de índole crónica, se utilizarán en ambos la misma especie y cepa.
Número y sexo
Se utilizarán al menos 20 animales (10 hembras y 10 machos) para cada concentración de exposición. Las hembras serán nulíparas y no estarán preñadas. Si está previsto el sacrificio de algunos animales durante la prueba, debe aumentarse su número en una cantidad igual a la de los que vayan a sacrificarse antes de terminar el estudio. Además puede tratarse a un grupo satélite de 20 animales (10 de cada sexo) con la dosis más alta durante 90 días para observar en ellos la reversibilidad, la persistencia o la aparición tardía de efectos tóxicos durante 28 días tras el tratamiento.
Concentración de exposición
Son necesarias al menos tres concentraciones, y un control o control del vehículo (correspondiente a la concentración del vehículo en el nivel de exposición máximo). Salvo por la omisión de la sustancia estudiada, los animales del grupo de control recibirán un trato idéntico al de los integrantes del grupo de prueba. La dosis más alta debe originar efectos tóxicos, pero producir pocos fallecimientos, o ninguno. Cuando se disponga de una estimación válida de la exposición humana, la dosis mínima será superior a este valor. Lo ideal sería que la dosis media produjera el efecto tóxico mínimo observable. Si se emplea más de una dosis intermedia, la diferencia entre dosis será suficiente para que los efectos tóxicos resultantes sean escalonados. En los grupos con dosis bajas e intermedias, así como en los controles, la mortalidad debe ser baja, a fin de permitir una evaluación significativa de los resultados.
Período de exposición
La exposición diaria será de 6 horas tras la obtención de las concentraciones en la cámara de exposición. Cabe utilizar otros períodos para satisfacer necesidades especiales.
Equipo
Los animales se someterán al estudio en un dispositivo de inhalación capaz de mantener un flujo de aire continuo de al menos 12 renovaciones de aire a la hora, y de garantizar un contenido de oxígeno apropiado y una distribución uniforme del producto estudiado en el aire. Cuando se utilice una cámara, deberá ser de características tales que permita un hacinamiento mínimo de los animales y su exposición óptima a la sustancia objeto de ensayo. Como norma general para garantizar la estabilidad de la atmósfera de la cámara, el volumen total de los animales de experimentación no debe exceder del 5 % del volumen de la cámara empleada. También cabe recurrir a sistemas con exposición oronasal, de la cabeza sola o de todo el cuerpo en cámara individual; los dos primeros reducen la penetración por otras vías.
Período de observación
Es preciso observar a diario a los animales de experimentación en busca de signos de toxicidad. Hay que tomar nota del momento del fallecimiento y de los de aparición y desaparición de signos de toxicidad.
Procedimiento
Se expone a los animales diariamente a la sustancia estudiada, a razón de 5 o 7 días a la semana, durante un período de 90 días. Los animales de grupos satélites destinados a observaciones complementarias deben mantenerse 28 días más sin tratamiento para apreciar la recuperación de los efectos tóxicos, o su persistencia. La temperatura a la que se efectúa la prueba debe mantenerse, con variaciones de ± 3 en 22 oC. La humedad relativa ideal sería la comprendida entre el 30 y el 70 %, pero en determinados casos (por ejemplo, pruebas con aerosoles) quizá no sea factible. Durante la exposición, se suprimirán el alimento y la bebida.
Se utilizará un sistema de inhalación dinámico que disponga de un sistema apropiado de control analítico de la concentración. Se recomienda practicar un primer ensayo para determinar las concentraciones de exposición adecuadas. Se ajustará el flujo de aire de forma que se garanticen unas condiciones de exposición homogéneas en toda la cámara. El sistema permitirá obtener condiciones de exposición estables con la mayor rapidez posible.
Se determinarán o controlarán:
|
a) |
El flujo de aire (continuamente). |
|
b) |
La concentración real de la sustancia estudiada, medida en la zona de respiración. Durante el período de exposición diaria, la concentración no diferirá en más del ± 15 % del valor medio. No obstante, cuando se trate de polvos y aerosoles, quizá no sea posible esta precisión, y podrá aceptarse una desviación mayor. Se mantendrán durante la totalidad del estudio lo más constante posible las concentraciones diarias. Al ajustar el sistema generador, se practicará un análisis granulométrico de las partículas, para determinar la estabilidad de las concentraciones de aerosol. Durante la exposición, se practicarán con la frecuencia necesaria análisis para determinar la estabilidad de la distribución granulométrica. |
|
c) |
Temperatura y humedad. |
|
d) |
Durante la exposición y después de ella, se practican y registran sistemáticamente observaciones; se llevarán fichas individuales de cada animal. Es preciso observar a diario a todos los animales y registrar los signos de toxicidad, incluidos el momento de comienzo, el grado y la duración. Entre las observaciones deben figurar las de modificaciones de la piel y el pelo, los ojos, las membranas mucosas, los sistemas respiratorio, circulatorio, autonómico y nervioso central, la actividad somatomotriz y el comportamiento. Se determinarán cada semana el consumo de alimento y el peso de los animales. Es necesaria una observación periódica de los animales para evitar la pérdida de alguno de ellos por causas como canibalismo, autólisis de tejidos o enjaulamiento erróneo. Al final del período de prueba, se realiza la autopsia a todos los animales supervivientes. Los animales moribundos deben retirarse y someterse a autopsia. |
En todos los animales, incluidos los controles, se practican habitualmente los exámenes siguientes:
|
a) |
Exploración oftalmológica con un oftalmoscopio o equipo apropiado equivalente, antes de administrar la sustancia estudiada y al final del estudio, preferiblemente en todos los animales, pero por lo menos en los grupos con dosis altas y de control. Si se apreciaran alteraciones oculares, se examinará a todos los animales. |
|
b) |
Pruebas hematológicas al final del período de estudio: hematocrito, concentración de hemoglobina, recuento eritrocitario, recuento y fórmula leucocitarios, y determinación de la coagulación por pruebas como tiempo de coagulación, tiempo de protrombina, tiempo de tromboplastina o recuento de plaquetas. |
|
c) |
Determinaciones bioquímicas clínicas en la sangre al final del período de prueba. Se consideran aspectos de interés en todos los estudios el balance electrolítico, el metabolismo de hidratos de carbono y las funciones hepática y renal. En la selección de las pruebas concretas influirán las observaciones sobre el modo de acción de la sustancia. Se sugiere la determinación de los parámetros siguientes: calcio, fósforo, cloruro, sodio, potasio, glucosa en ayunas (con período de ayuno apropiado a cada especie), transaminasas glutamicopirúvica (4) y glutamicooxaloacética (5) séricas, ornitindescarboxilasa, gammaglutamiltranspeptidasa, nitrógeno ureico, albúmina, creatinina plasmática, bilirrubina total y proteínas séricas totales. Entre otras determinaciones que pueden ser necesarias para una evaluación toxicológica adecuada figuran los análisis de lípidos, hormonas, equilibrio acidobásico, metahemoglibina y actividad de colinesterasa. Siempre que sea preciso, se emplearán otras pruebas bioquímicas clínicas para ampliar la investigación de los efectos observados. |
|
d) |
El análisis de orina no es necesario de forma sistemática, sino solo en caso de efectos tóxicos probables o manifiestos. |
Si los datos de referencia previos son insuficientes, habrá que considerar la posible determinación de los parámetros hematológicos y de bioquímica clínica antes de iniciar la administración.
Autopsia
Se practicará en todos los animales una autopsia completa que comprenda la inspección de la superficie externa del cuerpo, todos sus orificios y las cavidades craneal, torácica y abdominal y sus contenidos. Hígado, riñones, suprarrenales y testículos deben pesarse frescos lo antes posible tras la disección, a fin de evitar la desecación. Se conservarán en un medio adecuado para un posible examen histopatológico posterior los órganos y tejidos siguientes: todas las lesiones macroscópicas, los pulmones —que se extirparán intactos, se pesarán y se tratarán con un fijador adecuado para garantizar el mantenimiento de su estructura (se considera método eficaz la perfusión con el fijador)—, los tejidos nasofaríngeos, el cerebro —incluidos cortes de bulbo/protuberancia—, corteza cerebelosa y encefálica, pulmones, corazón, aorta, glándulas salivales, hígado, bazo, riñones, suprarrenales, páncreas, gónadas, útero, (órganos genitales accesorios), (piel), vesícula biliar (si existe), esófago, estómago, duodeno, yeyuno, íleon, ciego, colon, recto, vejiga urinaria, ganglio linfático representativo (glándula mamaria femenina), (musculatura del muslo), nervio periférico, (ojos), esternón con médula ósea, (fémur, incluida superficie articular), y (médula espinal a tres niveles: cervical, mesotorácico y lumbar). Los tejidos que aparecen entre paréntesis solo se examinarán si así lo aconsejan los signos de toxicidad o afectación del órgano efector.
Examen histopatológico
|
a) |
Se sometarán a un examen histopatológico completo el aparato respiratorio y otros órganos y tejidos de los animales de los grupos de control y con la dosis máxima. |
|
b) |
Se examinarán todas las lesiones macroscópicas. |
|
c) |
Se examinarán los órganos efectores de los animales de grupos con otras dosis. |
|
d) |
Se someterán también a examen histopatológico los pulmones de los animales pertenecientes a los grupos con dosis baja e intermedia, por constituir un medio conveniente de valoración del estado de salud de los animales. Es posible que en los animales de estos grupos no sean necesarios por sistema exámenes histopatológicos complementarios, pero siempre se llevarán a cabo en los órganos de los del grupo con dosis máxima que presenten indicios de lesiones. |
|
e) |
Cuando se utilice un grupo satélite, se practicará un examen histopatológico de los tejidos y órganos que presenten signos de toxicidad en los grupos tratados. |
2. RESULTADOS
Los resultados se resumirán en forma de tabla, y mostrarán para cada grupo de prueba el número de animales al comienzo del ensayo, el de los que presenten lesiones y el porcentaje de animales con cada tipo de lesión. Los resultados se evaluarán por un método estadístico apropiado. Puede usarse cualquier método estadístico reconocido.
3. INFORME
3.1. DATOS DEL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
especie, cepa, origen, condiciones ambientales, alimentación, |
|
— |
condiciones de la prueba: Descripción del dispositivo de exposición, incluidos diseño, tipo, dimensiones, fuente de aire, sistema de generación de partículas y aerosoles, método de acondicionamiento del aire, tratamiento del aire evacuado y, cuando se emplee, método de alojamiento de los animales en la cámara de prueba. Se describirá el equipo empleado para determinar la temperatura, la humedad y, en su caso, la estabilidad de las concentraciones de aerosol o el tamaño de las partículas. Datos de exposición: Se presentarán en forma de tabla en la que figuren tanto los valores medios como una medida de la variabilidad (por ejemplo, desviación estándar), y comprenderán:
|
|
— |
datos de respuesta tóxica en función del sexo y la concentración, |
|
— |
dosis carente de efectos, si es posible, |
|
— |
momento de la muerte durante el estudio, o indicación de que los animales sobrevivieron a la experiencia, |
|
— |
descripción de los efectos tóxicos o de otro tipo, |
|
— |
momento de observación de cada signo anómalo y evolución de este, |
|
— |
datos sobre alimentación y peso, |
|
— |
hallazgos oftalmológicos, |
|
— |
pruebas hematológicas practicadas y sus resultados completos, |
|
— |
pruebas de bioquímica clínica empleadas y sus resultados completos (incluidos los del análisis de orina, si procede), |
|
— |
hallazgos de autopsia, |
|
— |
descripción detallada de los hallazgos histopatológicos, |
|
— |
tratamiento estadístico de los resultados, si es posible, |
|
— |
comentario de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.30. ENSAYO DE TOXICIDAD CRÓNICA
l. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIONES
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
La sustancia objeto de estudio se administra normalmente 7 días a la semana, por una vía apropiada, a varios grupos de animales de experimentación, a razón de una dosis por grupo, durante una parte importante de su existencia. Durante la exposición a la sustancia estudiada, y después de ella, se observa a diario a los animales para apreciar posibles signos de toxicidad.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
Preparativos
Se mantiene a los animales en las condiciones de alojamiento y alimentación experimentales durante al menos 5 días previos a la prueba. Antes de comenzarla, se distribuye al azar a los animales, que serán jóvenes y sanos, para formar grupos de tratamiento y de control.
Condiciones del ensayo
Animales de experimentación
La especie preferida es la rata,
aunque pueden utilizarse otras (de roedores o no roedores) basándose en los resultados de estudios previos. Deben emplearse animales jóvenes y sanos, de cepas de laboratorio de uso habitual, y la administración se iniciará lo antes posible tras el destete.
Al principio del estudio, el peso de los animales no debe diferir en un porcentaje superior al ± 20 % del valor medio. Cuando se practique un estudio oral subcrónico previo a otro de índole crónica, se utilizarán en ambos las mismas especie/raza y cepa.
Número y sexo
En roedores, se utilizarán al menos 40 animales (20 hembras y 20 machos) para cada dosis y grupo de control correspondiente. Las hembras serán nulíparas y no grávidas. Si está previsto el sacrificio de algunos animales durante la prueba, debe aumentarse su número en una cantidad igual a la de los que vayan a sacrificarse antes de terminar el estudio.
En no roedores, puede aceptarse un número menor de animales, aunque no inferior a cuatro por sexo y grupo.
Dosis y frecuencia de exposición
Se utilizarán al menos tres niveles posológicos, además del grupo de control correspondiente.
La dosis más alta debe originar signos definidos de toxicidad sin causar una mortalidad excesiva. La dosis más baja no debe provocar indicio alguno de toxicidad.
La o las dosis intermedias se situarán más o menos en un punto medio entre la alta y la baja.
En la selección de las dosis se tendrán en cuenta datos de pruebas y estudios de toxicidad precedentes.
Normalmente, la exposición será diaria. Si se administra la sustancia química en el agua de bebida o mezclada con el alimento, deberá estar continuamente disponible.
Controles
Se utilizará un grupo de control idéntico en todos los aspectos a los tratados, salvo por la no exposición a la sustancia probada.
En circunstancias especiales, como las concurrentes en estudios de inhalación con aerosoles o en caso de empleo de un emulgente de actividad biológica no caracterizada en estudios orales, se utilizará también un grupo de control negativo concurrente. Este grupo de control negativo se tratará de igual manera que los grupos de prueba, salvo en que los animales no se expondrán a la sustancia estudiada ni a ningún vehículo.
Vía de administración
Las dos vías de administración principales son la oral y la respiratoria (inhalación). La elección de una de ellas dependerá de las características físicas y químicas de la sustancia estudiada y de la vía probable de exposición en seres humanos.
El uso de la vía dérmica plantea problemas prácticos considerables. Normalmente, es posible deducir la toxicidad crónica general debida a la absorción percutánea de los resultados de la prueba hecha por vía oral y de la cantidad de sustancia absorbida por vía percutánea en pruebas de toxicidad percutánea previas.
Estudios orales:
Cuando la sustancia estudiada se absorba en el aparato gastrointestinal, y si la ingestión es una vía posible de exposición humana, se preferirá la vía oral de administración, salvo si existen contraindicaciones. Los animales pueden recibir la sustancia con la alimentación, disuelta en el agua de bebida o por medio de cápsulas. Lo ideal sería la administración los 7 días de la semana, ya que con 5 dosis a la semana es posible una recuperación o la reducción de la toxicidad en el período exento de dosis, con la consiguiente influencia en el resultado y la evaluación posterior. Sin embargo, y por motivos esencialmente prácticos, se considera aceptable la administración 5 días a la semana,
Estudios de inhalación:
Dado que los estudios de inhalación plantean problemas técnicos de mayor complejidad que las demás vías de administración, ofrecemos aquí una orientación más detallada sobre este modo de administración. Hay que señalar también que la instilación intratraqueal constituye un método válido en situaciones determinadas.
Las exposiciones prolongadas suelen adecuarse a las condiciones de exposición humana proyectadas; así, se expone a los animales 5 días a la semana (exposición intermitente) durante 6 horas diarias tras equilibrado de las concentraciones en la cámara de prueba, o bien los 7 días de la semana (exposición permanente) con una exposición diaria de 22 a 24 horas, dedicando alrededor de una hora a la alimentación de los animales (horario regular) y el mantenimiento de la cámara.
En ambos casos, los animales suelen exponerse a concentraciones fijas de la sustancia objeto de estudio. Una diferencia importante entre la exposición intermitente y la permanente es que con la primera existe un período de 17 a 18 horas en que los animales pueden recuperarse de los efectos de cada exposición diaria, con un período de recuperación aún mayor durante los fines de semana.
La elección de la exposición intermitente o permanente dependerá de los objetivos del estudio y de la exposición humana que pretenda simularse. No obstante, hay que tener en cuenta ciertas dificultades técnicas. Por ejemplo, es posible que las ventajas de la exposición permanente en la simulación de condiciones ambientales queden contrapesadas por la necesidad de suministrar alimento y bebida durante la exposición, así como por la necesidad de técnicas de generación de aerosoles y vapores, y de control, más complicadas (y fiables).
Cámaras de exposición
Los animales se someterán al estudio en un dispositivo de inhalación capaz de mantener un flujo de aire continuo de, al menos, 12 renovaciones de aire a la hora, y de garantizar un contenido de oxígeno apropiado y una distribución uniforme del producto estudiado en el aire. Las cámaras de control y de exposición serán de construcción y diseño idénticos para garantizar condiciones de exposición comparables en todos los aspectos, salvo en la exposición a la sustancia probada. Por regla general, se mantiene una ligera presión negativa en el interior de la cámara para impedir el escape de la sustancia estudiada a la zona circundante. Se evitará el hacinamiento de los animales en las cámaras. Como norma general, para garantizar la estabilidad de la atmósfera de la cámara, el volumen total de los animales de experimentación no debe exceder del 5 % del volumen de la cámara empleada.
Se practicarán determinaciones o controles de:
|
i) |
Flujo del aire: el flujo de aire en la cámara se controlará preferiblemente de modo constante. |
|
ii) |
Concentración: durante el período diario de exposición, la concentración de la sustancia estudiada no diferirá en más del ± 15 % del valor medio. |
|
iii) |
Temperatura y humedad: en roedores, la temperatura se mantendrá a 22 oC (± 2 oC), y la humedad en el interior de la cámara del orden del 30 al 70 %, salvo cuando se emplee agua para suspender la sustancia estudiada en la atmósfera de la cámara. Ambos factores se controlarán preferiblemente de modo continuo. |
|
iv) |
Análisis granulométrico de las partículas: en las atmósferas de la cámara que exijan la utilización de aerosoles líquidos o sólidos, se determinará la distribución de las partículas por tamaños. Las partículas de los aerosoles serán de un tamaño respirable para el animal de experimentación utilizado. Se recogerán muestras de las atmósferas de la cámara en la zona de respiración de los animales. La muestra de aire estará acorde con la distribución de las partículas a las que se exponga a los animales y será representativa, sobre una base gravimétrica, de la totalidad del aerosol en suspensión, incluso aunque gran parte de este no sea respirable. Los análisis granulométricos se efectuarán con frecuencia durante la adaptación del sistema generador para garantizar la estabilidad del aerosol, y con posterioridad, cuando se consideren necesarios para determinar debidamente la constancia de la distribución de las partículas a que se expone a los animales. |
Duración del estudio
La duración del período de administración será de, al menos, 12 meses.
Procedimiento
Observaciones
Se realizará, al menos una vez al día, una exploración clínica detenida. Se practicarán diariamente observaciones complementarias, y se adoptarán medidas apropiadas para reducir al mínimo las pérdidas de animales del estudio, en forma, por ejemplo, de autopsia o refrigeración de los animales encontrados muertos, y de aislamiento o sacrificio de los débiles o moribundos. Son necesarias observaciones detenidas para apreciar el comienzo y la evolución de efectos tóxicos, así como para reducir las pérdidas de animales por enfermedad, autólisis o canibalismo.
Se anotarán para todos los animales los signos clínicos, incluidas tanto las alteraciones neurológicas y oculares como la mortalidad. Se registrarán el momento de comienzo y la evolución de los procesos tóxicos, incluidos los presuntos tumores.
Se determinará y anotará el peso de cada animal una vez a la semana durante las 13 primeras del período de prueba, y al menos una vez cada 4 semanas con posterioridad. Se determinará la ingestión de alimento cada semana durante las 13 primeras del estudio, y luego a intervalos aproximadamente trimestrales, salvo que el estado de salud o las modificaciones del peso aconsejen otra actitud.
Examen hematológico
Se realizará un examen hematológico (por ejemplo, contenido de hemoglobina, hematocrito, recuentos de hematíes y leucocitos, plaquetas u otras medidas de la capacidad de coagulación) a los 3 y a los 6 meses, y después con intervalos aproximados de 6 y, al final, sobre muestras de sangre recogidas de todos los no roedores y de 10 ratas/sexo de todos los grupos. A ser posible, las muestras procederán de las mismas ratas en cada intervalo. Además, a los no roedores se les extraerá una muestra antes de la prueba.
Si las observaciones clínicas indican un menoscabo de la salud de los animales durante el estudio, se practicará un recuento con fórmula leucocitaria de los animales afectados.
Se averiguará la fórmula leucocitaria en muestras de los animales del grupo con dosis máxima y de los controles. Solo se determinará en los grupos con dosis menores si se aprecia una discrepancia importante entre el grupo con dosis máxima y los controles, o si lo aconsejan los hallazgos patológicos.
Análisis de orina
Se recogerán para su análisis muestras de orina de todos los animales no roedores y de 10 ratas/sexo de todos los grupos, a ser posible de las mismas ratas que sirvan para el examen hematológico y respetando intervalos idénticos. Se realizarán las determinaciones siguientes en animales individuales, o en una muestra acumulada/sexo/grupo en el caso de los roedores:
|
— |
aspecto: volumen y densidad en cada animal, |
|
— |
proteínas, glucosa, cetonas, sangre oculta (semicuantitativamente), |
|
— |
microscopía del sedimento (semicuantitativamente). |
Química clínica
Con intervalos aproximados de 6 meses y a la conclusión, se extraen muestras de sangre para determinaciones de química clínica de todos los no roedores y de 10 ratas/sexo de todos los grupos, a ser posible de las mismas ratas en cada intervalo. Además, se recogerá en todos los no roedores una muestra previa a la prueba. Se prepara plasma a partir de estas muestras, y se realizan las determinaciones siguientes:
|
— |
concentración de proteínas totales, |
|
— |
concentración de albúmina, |
|
— |
pruebas de función hepática (como actividad de fosfatasa alcalina, transaminasa glutamicopirúvica (4) y transaminasa glutamicooxaloacética (5), gammaglutamiltranspeptidasa, ornitindescarboxilasa, |
|
— |
metabolismo de carbohidratos, como glucemia en ayunas, |
|
— |
pruebas de función renal, como nitrógeno ureico en sangre. |
Autopsia
Se practicará una autopsia completa en todos los animales, incluidos los fallecidos durante el experimento o sacrificados al encontrarse moribundos. Antes del sacrificio, se recogerán de todos los animales muestras de sangre para la realización de recuentos sanguíneos con fórmula leucocitaria. Se conservarán todas las lesiones macroscópicas visibles, así como los tumores o lesiones que se sospeche son tumores. Se intentarán relacionar las observaciones macroscópicas con los hallazgos microscópicos.
Se conservarán para examen histopatológico todos los órganos y tejidos, habitualmente los siguientes: cerebro (6) (bulbo/protuberancia, corteza cerebelosa y encefálica), hipófisis, tiroides, (incluidas paratiroides), timo, pulmones (incluida tráquea), corazón, aorta, glándulas salivales, hígado (6), bazo, riñones (6), suprarrenales (6), esófago, estómago, duodeno, yeyuno, íleon, ciego, colon, recto, útero, vejiga urinaria, ganglios linfáticos, páncreas, gónadas (6), órganos genitales accesorios, glándula mamaria femenina, piel, musculatura del muslo, nervio periférico, médula espinal (cervical, dorsal, lumbar), esternón con médula ósea y fémur (incluida articulación) y ojos. El inflado de pulmones y vejiga urinaria con un fijador es el medio óptimo de conservación de estos tejidos; por otro lado, el inflado de los pulmones es esencial en los estudios de inhalación para efectuar un examen histopatológico apropiado. En estudios especiales, como los de inhalación, se examinará la totalidad del aparato respiratorio, incluidas nariz, faringe y laringe.
Si se realizan otros exámenes clínicos, la información con ellos obtenida deberá estar disponible antes del examen microscópico, ya que puede suponer una orientación importante para el anatomopatólogo.
Examen histopatológico
Se examinarán al microscopio todas las alteraciones visibles, y en particular los tumores y demás lesiones aparecidas en cualquier órgano. Se recomiendan además los procedimientos siguientes:
|
a) |
examen microscópico de todos los órganos y tejidos conservados, con descripción completa de todas las lesiones encontradas en:
|
|
b) |
se examinarán también los órganos o tejidos de animales con dosis mínima que muestren anomalías causadas de forma indudable o posible por la sustancia objeto de estudio; |
|
c) |
cuando el resultado de la prueba indique una reducción sustancial del período vital de los animales o la inducción de efectos capaces de influir en la respuesta tóxica, se someterá al examen citado en el párrafo anterior a los animales tratados con la dosis inmediatamente inferior; |
|
d) |
es indispensable disponer de información sobre la incidencia de las lesiones que afectan normalmente a la cepa de animales en condiciones iguales a las de la prueba (es decir, datos de control publicados) para evaluar correctamente el significado de las modificaciones observadas en los animales tratados. |
2. RESULTADOS
Los resultados se resumirán en forma de tabla y mostrarán, para cada grupo de prueba, el numero de animales al comienzo del ensayo, el de los que presenten lesiones y el porcentaje de animales con cada tipo de lesión. Los resultados se evaluarán por un método estadístico apropiado. Puede usarse cualquier método estadístico reconocido.
3. INFORME
3.1. DATOS DEL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
especie, cepa, origen, condiciones ambientales, alimentación, |
|
— |
condiciones de la prueba: Descripción del dispositivo de exposición, incluidos diseño, tipo, dimensiones, fuente de aire, sistema de generación de partículas y aerosoles, método de acondicionamiento del aire, tratamiento del aire evacuado y, cuando se emplee, método de alojamiento de los animales en la cámara de prueba. Se describirá el equipo empleado para determinar la temperatura, la humedad y, en su caso, la estabilidad de las concentraciones de aerosol o el tamaño de las partículas. Datos de exposición: Se presentarán en forma de tabla en la que figuren tanto los valores medios como una medida de la variabilidad (por ejemplo, desviación estándar), y comprenderán:
|
|
— |
dosis (incluido el vehículo, si se emplea) y concentraciones, |
|
— |
datos de respuesta tóxica en función del sexo y la dosis, |
|
— |
dosis carente de efectos, |
|
— |
momento de la muerte durante el estudio, o indicación de que los animales sobrevivieron a la experiencia, |
|
— |
descripción de los efectos tóxicos o de otro tipo, |
|
— |
momento de observación de cada signo anómalo y evolución de este, |
|
— |
datos sobre alimentación y peso, |
|
— |
hallazgos oftalmológicos, |
|
— |
pruebas hematológicas practicadas y sus resultados completos, |
|
— |
pruebas de bioquímica clínica empleadas y sus resultados completos (incluidos los del análisis de orina, si procede), |
|
— |
hallazgos de autopsia, |
|
— |
descripción detallada de los hallazgos histopatológicos, |
|
— |
tratamiento estadístico de los resultados, si es posible, |
|
— |
comentario de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.31. ESTUDIO DE TOXICIDAD PARA EL DESARROLLO PRENATAL
1. MÉTODO
El presente método reproduce las directrices de ensayo de la OCDE TG 414 (2001).
1.1. INTRODUCCIÓN
El presente ensayo de toxicidad para el desarrollo tiene por objeto proporcionar información general sobre los efectos de la exposición prenatal en la hembra grávida sometida a ensayo y en el organismo intrauterino en desarrollo; incluye, en particular, la evaluación de los efectos sobre la madre, así como la muerte, anomalías estructurales y alteraciones del crecimiento del feto. Pese a que las deficiencias funcionales constituyen un aspecto importante del crecimiento, no se contemplan en el presente método de ensayo. Pueden investigarse al margen o como complemento de este método en un estudio de neurotoxicidad para el desarrollo. Este último y el estudio de toxicidad para la reproducción en dos generaciones pueden emplearse para detectar deficiencias funcionales y otros efectos postnatales.
En algunos casos particulares, puede ser necesario adaptar el protocolo de este ensayo, en función, por ejemplo, de las propiedades fisicoquímicas o toxicológicas de la sustancia de ensayo. La adaptación será aceptable si se dispone de datos científicos convincentes que indiquen que de esa manera el ensayo proporcionará mayor información. En tal caso, esos datos científicos deberán incluirse minuciosamente en el informe del ensayo.
1.2. DEFINICIONES
Toxicología del desarrollo: estudio de los efectos adversos sobre un organismo en desarrollo, que pueden producirse tras la exposición antes de la concepción y durante el desarrollo prenatal y postnatal hasta la maduración sexual. Los principales signos de la toxicidad para el desarrollo son la muerte del organismo, las anomalías estructurales, la alteración del crecimiento y las deficiencias funcionales. Antiguamente, la toxicología del desarrollo solía denominarse teratología.
Efecto adverso: toda alteración respecto a una situación de referencia, relacionada con el tratamiento, que disminuya la capacidad del organismo para sobrevivir, reproducirse o adaptarse al entorno. La toxicología del desarrollo, considerada en su sentido más amplio, incluye todos los efectos que afectan al desarrollo normal del producto de la concepción, antes y después del nacimiento.
Alteración del crecimiento: alteración que afecte al peso o el tamaño del cuerpo o los órganos de la descendencia.
Alteraciones (anomalías): alteraciones estructurales en el desarrollo, que incluyen las malformaciones y las variaciones (28).
Malformación/anomalía grave: cambio estructural que se considere perjudicial (también puede resultar letal) para el animal y, por lo general, infrecuente.
Variación/anomalía leve: cambio estructural que se considere poco o nada perjudicial para el animal; puede tener carácter transitorio y ser relativamente frecuente en la población de control.
Producto de la concepción: conjunto de los derivados de un óvulo fecundado, en cualquier fase del desarrollo, desde la fecundación hasta el nacimiento, incluidas las membranas extraembrionarias y el embrión o el feto.
Implantación (anidación): fijación del blastocisto en la túnica epitelial del útero, que incluye la penetración por el epitelio uterino y la inserción en el endometrio.
Embrión: primera fase del desarrollo de un organismo, en particular, la fase de desarrollo de un huevo fecundado desde que aparece el eje mayor hasta que quedan representadas las estructuras principales.
Embriotoxicidad: novicidad para la estructura, el desarrollo, el crecimiento y/o la viabilidad normales de un embrión.
Feto: descendencia nonata en el período postembrionario.
Fetotoxicidad: novicidad para la estructura, el desarrollo, el crecimiento y/o la viabilidad normales de un feto.
Aborto: expulsión prematura del útero de los productos de la concepción (embrión o feto no viable).
Resorción: absorción de un producto de la concepción que muere una vez implantado en el útero.
Resorción precoz: signos de implantación sin embrión ni feto reconocible.
Resorción tardía: embrión o feto muerto, que presenta cambios degenerativos externos.
NOAEL: sigla inglesa referente a la dosis de exposición sin efectos adversos observados, es decir, la dosis o grado de exposición más alto al que no se observa ningún efecto adverso debido al tratamiento.
1.3. SUSTANCIA DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO DE ENSAYO
Por lo general, se administra la sustancia de ensayo a las hembras grávidas al menos desde la implantación hasta la víspera del sacrificio, que debe llevarse a cabo tan cerca como sea posible de la fecha probable del parto, pero no demasiado tarde, para evitar la pérdida de datos en caso de parto prematuro. El método de ensayo no se refiere únicamente al período de la organogénesis (del 5° al 15° día en los roedores y del 6° al 18° en el conejo), sino que estudia asimismo los efectos a lo largo de toda la gestación, desde la fase previa a la implantación, en su caso, hasta la víspera de la cesárea. Las hembras se sacrifican poco antes de la cesárea. Se estudia el contenido uterino y se examinan los fetos para detectar las anomalías externas visibles y los cambios en los tejidos blandos y el esqueleto.
1.5. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
1.5.1. Selección de la especie animal
Se recomienda realizar el ensayo con la especie más adecuada y utilizar las especies y cepas de laboratorio más comunes en los ensayos de toxicidad para el desarrollo prenatal. El roedor idóneo es la rata y, entre los no roedores, el conejo. Deberá justificarse el empleo de otra especie.
1.5.2. Alojamiento y alimentación
El cuarto de experimentación ha de estar a una temperatura de 22 °C (± 3°) en el caso de los roedores y de 18 °C (+ 3°) en el caso de los conejos, y con una humedad relativa mínima del 30 % y preferiblemente inferior al 70 %, salvo durante la limpieza del local, si bien lo ideal es que esté comprendida entre el 50 y el 60 %. Se aplica una iluminación artificial en una secuencia de 12 horas de luz y 12 horas de oscuridad. Se proporciona una dieta alimentaria corriente para animales de laboratorio y agua potable a voluntad.
El apareamiento debe llevarse a cabo en jaulas adecuadas. Si bien es preferible colocar a los animales apareados en jaulas individuales, también pueden alojarse en grupos reducidos.
1.5.3. Preparación de los animales
Deben emplearse animales sanos, que se hayan mantenido al menos 5 días en las condiciones de laboratorio para su aclimatación y que no hayan sido sometidos a experimentos previos. Se caracteriza la especie, cepa, procedencia, sexo, peso y/o edad de los animales de experimentación. El peso y la edad de los animales de experimentación debe ser lo más uniforme posible en todos los lotes de ensayo. Deben emplearse hembras adultas jóvenes y nulíparas para cada dosis. Las hembras han de aparearse con machos de la misma especie y cepa y debe evitarse el apareamiento entre animales hermanos. En el caso de los roedores, el día 0 de la gestación es el día en que se observa un tapón vaginal y/o la presencia de semen; en el caso de los conejos, suele ser el día del coito o de la inseminación artificial, si se emplea esta técnica. Las hembras apareadas se reparten al azar entre los lotes tratados y los de control. Las jaulas se disponen de forma que se reduzcan al mínimo los posibles efectos debidos al enjaulamiento. Se asigna a cada animal un número de identificación distinto. Si el apareamiento se efectúa por grupos, los animales de un mismo grupo se repartirán de forma uniforme entre los distintos lotes. Asimismo, las hembras inseminadas por un mismo macho se repartirán de manera uniforme entre los lotes.
1.6. PROCEDIMIENTO
1.6.1. Número y sexo de los animales
Cada lote de ensayo y de control deberá estar integrado por una cantidad de hembras que permita practicar la autopsia de unos 20 ejemplares que presenten puntos de implantación. Los lotes de menos de 16 hembras con puntos de implantación pueden resultar inadecuados. La mortalidad materna no invalida necesariamente el estudio, siempre y cuando no supere el 10 % aproximadamente.
1.6.2. Preparación de las dosis
Si se emplea un aditivo u otro vehículo para facilitar la dosificación, deberán tenerse presentes los efectos sobre la absorción, distribución, metabolismo y retención o excreción de la sustancia de ensayo, los efectos sobre las propiedades químicas de esta que puedan modificar su toxicidad, y los efectos sobre el consumo de alimentos y agua y sobre el estado nutricional de los animales. El vehículo no debe ser tóxico para el desarrollo ni afectar a la reproducción.
1.6.3. Posología
Por lo general, la sustancia de ensayo se administra todos los días desde la implantación (por ejemplo, el quinto día después del apareamiento) hasta la víspera del día en que esté prevista la cesárea. Si los estudios preliminares disponibles no señalan un riesgo elevado de pérdidas preimplantatorias, puede administrarse el tratamiento durante todo el período de gestación, desde el apareamiento hasta la víspera de la cesárea. Se sabe que el estrés y la manipulación indebida durante la gestación pueden ocasionar pérdidas prenatales. Con objeto de evitar las pérdidas prenatales independientes del tratamiento, debe evitarse la manipulación innecesaria de las hembras grávidas y el estrés debido a factores externos como el ruido.
Se emplean al menos tres dosis de ensayo y un lote de control en paralelo. Los animales sanos se distribuyen al azar entre el lote de control y los lotes de ensayo. Las dosis han de ser progresivas para obtener una gradación de efectos tóxicos. A menos que las características fisicoquímicas o las propiedades biológicas de la sustancia de ensayo impongan restricciones, la dosis superior debe seleccionarse con el propósito de inducir efectos tóxicos para el desarrollo y/o en la madre (signos clínicos o disminución del peso corporal), pero sin llegar a provocar la muerte ni un sufrimiento intenso. Al menos una dosis intermedia debe producir los efectos tóxicos mínimos observables. La dosis mínima no debe resultar tóxica en absoluto para la madre ni para el desarrollo. Se selecciona una serie de dosis decrecientes para poner de manifiesto las respuestas en función de la dosis y la dosis sin efectos adversos observados (NOAEL). Los intervalos del doble al cuádruple suelen ser óptimos para establecer las dosis decrecientes y a menudo es preferible añadir un cuarto lote de ensayo en lugar de utilizar intervalos muy amplios (por ejemplo, con un factor superior a 10) entre dosis. Si bien es cierto que se trata de determinar la NOAEL en la madre, los estudios que no permiten determinarla también son aceptables (1).
Las dosis deben establecerse tomando en consideración todos los datos disponibles sobre la toxicidad y la información complementaria relativa al metabolismo y la toxicocinética de la sustancia de ensayo o productos afines. Dicha información servirá asimismo para justificar la pertinencia de la gama de dosis.
Se emplea un lote de control en paralelo, que no recibe la sustancia de ensayo, pero sí el vehículo en caso de que este se utilice para la sustancia. Debe administrarse el mismo volumen de sustancia de ensayo o de vehículo a todos los lotes. Los animales del lote o lotes de control deben tratarse de la misma manera que los de los lotes de ensayo. Los lotes de control a los que se administra el vehículo han de recibir el mayor volumen que se haya utilizado (el que se administre al lote tratado con la dosis inferior).
1.6.4. Ensayo límite
Si en un ensayo con una sola dosis equivalente, al menos, a 1 000 mg/kg peso corporal/día, administrada por vía oral y siguiendo el procedimiento descrito en el presente estudio no se produce ningún efecto tóxico observable en las hembras grávidas ni en la progenie y si, a la luz de los datos disponibles (por ejemplo, sobre sustancias con características estructurales y/o metabólicas similares), no cabe esperar efectos tóxicos, puede considerarse innecesario realizar un estudio completo con tres dosis. Según el grado previsto de exposición humana, puede ser preciso administrar una dosis oral superior en el ensayo límite. Si se trata de otras vías de administración, como la inhalación o la aplicación cutánea, suelen ser las características fisicoquímicas de la sustancia de ensayo las que determinan y limitan el grado máximo de exposición que puede alcanzarse (por ejemplo, la aplicación cutánea no debe producir toxicidad local grave).
1.6.5. Administración de las dosis
La sustancia de ensayo y el vehículo suelen administrarse mediante intubación oral. Si se opta por otra vía de administración, deberá motivarse y justificarse, y procederse a las modificaciones pertinentes (2) (3) (4). La sustancia de ensayo debe administrarse todos los días a la misma hora más o menos.
Por lo general, la dosis que se administra a cada animal se calcula con arreglo a la última determinación del peso corporal, si bien conviene ser prudente al adaptar la dosis en el último período de la gestación. Debe considerarse la información disponible para seleccionar la dosis de manera que se evite una toxicidad excesiva para la madre. Si, a pesar de todo, se observa una toxicidad excesiva en las madres tratadas, deberán sacrificarse por métodos compasivos. Si varias hembras grávidas presentan signos de toxicidad excesiva, se contemplará la posibilidad de sacrificar todo el lote tratado con esa dosis. Si la sustancia de ensayo se administra por sonda, debe hacerse en una sola dosis y con una sonda gástrica o una cánula de intubación adecuada. El volumen máximo de líquido que puede administrarse de una sola vez depende del tamaño del animal y no debe superar 1 ml/l00 g de peso corporal, salvo en el caso de las soluciones acuosas, en que puede llegarse a 2 ml/l00 g de peso corporal. En caso de que se emplee aceite de maíz como vehículo, el volumen no debe superar 0,4 ml/l00 g de peso corporal. La variabilidad del volumen de ensayo debe reducirse al mínimo ajustando la concentración para que el volumen sea constante en todas las dosis.
1.6.6. Observación de las madres
Las observaciones clínicas y el correspondiente registro deben efectuarse al menos una vez al día, preferentemente a la misma hora y teniendo en cuenta el período más agudo de los efectos previstos tras la administración. Se registra el estado clínico de los animales y, en particular, los animales muertos o moribundos, las alteraciones pertinentes del comportamiento y todos los signos de toxicidad manifiesta.
1.6.7. Peso corporal y consumo de alimentos
Los animales deben pesarse el día 0 de la gestación o, a más tardar, el día 3 si se trata de animales ya apareados procedentes de un criador externo; a continuación, el primer día de tratamiento, al menos cada 3 días durante el período de administración y, por último, el día del sacrificio.
Se registra el consumo de alimentos cada 3 días, coincidiendo con los días en que se pesen los animales.
1.6.8. Autopsia
Se sacrifican las hembras la víspera del día previsto para al parto. Aquellas que presenten signos de aborto o parto prematuro antes de la fecha en que esté previsto darles muerte serán sacrificadas y sometidas a un examen macroscópico exhaustivo.
Tras el sacrificio o la muerte en el transcurso del estudio, las hembras se someten a un examen macroscópico para detectar cualquier cambio patológico o anomalía estructural. Con objeto de minimizar los sesgos, es preferible efectuar la observación de las madres durante la cesárea y el examen posterior del feto sin conocer la dosis administrada.
1.6.9. Examen del contenido uterino
Inmediatamente después del sacrificio o tan pronto como sea posible tras la muerte del animal, se extrae el útero y se comprueba el estado de gravidez. También se examinarán los úteros que no parezcan grávidos (por ejemplo, mediante tinción de sulfuro amónico en el caso de los roedores y tinción de Salewski u otro método adecuado, en el de los conejos) para confirmar la ausencia de gravidez (5).
Se pesan los úteros grávidos con el cuello, salvo los de las hembras que hayan muerto durante el estudio.
Se cuenta el número de cuerpos amarillos en las hembras grávidas.
Se examina el contenido uterino para determinar el número de embriones o fetos muertos y de fetos viables. Es preciso describir el grado de resorción para calcular la fecha aproximada de la muerte del producto de la concepción (véase el apartado 1.2).
1.6.10. Examen de los fetos
Se determina el sexo y el peso corporal de cada feto.
Se examinan todos los fetos para ver si presentan alteraciones externas (6).
Se ve, asimismo, si presentan alteraciones del esqueleto o los tejidos blandos (por ejemplo, variaciones y malformaciones o anomalías) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16) (17) (18) (19) (20) (21) (22) (23) (24). Es aconsejable, pero no indispensable, clasificar las alteraciones fetales. Si se efectúa la clasificación, deben establecerse claramente los criterios que definan cada clase. Se estudiará detenidamente si ha habido alteraciones en el desarrollo del tracto genital.
En el caso de los roedores, se prepara aproximadamente la mitad de cada camada para estudiar las alteraciones del esqueleto. El resto se prepara para el estudio de las alteraciones de los tejidos blandos, que se efectuará en cortes seriados realizados por métodos reconocidos o apropiados o mediante técnicas finas de disección macroscópica.
Si se han empleado no roedores, por ejemplo conejos, todos los fetos se someten a examen para detectar posibles alteraciones de los tejidos blandos y el esqueleto. Se diseccionan cuidadosamente los cuerpos de los fetos para buscar alteraciones de los tejidos blandos; esta operación puede incluir técnicas que permitan examinar más exhaustivamente la estructura cardíaca interna (25). Se toman las cabezas de la mitad de los fetos analizados de esta manera y se preparan para evaluar las alteraciones de los tejidos blandos (en particular, ojos, cerebro, conductos nasales y lengua), mediante técnicas clásicas de cortes seriados (26) u otro método que presente la misma sensibilidad. Se preparan los cuerpos de esos fetos y los de los fetos intactos restantes para el estudio de las alteraciones del esqueleto mediante los métodos descritos para los roedores.
2. RESULTADOS
2.1. TRATAMIENTO DE LOS RESULTADOS
Deben proporcionarse datos de cada madre y su progenie y resumirse en un cuadro que recoja, para cada lote de ensayo, el número de animales al inicio del ensayo, el número de animales hallados muertos durante el mismo o sacrificados por razones compasivas, el momento de la muerte o sacrificio compasivo, el número de hembras grávidas, el número de animales que presenten signos de toxicidad, una descripción de dichos signos (con inclusión del momento de su aparición, duración y gravedad), los tipos de observaciones de los embriones o fetos y todos los datos pertinentes sobre la camada.
Los resultados numéricos deben evaluarse mediante un método estadístico adecuado y comúnmente aceptado, tomando la camada como unidad para el análisis de los datos. La elección de los métodos estadísticos debe efectuarse en la fase de diseño del estudio y justificarse. Deben registrarse también los datos relativos a los animales que no sobrevivan hasta la fecha programada para el sacrificio y, si procede, pueden incluirse en las medias de los lotes. La pertinencia de dichos datos y, por tanto, la decisión de incluirlos o no en las medias de los lotes, debe valorarse caso por caso y justificarse.
2.2. EVALUACIÓN DE LOS RESULTADOS
Los resultados del estudio de toxicidad para el desarrollo prenatal deben evaluarse respecto a los efectos observados. La evaluación debe incluir la información siguiente:
|
— |
resultados de los ensayos en las madres y los embriones o fetos, incluida la evaluación de la relación, o la ausencia de relación, entre la exposición de los animales a la sustancia de ensayo y la incidencia y gravedad de todos los efectos observados, |
|
— |
criterios aplicados, en su caso, para clasificar las alteraciones fetales externas, de los tejidos blandos y del esqueleto, |
|
— |
si procede, datos previos relativos a los controles para afinar la interpretación de los resultados del estudio, |
|
— |
cifras empleadas en el cálculo de todos los índices o porcentajes, |
|
— |
análisis estadístico apropiado de los resultados del estudio e inclusión de información suficiente sobre el método empleado, de manera que un revisor o estadístico independiente pueda reevaluarlo y reconstruirlo. |
Si el estudio no pone de manifiesto ningún efecto tóxico, debe valorarse la pertinencia de efectuar experimentos complementarios para determinar la absorción y la biodisponibilidad de la sustancia de ensayo.
2.3. INTERPRETACIÓN DE LOS RESULTADOS
Los estudios de toxicidad para el desarrollo prenatal proporcionan información sobre los efectos de la exposición repetida a una sustancia administrada durante la gravidez en las madres y en el desarrollo intrauterino de su progenie. Los resultados del estudio han de interpretarse a la luz de los de los estudios de toxicidad subcrónica, reproducción, toxicocinéticos y de otro tipo. Dado que el estudio se centra tanto en la toxicidad general para la madre como en la toxicidad para el desarrollo, los resultados permiten distinguir en cierta medida los efectos sobre el desarrollo que se producen en ausencia de toxicidad general de los que se manifiestan únicamente a dosis que también son tóxicas para la madre (27).
3. INFORME
3.1. INFORME DEL ENSAYO
El informe del ensayo debe incluir la información siguiente:
|
|
Sustancia de ensayo:
|
|
|
Vehículo (si procede):
|
|
|
Animales sometidos a ensayo:
|
|
|
Condiciones de ensayo:
|
|
|
Resultados: |
|
|
Datos relativos a la toxicidad para la madre en función de las dosis, indicando al menos lo siguiente:
|
|
|
Datos relativos al desarrollo por dosis y camada (con implantaciones), en particular:
|
|
|
Datos relativos al desarrollo por dosis y camada (con fetos vivos), en particular:
|
|
|
Discusión de los resultados. |
|
|
Conclusiones. |
4. REFERENCIAS
|
(1) |
Kavlock R.J. et al. (1996) A Simulation Study of the Influence of Study Design on the Estimation of Benchmark Doses for Developmental Toxicity. Risk Analysis 16; 399-410. |
|
(2) |
Kimmel, C.A. and Francis, E.Z. (1990) Proceedings of the Workshop on the Acceptability and Interpretation of Dermal Developmental Toxicity Studies. Fundamental and Applied Toxicology 14; 386-398. |
|
(3) |
Wong, B.A., et al. (1997) Developing Specialized Inhalation Exposure Systems to Address Toxicological Problems. CIIT Activities 17; 1-8. |
|
(4) |
US Environmental Protection Agency (1985) Subpart E-Specific Organ/Tissue Toxicity, 40 CFR 798.4350: Inhalation Developmental Toxicity Study. |
|
(5) |
Salewski, E. (1964) Faerbermethode zum Makroskopischen Nachweis von Implantations Stellen am Uterusder Ratte. Naunyn-Schmeidebergs Archivfur Pharmakologie und Experimentelle Pathologie 247:367. |
|
(6) |
Edwards, J.A. (1968) The external Development of the Rabbit and Rat Embryo. In Advances in Teratology. D.H.M. Woolam (ed.) Vol. 3. Academic Press, NY. |
|
(7) |
Inouye, M. (1976) Differential Staining of Cartilage and Bone in Fetal Mouse Skeleton by Alcian Blue and Alizarin Red S. Congenital Anomalies 16; 171-173. |
|
(8) |
Igarashi, E. et al. (1992) Frequency Of Spontaneous Axial Skeletal Variations Detected by the Double Staining Techniquefor Ossified and Cartilaginous Skeleton in Rat Foetuses. Congenital Anomalies 32; :381-391. |
|
(9) |
Kimmel, C.A. et al. (1993) Skeletal Development Following Heat Exposure in the Rat. Teratology 47:229-242. |
|
(10) |
Marr, M.C. et al. (1988) Comparison of Single and Double Staining for Evaluation of Skeletal Development: The Effects of Ethylene Glycol (EG) in CD Rats. Teratology 37; 476. |
|
(11) |
Barrow, M.V. and Taylor, W.J. (1969) A Rapid Method forDetecting Malformations in Rat Foetuses. Journal of Morphology 127:291-306. |
|
(12) |
Fritz, H. (1974) Prenatal Ossification in Rabbits ss Indicative of Foetal Maturity. Teratology 11; 313-320. |
|
(13) |
Gibson, J.P. et al. (1966) Use of the Rabbit in Teratogenicity Studies. Toxicology and Applied Pharmacology 9;:398-408. |
|
(14) |
Kimmel, C.A. and Wilson, J.G. (1973) Skeletal Deviation in Rats: Malformations or Variations? Teratology 8; 309-316. |
|
(15) |
Marr, M.C. et al. (1992) Developmental Stages of the CD (Sprague-Dawley) Rat Skeleton after Maternal Exposure to Ethylene Glycol. Teratology 46; 169-181. |
|
(16) |
Monie, I.W. et al. (1965) Dissection Procedures for Rat Foetuses Permitting Alizarin Red Staining of Skeleton and Histological Study of Viscera. Supplement to Teratology Workshop Manual, pp. 163-173. |
|
(17) |
Spark, C. and Dawson, A.B. (1928) The Order and Time of appearance of Centers of Ossification in the Fore and Hind Limbs of the Albino Rat, with Special Reference to the Possible Influence of the Sex Factor. American Journal of Anatomy 41; 411-445. |
|
(18) |
Staples, R.E. and Schnell, V.L. (1964) Refinements in Rapid Clearing Technique in the KOH-Alizarin Red S Method for Fetal Bone. Stain Technology 39; 61 -63. |
|
(19) |
Strong, R.M. (1928) The Order Time and Rate of Ossification of the Albino Rat (Mus Norvegicus Albinus) Skeleton. American Journal of Anatomy 36; 313-355. |
|
(20) |
Stuckhardt, J.L. and Poppe, S.M. (1984) Fresh Visceral Examination of Rat and Rabbit Foetuses Used in Teratogenicity Testing. Teratogenesis, Carcinogenesis, and Mutagenesis 4; 181-188. |
|
(21) |
Walker, D.G. and Wirtschafter, Z.T. (1957) The Genesis of the Rat Skeleton. Thomas, Springfield, IL. |
|
(22) |
Wilson, J.G. (1965) Embryological Considerations in Teratology. In Teratology: Principies and Techniques, Wilson J.G. and Warkany J. (eds). University of Chicago, Chicago, IL, pp 251-277. |
|
(23) |
Wilson, J.G. and Fraser, F.C. (eds). (1977) Handbook of Teratology, Vol. 4. Plenum, NY. |
|
(24) |
Vamagy, L. (1980) Use of Recent Fetal Bone Staining Techniques in the Evaluation of Pesticida Teratogenicity. Acta Vet. Acad. Sci. Hung. 28; 233-239. |
|
(25) |
Staples, R.E. (1974) Detection of visceral Alterations in Mammalian Foetuses. Teratology 9; 37-38. |
|
(26) |
Van Julsingha, E.B. and C.G. Bennett (1977) A Dissecting Procedure for the Detection of Anomalies in the Rabbit Foetal Head. In: Methods in Prenatal Toxicology Neubert, D., Merker, H.J. and Kwasigroch, T.E. (eds.). University of Chicago, Chicago, IL, pp. 126-144. |
|
(27) |
US Environmental Protection Agency (1991) Guidelines for Developmental Toxicity Risk Assessment. Federal Register 56; 63798-63826. |
|
(28) |
Wise, D.L. et al. (1997) Terminology of Developmental Abnornialities in Common Laboratory Mammals (Version 1) Teratology 55; 249-292. |
---
B.32. ENSAYO DE CARCINOGÉNESIS
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIONES
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
La sustancia objeto de estudio se administra normalmente 7 días a la semana, por una vía apropiada, a varios grupos de animales de experimentación, a razón de una dosis por grupo, durante una parte importante de su existencia. Durante la exposición a la sustancia estudiada, y después de ella, se observa a diario a los animales para apreciar posibles signos de toxicidad, y, en particular, la aparición de tumores.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO PREPARATIVOS
Se mantiene a los animales en las condiciones de alojamiento y alimentación experimentales durante al menos 5 días previos a la prueba. Antes de comenzarla, se distribuye al azar a los animales, que serán jóvenes y sanos, para formar grupos de tratamiento y de control.
Animales de experimentación
La especie preferida es la rata, aunque pueden utilizarse otras (de roedores y no roedores) basándose en los resultados de estudios previos. Deben emplearse animales jóvenes y sanos de cepas de laboratorio de uso habitual, y la administración se iniciará lo antes posible tras el destete.
Al principio del estudio, el peso de los animales no debe diferir en un porcentaje superior al ± 20 % del valor medio. Cuando se practique un estudio oral subcrónico previo a otro de índole crónica, se utilizarán en ambos las mismas especie/raza y cepa.
Número y sexo
En roedores, se utilizarán al menos 100 animales (50 hembras y 50 machos) para cada dosis y grupo de control correspondiente. Las hembras serán nulíparas y no grávidas. Si está previsto el sacrificio de algunos animales durante la prueba, debe aumentarse su número en una cantidad igual a la de los que vayan a sacrificarse antes de terminar el estudio.
Dosis y frecuencia de exposición
Se utilizarán al menos 3 niveles posológicos, además del grupo de control correspondiente. La dosis más alta debe originar signos de toxicidad mínimos, como una leve disminución de la ganancia de peso corporal (inferior al 10 %), sin alterar de manera notable la duración normal de la existencia a causa de efectos distintos de tumores.
La dosis más baja no debe alterar el crecimiento, el desarrollo ni la longevidad normales del animal, ni generar indicio alguno de toxicidad. En general, no será inferior al 10 % de la dosis alta.
La o las dosis intermedias se situarán más o menos en un punto medio entre la alta y la baja.
En la selección de las dosis se tendrán en cuenta datos de pruebas y estudios de toxicidad precedentes.
Normalmente, la exposición será diaria.
Si se administra la sustancia química en el agua de bebida o mezclada con el alimento, deberá estar continuamente disponible.
Controles
Se utilizará un grupo de control idéntico en todos los aspectos a los tratados, salvo por la no exposición a la sustancia probada.
En circunstancias especiales, como las concurrentes en estudios de inhalación con aerosoles o en caso de empleo de un emulgente de actividad biológica no caracterizada en estudios orales, se utilizará otro grupo de control que no se expondrá al vehículo.
Vía de administración
Las tres vías de administración principales son la oral, la dérmica y la respiratoria (inhalación). La elección de una de ellas dependerá de las características físicas y químicas de la sustancia estudiada y de la vía probable de exposición en seres humanos.
1.6.5.1. Estudios orales
Cuando la sustancia estudiada se absorba en el aparato gastrointestinal, y si la ingestión es una vía posible de exposición humana, se preferirá la vía oral de administración, salvo si existen contraindicaciones. Los animales pueden recibir la sustancia con la alimentación, disuelta en el agua de bebida o por medio de cápsulas.
Lo ideal sería la administración los 7 días de la semana, ya que con 5 dosis semanales es posible una recuperación o la reducción de la toxicidad en el período exento de dosis, con la consiguiente influencia en el resultado y la evaluación posterior. Sin embargo, y por motivos esencialmente prácticos, se considera aceptable la administración 5 días a la semana.
1.6.5.2. Estudios dérmicos
Es posible optar por la exposición cutánea mediante aplicación sobre la piel con un pincel para simular una vía importante de exposición del ser humano, así como modo de sistema modelo para la inducción de lesiones cutáneas.
1.6.5.3. Estudios de inhalación
Dado que los estudios de inhalación plantean problemas técnicos de mayor complejidad que las demás vías de administración, ofrecemos aquí una orientación más detallada sobre este modo de administración. Hay que señalar también que la instilación intratraqueal constituye un método válido en situaciones determinadas.
Las exposiciones prolongadas suelen adecuarse a las condiciones de exposición humana proyectadas; así, se expone a los animales 5 días a la semana (exposición intermitente) durante 6 horas diarias tras equilibrado de las concentraciones en la cámara de prueba, o bien los 7 días de la semana (exposición permanente) a una exposición diaria de 22 a 24 horas, dedicando alrededor de una hora a la alimentación de los animales (horario regular) y el mantenimiento de la cámara. En ambos casos, los animales suelen exponerse a concentraciones fijas de las sustancia objeto de estudio. Una diferencia es que con la primera existe un período de 17 a 18 horas en que los animales pueden recuperarse de los efectos de cada exposición diaria, con un período de recuperación aún mayor durante los fines de semana.
La elección de la exposición intermitente o permanente dependerá de los objetivos del estudio y de la exposición humana que pretenda simularse. No obstante, hay que tener en cuenta ciertas dificultades técnicas. Por ejemplo, es posible que las ventajas de la exposición permanente en la simulación de condiciones ambientales queden contrapesadas por la necesidad de suministrar alimento y bebida durante la exposición, así como por la necesidad de técnicas de generación de aerosoles y vapores, y de técnicas de control más complicadas (y fiables).
Cámaras de exposición
Los animales se someterán al estudio en un dispositivo de inhalación capaz de mantener un flujo de aire continuo de al menos 12 renovaciones de aire a la hora, y de garantizar un contenido de oxígeno apropiado y una distribución uniforme del producto estudiado en el aire. Las cámaras de control y de exposición serán de construcción y diseño idénticos para garantizar condiciones de exposición comparables en todos los aspectos, salvo en la exposición a la sustancia probada. Por regla general, se mantiene una ligera presión negativa en el interior de la cámara para impedir el escape de la sustancia estudiada a la zona circundante. Se evitará el hacinamiento de los animales en las cámaras. Como norma general, para garantizar le estabilidad de la atmósfera de la cámara, el volumen total de los animales de experimentación no debe exceder del 5 % del volumen de la cámara empleada.
Se practicarán determinaciones o controles de:
|
i) |
Flujo de aire: el flujo de aire en la cámara se controlará preferiblemente de modo constante. |
|
ii) |
Concentración: durante el período diario de exposición, la concentración de la sustancia estudiada no diferirá en más del ± 15 % del valor medio. Durante todo el estudio, las concentraciones se mantendrán lo más constantes posible de un día a otro. |
|
iii) |
Temperatura y humedad: en roedores, la temperatura se mantendrá a 22 oC (± 2 oC), y la humedad en el interior de la cámara del orden del 30 al 70 %, salvo cuando se emplee agua para suspender la sustancia estudiada en la atmósfera de la cámara. Ambos factores se controlarán preferiblemente de modo continuo. |
|
iv) |
Análisis granulométrico de las partículas: en las atmósferas de la cámara que exijan la utilización de aerosoles líquidos o sólidos, se determinará la distribución de las partículas por tamaños. Las partículas de los aerosoles serán de un tamaño respirable para el animal de experimentación utilizado. Se recogerán muestras de las atmósferas de la cámara en la zona de respiración de los animales. La muestra de aire estará acorde con la distribución de las partículas a las que se exponga a los animales y será representativa, sobre una base gravimétrica, de la totalidad del aerosol en suspensión, incluso aunque gran parte de él no sea respirable. Los análisis granulométricos se efectuarán con frecuencia durante la adaptación del sistema generador para garantizar la estabilidad del aerosol, y con posterioridad cuando se consideren necesarios para determinar debidamente la constancia de la distribución de las partículas a que se expone a los animales. |
Duración del estudio
Una prueba de carcinogénesis debe abarcar la mayor parte del período de existencia de los animales a ella sometidos: 18 meses en ratones y hámsteres y 24 meses en ratas; sin embargo, en determinadas cepas de animales de mayor longevidad o índice de tumoración espontánea bajo, se prolongará hasta 24 meses en ratones y hámsteres y hasta 30 en ratas. También es posible dar por concluido un estudio ampliado cuando el número de supervivientes en el grupo con dosis más baja o de control alcance el 25 %. Cuando en una prueba se observen respuestas aparentemente diferentes en cada sexo, se les estudiará por separado. Si solo fallecieran prematuramente los animales del grupo con dosis alta, por causas evidentes de toxicidad, no es obligada la conclusión del estudio, siempre y cuando las manifestaciones tóxicas no provoquen problemas en los demás grupos. Para considerar aceptable una prueba negativa, es preciso que no se pierda más del 10 % de los animales de cualquier grupo por causa de autólisis, canibalismo o problemas de organización, y que la supervivencia en todos los grupos no sea inferior al 50 %, en ratones y hámsteres a los 18 meses y a los 24 en ratas.
Procedimiento
Observaciones
Las observaciones diarias de los animales en sus jaulas deben incluir los cambios en la piel y el pelo, los ojos y las membranas mucosas, así como en los sistemas respiratorio, circulatorio, nervioso autónomo y central, la actividad somatomotriz y el comportamiento.
Son necesarias observaciones periódicas de los animales para evitar en lo posible la pérdida de alguno de ellos por causa de canibalismo, autólisis de tejidos o enjaulamiento erróneo. Los animales moribundos se retirarán y someterán a autopsia.
Se registrarán los signos clínicos y la mortalidad para todos los animales. Ha de prestarse una atención especial a la tumorogénesis; se registrarán el momento de aparición y la localización, dimensiones, aspecto y progresión de todo tumor visible macroscópica o palpablemente.
Se determinará el consumo de alimento (y el de agua cuando se administre en ella la sustancia estudiada) semanalmente durante las 13 primeras semanas del estudio, y luego a intervalos aproximadamente trimestrales, salvo si aconsejaran otra cosa el estado de salud o las alteraciones del peso corporal.
Se determinará y anotará el peso de cada animal una vez a la semana durante las 13 primeras del período de prueba, y al menos una vez cada 4 semanas con posterioridad.
Exámenes clínicos
Hematología
Si las observaciones indicaran un menoscabo de la salud de los animales durante el estudio, se determinará la fórmula leucocitaria de los animales afectados.
A los 12 y 18 meses y antes del sacrificio, se obtiene de los animales un frotis sanguíneo. Se determinará la fórmula leucocitaria en muestras de los animales de los grupos con dosis máxima y de control. Si estos datos, especialmente los obtenidos antes del sacrificio, o los obtenidos del examen histopatológico así lo aconsejan, se determinarán también las fórmulas leucocitarias en el grupo o grupos inmediatamente inferiores.
Autopsia
Se practicará una autopsia completa en todos los animales, incluidos los fallecidos durante el experimento o sacrificados al encontrarse moribundos. Se conservarán todos los tumores o lesiones macroscópicas visibles, así como las lesiones que se sospeche son tumores.
Se conservarán en medios adecuados para un posible examen histopatológico los órganos y tejidos siguientes: cerebro (bulbo/protuberancia, corteza cerebelosa y encefálica), hipófisis, tiroides, paratiroides, timo, pulmones y tráquea, corazón, aorta, glándulas salivares, hígado, bazo, riñones, suprarrenales, páncreas, gónadas, útero, órganos genitales secundarios, piel, esófago, estómago, duodeno, yeyuno, íleon, ciego, colon, recto, vejiga urinaria, ganglio linfático representativo, glándula mamaria femenina, musculatura del muslo, nervio periférico, médula espinal (cervical, dorsal, lumbar), esternón con médula ósea, fémur (incluida articulación) y ojos.
El inflado de pulmones y vejiga urinaria con un fijador es el medio óptimo de conservación de estos tejidos; por otro lado, el inflado de los pulmones es esencial en los estudios de inhalación para efectuar un examen histopatológico apropiado. En estudios especiales, como los de inhalación, se estudiará la totalidad del aparato respiratorio, incluidas nariz, faringe y laringe.
Examen histopatológico
|
a) |
Se realizará un examen histopatológico completo de los órganos y tejidos de todos los animales fallecidos o sacrificados durante la prueba, y de los miembros de los grupos de control y con la dosis máxima. |
|
b) |
Se examinarán al microscopio en todos los grupos todos los tumores macroscópicos visibles o las lesiones que se sospeche son tumores. |
|
c) |
Si existe una diferencia significativa en la incidencia de lesiones neoplásicas entre el grupo con dosis máxima y el de control, se practicará un examen histopatológico sobre el órgano o tejido de que se trate en los demás grupos. |
|
d) |
Si la supervivencia del grupo con dosis máxima es notablemente inferior a la observada en el de control, se examinará detenidamente el grupo con dosis inmediatamente inferior. |
|
c) |
Si se apreciaran en el grupo con dosis máxima indicios de la inducción de efectos tóxicos o de otro tipo capaces de influir en una respuesta neoplásica se examinará detenidamente el grupo con dosis immediatamente inferior. |
2. RESULTADOS
Los resultados se resumirán en forma de tabla, y mostrarán para cada grupo de prueba el número de animales al comienzo del ensayo, el de los que han mostrado tumores apreciables durante el mismo, el momento del descubrimiento y el número de animales en que se encontraron tumores después del sacrificio. Los resultados se evaluarán por un método estadístico apropiado. Puede usarse cualquier método estadístico reconocido.
3. INFORME
3.1. DATOS DEL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
especie, cepa, origen, condiciones ambientales, alimentación, |
|
— |
condiciones de la prueba: Descripción del dispositivo de exposición, incluidos diseño, tipo, dimensiones, fuente de aire, sistema de generación de partículas y aerosoles, método de acondicionamiento del aire, tratamiento del aire evacuado y, cuando se emplee, método de alojamiento de los animales en la cámara de prueba. Se describirá el equipo empleado para determinar la temperatura, la humedad y, en su caso, la estabilidad de las concentraciones de aerosol o el tamaño de las partículas. Datos de exposición: Se presentarán en forma de tabla en la que figuren tanto los valores medios como una medida de la variabilidad (por ejemplo, desviación estándar), y comprenderán:
|
|
— |
dosis (incluido el vehículo, si se emplea) y concentraciones, |
|
— |
datos de la incidencia de tumores en función del sexo, la dosis y el tipo tumoral, |
|
— |
momento de la muerte durante el estudio, o indicación de que los animales sobrevivieron a la experiencia, |
|
— |
datos de respuesta tóxica en función del sexo y la dosis, |
|
— |
descripción de los efectos tóxicos o de otro tipo, |
|
— |
momento de observación de cada signo anómalo y evolución de este, |
|
— |
datos sobre alimentación y peso, |
|
— |
pruebas hematológicas practicadas y sus resultados completos, |
|
— |
hallazgos de autopsia, |
|
— |
descripción detallada de los hallazgos histopatológicos, |
|
— |
tratamiento estadístico de los resultados, con descripción de los métodos empleados, |
|
— |
comentario de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.33. ENSAYO COMBINADO DE TOXICIDAD CRÓNICA Y CARCINOGÉNESIS
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIONES
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
El objetivo de una prueba combinada de toxicidad crónica y carcinogénesis es determinar los efectos crónicos y cancerígenos de una sustancia en una especie de mamífero tras exposición prolongada.
A este fin, se completa una prueba de carcinogénesis con un grupo satélite tratado y otro de control, por lo menos. La dosis empleada para el grupo satélite con dosis máxima puede ser superior a la utilizada para idéntico grupo en la prueba de carcinogénesis. Los animales de la prueba de carcinogénesis se examinan tanto en busca de signos de toxicidad general como de una respuesta cancerígena. Los animales del grupo satélite tratado se examinan en busca de signos de toxicidad general.
La sustancia objeto de estudio se administra normalmente 7 días a la semana, por una vía apropiada, a varios grupos de animales de experimentación, a razón de una dosis por grupo, durante una parte importante de su existencia. Durante la exposición a la sustancia estudiada, y después de ella, se observa a diario a los animales para apreciar posibles signos de toxicidad.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
Se mantiene a los animales en las condiciones de alojamiento y alimentación experimentales durante al menos 5 días previos a la prueba. Antes de comenzarla, se distribuye al azar a los animales, que serán jóvenes y sanos, para formar grupos de tratamiento y de control.
Animales de experimentación
La especie preferida es la rata, aunque pueden utilizarse otras (roedores o no roedores) basándose en los resultados de estudios previos. Deben emplearse animales jóvenes y sanos de cepas de laboratorio de uso habitual, y la administración se iniciará lo antes posible tras el destete.
Al principio del estudio, el peso de los animales no debe diferir en un porcentaje superior al ± 20 % del valor medio. Cuando se practique un estudio oral subcrónico previo a otro de índole crónica, se utilizarán en ambos las mismas especie/raza y cepa.
Número y sexo
En roedores, se utilizarán al menos 100 animales (50 hembras y 50 machos) para cada dosis y grupo de control correspondiente. Las hembras serán nulíparas y no grávidas. Si está previsto el sacrificio de algunos animales durante la prueba, debe aumentarse su número en una cantidad igual a la de los que vayan a sacrificarse antes de terminar el estudio.
El grupo o grupos satélites tratados para la evaluación de alteraciones patológicas distintas de los tumores contendrán 20 animales de cada sexo, en tanto que el grupo de control satélite constará de 10 animales de cada sexo.
Dosis y frecuencia de exposición
Para el estudio de la carcinogénesis se utilizarán al menos tres niveles posológicos, además del grupo de control correspondiente. La dosis más alta debe originar signos de toxicidad mínima, como una disminución en la ganancia de peso corporal (inferior al 10 %), sin alterar en medida notable la duración normal de la existencia por causa de efectos distintos de tumores.
La dosis más baja no debe alterar el crecimiento, el desarrollo ni la longevidad normales del animal, ni generar indicio alguno de toxicidad. En general, no será inferior al 10 % de la dosis máxima.
La o las dosis intermedias se situarán más o menos en un punto medio entre la alta y la baja.
En la selección de las dosis se tendrán en cuenta datos de pruebas y estudios de toxicidad precedentes.
Para estudiar la toxicidad crónica, se incluirán en la prueba grupos tratados adicionales y un grupo satélite de control correspondiente. La dosis máxima de los animales satélites tratados producirá signos definidos de toxicidad.
Normalmente, la frecuencia de exposición será diaria.
Si se administra la sustancia química en el agua de bebida o mezclada con el alimento, deberá estar continuamente disponible.
Controles
Se utilizará un grupo de control idéntico en todos los aspectos a los tratados, salvo por la no exposición a la sustancia probada.
En circunstancias especiales, como las concurrentes en estudios de inhalación con aerosoles o en caso de empleo de un emulgente de actividad biológica no caracterizada en estudios orales, se utilizará también un grupo de control negativo concurrente. Este grupo de control negativo se tratará de igual manera que los grupos de prueba, salvo en que los animales no se expondrán a la sustancia estudiada ni a ningún vehículo.
Vía de administración
Las tres vías de administración principales son la oral, la dérmica y la respiratoria (inhalación). La elección de una de ellas dependerá de las características físicas y químicas de la sustancia estudiada y de la vía probable de exposición en seres humanos.
Estudios orales
Cuando la sustancia estudiada se absorba en el aparato gastrointestinal, se preferirá la vía oral de administración. Los animales pueden recibir la sustancia con la alimentación, disuelta en el agua de bebida o por medio de cápsulas.
Lo ideal sería la administración los 7 días de la semana, ya que con 5 dosis a la semana es posible una recuperación o la reducción de la toxicidad en el período exento de dosis, con la consiguiente influencia en el resultado y la evaluación posterior. Sin embargo, y por motivos esencialmente prácticos, se considera aceptable la administración 5 días a la semana.
Estudios dérmicos
Es posible optar por la exposición cutánea mediante aplicación sobre la piel con un pincel para simular una vía importante de exposición del ser humano, así como a modo de sistema modelo para la inducción de lesiones cutáneas.
Estudios de inhalación
Dado que los estudios de inhalación plantean problemas técnicos de mayor complejidad que las demás vías de administración, ofrecemos aquí una orientación más detallada sobre este modo de administración. Hay que señalar también que la instilación intratraqueal constituye un método válido en situaciones determinadas.
Las exposiciones prolongadas suelen adecuarse a las condiciones de exposición humana proyectadas; así, se expone a los animales 5 días a la semana (exposición intermitente) durante 6 horas diarias tras equilibrado de las concentraciones en la cámara de prueba, o bien los 7 días de la semana (exposición permanente) a una exposición diaria de 22 a 24 horas, dedicando alrededor de una hora a la alimentación de los animales (horario regular) y el mantenimiento de la cámara. En ambos casos, los animales suelen exponerse a concentraciones fijas de la sustancia objeto de estudio. Una diferencia importante entre la exposición intermitente y la permanente es que con la primera existe un período de 17 a 18 horas en que los animales pueden recuperarse de los efectos de cada exposición diaria, con un período de recuperación aún mayor durante los fines de semana.
La elección de la exposición intermitente o permanente dependerá de los objetivos del estudio y de la exposición humana que pretenda simularse. No obstante, hay que tener en cuenta ciertas dificultades técnicas. Por ejemplo, es posible que las ventajas de la exposición permanente en la simulación de condiciones ambientales queden contrapesadas por la necesidad de suministrar alimento y bebida durante la exposición, así como por la necesidad de técnicas de generación de aerosoles y vapores, y de técnicas de control, más complicadas (y fiables).
Cámaras de exposición
Los animales se someterán al estudio en un dispositivo de inhalación capaz de mantener un flujo de aire continuo de al menos 12 renovaciones de aire a la hora, y de garantizar un contenido de oxígeno apropiado y una distribución uniforme del producto estudiado en el aire. Las cámaras de control y de exposición serán de construcción y diseño idénticos para garantizar condiciones de exposición comparables en todos los aspectos, salvo en la exposición a la sustancia probada. Por regla general, se mantiene una ligera presión negativa en el interior de la cámara para impedir el escape de la sustancia estudiada a la zona circundante. Se evitará el hacinamiento de los animales en las cámaras. Como norma general, para garantizar la estabilidad de la atmósfera de la cámara, el volumen total de los animales de experimentación no debe exceder del 5 % del volumen de la cámara empleada.
Se practicarán determinaciones o controles de:
|
i) |
Flujo del aire: el flujo de aire en la cámara se controlará preferiblemente de modo constante. |
|
ii) |
Concentración: durante el período diario de exposición, la concentración de la sustancia estudiada no diferirá en más del ± 15 % del valor medio. |
|
iii) |
Temperatura y humedad: en roedores, la temperatura se mantendrá a 22 oC (± 2 oC), y la humedad en el interior de la cámara del orden del 30 al 70 %, salvo cuando se emplee agua para suspender la sustancia estudiada en la atmósfera de la cámara. Ambos factores se controlarán preferiblemente de modo continuo. |
|
iv) |
Análisis granulométrico de las partículas: en las atmósferas de la cámara que exijan la utilización de aerosoles líquidos o sólidos, se determinará la distribución de las partículas por tamaños. Las partículas de los aerosoles serán de un tamaño respirable para el animal de experimentación utilizado. Se recogerán muestras de las atmósferas de la cámara en la zona de respiración de los animales. La muestra de aire estará acorde con la distribución de las partículas a las que se exponga a los animales y será representativa, sobre una base gravimétrica, de la totalidad del aerosol en suspensión, incluso aunque gran parte de este no sea respirable. Los análisis granulométricos se efectuarán con frecuencia durante la adaptación del sistema generador para garantizar la estabilidad del aerosol, y con posterioridad cuando se consideren necesarios para determinar debidamente la constancia de la distribución de las partículas a que se expone a los animales. |
Duración del estudio
La parte de la prueba dedicada a la carcinogénesis debe abarcar la mayor parte del período de existencia de los animales a ella sometidos: 18 meses en ratones y hámsteres, y 24 meses en ratas; sin embargo, en determinadas cepas de animales de mayor longevidad o de índice de tumoración espontánea bajo, se prolongará hasta los 24 meses en ratones y hámsteres, y los 30 en ratas. También es posible dar por concluido un estudio ampliado cuando el número de supervivientes en el grupo con dosis más baja o de control alcance el 25 %. Cuando en una prueba se observen respuestas aparentemente diferentes en cada sexo, se les estudiará por separado. Si solo fallecieran prematuramente los animales del grupo con dosis alta por causas evidentes de toxicidad, no es obligada la conclusión del estudio, siempre y cuando las manifestaciones tóxicas no provoquen problemas en los demás grupos. Para considerar aceptable una prueba negativa, es preciso que no se pierda más del 10 % de los animales de cualquier grupo por causa de autólisis, canibalismo o problemas de organización, y que la supervivencia en todos los grupos no sea inferior al 50 % a los 18 meses en ratones y hámsteres, y a los 24 en ratas.
Los grupos satélites de 20 animales tratados por sexo y 10 animales de control asociados por sexo, utilizados para estudiar la toxicidad crónica, se mantendrán en la prueba durante al menos 12 meses. Se programará el sacrificio de estos animales para determinar la posible existencia de patología relacionada con la sustancia estudiada y no complicada por alteraciones gerontológicas.
Procedimiento
Observaciones
Los animales deben observarse diariamente en sus jaulas, comprobando los cambios en la piel y el pelo, los ojos y las membranas mucosas así como en los sistemas respiratorio, circulatorio, nervioso autónomo y central, la actividad somatomotriz y el comportamiento.
Se efectuarán exploraciones clínicas a intervalos apropiados en los animales de los grupos satélites tratados.
Son necesarias observaciones periódicas de los animales para evitar en lo posible la pérdida de alguno por causa de canibalismo, autólisis de tejidos o enjaulamiento erróneo. Los animales moribundos se retirarán y someterán a autopsia.
Se anotarán para todos los animales los signos clínicos, incluidas tanto las alteraciones neurológicas y oculares como la mortalidad. Ha de prestarse una atención especial a la tumorogénesis; se registrarán el momento de aparición y la localización, dimensiones, aspecto y progresión de todo tumor visible macroscópico o palpable; se registrarán asimismo el momento de comienzo y la evolución de los procesos tóxicos.
Se determinará el consumo de alimento (y el de agua cuando se administre en ella la sustancia estudiada) semanalmente durante las 13 primeras semanas del estudio, y luego a intervalos aproximadamente trimestrales, salvo si aconsejaran otra actitud el estado de salud o las alteraciones del peso corporal.
Se determinará y anotará el peso de cada animal una vez a la semana durante las 13 primeras del período de prueba, y al menos una vez cada 4 semanas con posterioridad.
Exámenes clínicos
Hematología
Se realizará un examen hematológico (por ejemplo, contenido de hemoglobina, hematocrito, recuentos de hematíes y leucocitos, plaquetas u otras medidas de la capacidad de coagulación) a los 3 y los 6 meses, después con intervalos aproximados de 6 meses y, al final, sobre muestras de sangre recogidas de todos los no roedores y de 10 ratas/sexo de todos los grupos. A ser posible, las muestras procederán de las mismas ratas en cada intervalo.
Si las observaciones clínicas indican un menoscabo de la salud de los animales durante el estudio, se practicará un recuento con fórmula leucocitaria de los animales afectados. Se averiguará la fórmula leucocitaria en muestras de los animales del grupo con dosis máxima y de los controles. Solo se determinará en los grupos con dosis menores si se aprecia una discrepancia importante entre el grupo con dosis máxima y los controles, o si lo aconsejan los hallazgos patológicos.
Análisis de orina
Se recogerán para su análisis muestras de orina de todos los animales no roedores y de 10 ratas/sexo de todos los grupos, a ser posible de las mismas ratas que sirvan para el examen hematológico y respetando intervalos idénticos. Se realizarán las determinaciones siguientes en animales individuales, o en una muestra acumulada sexo/grupo en el caso de los roedores:
|
— |
aspecto: volumen y densidad en cada animal, |
|
— |
proteínas, glucosa, cetonas, sangre oculta (semicuantitativamente), |
|
— |
microscopía del sedimento (semicuantitativamente). |
Bioquímica
Con intervalos aproximados de 6 meses y a la conclusión, se extraen muestras de sangre para determinaciones de bioquímica de todos los no roedores y de 10 ratas/sexo de todos los grupos, a ser posible de las mismas ratas en cada intervalo. Además, se recogerá en todos los no roedores una muestra previa a la prueba. Se prepara plasma a partir de estas muestras, y se realizan las determinaciones siguientes:
|
— |
concentración de proteínas totales, |
|
— |
concentración de albúmina, |
|
— |
pruebas de función hepática (como actividad de fosfatasa alcalina, transaminasa glutamicopirúvica (4) y transaminasa glutamicooxaloacética (5), gammaglutamiltranspeptidasa, ornitindescarboxilasa, |
|
— |
metablismo de carbohidratos, como glucemia en ayunas, |
|
— |
pruebas de función renal, como nitrógeno ureico en sangre. |
Autopsia
Se practicará una autopsia completa en todos los animales, incluidos los fallecidos durante el experimento o sacrificados al encontrarse moribundos. Antes del sacrificio, se recogerán de todos los animales muestras de sangre para la realización de recuentos sanguíneos con fórmula leucocitaria. Se conservarán todas las lesiones macroscópicas visibles, así como los tumores o lesiones que se sospeche son tumores. Se intentarán relacionar las observaciones macroscópicas con los hallazgos microscópicos.
Se conservarán para examen histopatológico todos los órganos y tejidos, habitualmente los siguientes: cerebro (7) (bulbo/protuberancia, corteza cerebelosa y encefálica), hipófisis, tiroides, (incluida paratiroides), timo, pulmones (incluida tráquea), corazón, aorta, glándulas salivares, hígado (7), bazo, riñones (7), suprarrenales (7), esófago, estómago, duodeno, yeyuno, íleon, ciego, colon, recto, útero, vejiga urinaria, ganglios linfáticos, páncreas, gónadas (7), órganos genitales accesorios, glándula mamaria femenina, piel, musculatura del muslo, nervio periférico, médula espinal (cervical, dorsal, lumbar), esternón con médula ósea y fémur (incluida articulación) y otros.
Aunque el inflado de pulmones y vejiga urinaria con un fijador es el método óptimo de conservación de estos tejidos, el inflado de los pulmones es esencial en los estudios de inhalación para efectuar un examen histopatológico apropiado. En estudios especiales, como los de inhalación, se examinará la totalidad del aparato respiratorio, incluidas nariz, faringe y laringe.
Si se realizan otros exámenes clínicos, la información con ellos obtenida deberá estar disponible antes del examen microscópico, ya que puede suponer una orientación importante para el anatomopatólogo.
Examen histopatológico
En el apartado de toxicidad crónica:
Se practicará un examen detallado de los órganos conservados de todos los animales de los grupos satélites con dosis máxima y de control. Si se encontrara patología relacionada con la sustancia estudiada en el grupo satélite con dosis máxima, se someterán a un examen histopatológico completo y detallado los órganos efectores de los demás animales de cualquier otro grupo satélite tratado, así como los de los grupos tratados del apartado de carcinogénesis del estudio, a la conclusión de este.
En el apartado de carcinogénesis:
|
a) |
Se realizará un examen histopatológico completo de los órganos y tejidos de todos los animales fallecidos o sacrificados durante la prueba, y de los miembros de los grupos de control y con la dosis máxima. |
|
b) |
Se examinarán al microscopio, en todos los grupos, todos los tumores macroscópicos visibles o las lesiones, que se sospeche son tumores, aparecidas en cualquier órgano. |
|
c) |
Si existe una diferencia significativa en la incidencia de lesiones neoplásicas entre el grupo con dosis máxima y el de control, se practicará un examen histopatológico sobre el órgano, o tejido de que se trate, en los demás grupos. |
|
d) |
Si la supervivencia del grupo con dosis máxima es notablemente inferior a la observada en el de control, se examinará detenidamente el grupo con dosis inmediatamente inferior. |
|
e) |
Si se apreciaran en el grupo con dosis máxima indicios de la inducción de efectos tóxicos, o de otro tipo, capaces de influir en una respuesta neoplásica, se examinará detenidamente el grupo con dosis inmediatamente inferior. |
2. RESULTADOS
Los resultados se resumirán en forma de tabla y mostrarán, para cada grupo de prueba, el número de animales al comienzo del ensayo, el de los que han mostrado tumores apreciables durante el mismo, el momento del descubrimiento y el número de animales en que se encontraron tumores después del sacrificio. Los resultados se evaluarán por un método estadístico apropiado. Puede usarse cualquier método estadístico reconocido.
3. INFORME
3.1. DATOS DEL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
especie, cepa, origen, condiciones ambientales, alimentación, |
|
— |
condiciones de la prueba: Descripción del dispositivo de exposición, incluidos diseño, tipo, dimensiones, fuente de aire, sistema de generación de partículas y aerosoles, método de acondicionamiento del aire, tratamiento del aire evacuado y, cuando se emplee, método de alojamiento de los animales en la cámara de prueba. Se describirá el equipo empleado para determinar la temperatura, la humedad y, en su caso, la estabilidad de las concentraciones de aerosol o el tamaño de las partículas. Datos de exposición: Se presentarán en forma de tabla en la que figuren tanto los valores medios como una medida de la variabilidad (por ejemplo, desviación estándar), y comprenderán:
|
|
— |
dosis (incluido el vehículo, si se emplea) y concentraciones, |
|
— |
datos de la incidencia de tumores en función del sexo, la dosis y el tipo tumoral, |
|
— |
momento de la muerte durante el estudio, o indicación de que los animales sobrevivieron a la experiencia, |
|
— |
datos de la respuesta tóxica, por sexo y dosis, |
|
— |
descripción de los efectos tóxicos o de otro tipo, |
|
— |
momento de observación de cada signo anómalo y evaluación de este, |
|
— |
hallazgos oftalmológicos, |
|
— |
datos sobre alimentación y peso, |
|
— |
pruebas hematológicas practicadas y sus resultados completos, |
|
— |
pruebas de bioquímica clínica empleadas y sus resultados completos (incluidos los del análisis de orina, si procede), |
|
— |
hallazgos de autopsia, |
|
— |
descripción detallada de los hallazgos histopatológicos, |
|
— |
tratamiento estadístico de los resultados, si es posible, |
|
— |
comentario de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.34. ENSAYO DE REPRODUCCIÓN EN UNA GENERACIÓN
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIONES
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
Se administra la sustancia objeto de estudio, en dosis crecientes, a varios grupos de machos y hembras. Los machos deberán tratarse en la fase de crecimiento y durante al menos un ciclo de espermatogénesis completo (unos 56 días en el ratón y 70 días en la rata) para que la sustancia estudiada pueda provocar algún efecto adverso en la espermatogénesis.
Las hembras de la generación parental (P) recibirán tratamiento durante dos ciclos estrales completos, por lo menos, para que la sustancia estudiada pueda provocar algún efecto adverso en el estro. A continuación, se aparea a los animales. La sustancia ensayada se administra a los dos sexos durante el período de apareamiento, y luego únicamente a las hembras en los períodos de gestación y lactancia. El método deberá modificarse si se pretende administrar la sustancia por inhalación.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
Preparativos
Antes de la prueba, se distribuye al azar a los animales, que serán jóvenes y sanos, para formar grupos de tratamiento y de control. Los animales se mantienen en las condiciones de alojamiento y alimentación experimentales durante al menos 5 días previos a la prueba. Se recomienda la administración de la sustancia estudiada con la alimentación o en el agua de bebida, pero también son aceptables otras vías de administración. Todos los animales se tratarán por el mismo método durante el período experimental apropiado. Si se utiliza un vehículo u otros aditivos para facilitar la administración, estos no deberán ser tóxicos. Se administrará la sustancia los 7 días de la semana.
Animales de experimentación
Las especies preferidas son la rata y el ratón.
Han de utilizarse animales sanos, no sometidos a experimentación previa. No se utilizarán cepas de baja fecundidad. Se especificará la especie, la cepa, el sexo, el peso y la edad de los animales empleados.
Para evaluar debidamente la fecundidad, se estudiará tanto a los machos como a las hembras. Todos los animales tratados y de control deberán estar destetados antes del comienzo del tratamiento.
Número y sexo
Cada grupo tratado y de control comprenderá un número de animales suficiente para obtener unas 20 hembras grávidas a término o cerca de él.
El objetivo es conseguir gestaciones y progenie suficientes para permitir una evaluación significativa de la influencia de la sustancia en la fertilidad, la gestación y el comportamiento materno de los animales de la generación P así como en la lactancia, el crecimiento y el desarrollo de la generación F, desde la concepción al destete.
Condiciones del ensayo
Se suministrarán alimento y agua ad libitum. Al acercarse el momento del parto, se aislará a las hembras grávidas en jaulas individuales para partos o de maternidad, y pueden suministrárseles materiales de nidificación.
Dosis
Se utilizarán al menos tres grupos tratados y uno de control. Si se utiliza un vehículo para administrar la sustancia probada, el grupo de control recibirá este vehículo en el volumen máximo empleado. Si una sustancia objeto de estudio causa una reducción de la ingesta o del aprovechamiento de la alimentación, puede considerarse necesario el uso de un grupo de control emparejado. Lo ideal sería que, a menos que lo impidan la naturaleza física/química, o los efectos biológicos de la sustancia estudiada, la dosis máxima provoque toxicidad, pero no mortalidad, en los animales paternos (P). La o las dosis intermedias generarán efectos tóxicos mínimos atribuibles a la sustancia ensayada, y la dosis mínima no inducirá efectos adversos observables ni en los progenitores ni en la descendencia. Cuando la sustancia se administre por alimentación forzada o en cápsulas, la dosis dada a cada animal deberá basarse en el peso de cada uno y adaptarse semanalmente a las modificaciones que experimente. En las hembras grávidas, el tratamiento puede establecerse, si se desea, en función del peso corporal en el día 0 o 6 de gestación.
Prueba de límite
Cuando se trate de una sustancia dé toxicidad escasa, si una dosis de al menos 1 000 mg/kg no produce signo de alteración del rendimiento reproductor, serán necesarios estudios con otras dosis. Si un estudio preliminar con la dosis máxima muestra signos claros de toxicidad materna, pero ningún efecto adverso en la fertilidad, serán necesarios estudios con otras dosis.
Procedimiento
Planes experimentales
La administración diaria de la sustancia a los progenitores machos (P) se iniciará cuando tengan unas 5 a 9 semanas de edad, previo destete y aclimatación durante al menos 5 días. En las ratas, el tratamiento continúa durante 10 semanas antes del período de apareamiento (8 semanas en ratones). Los machos se sacrificarán y examinarán al final del período de apareamiento, o bien se les mantendrá con vida y en tratamiento por si se considerara conveniente la producción de una segunda camada; se les sacrificará y examinará en algún momento antes de finalizar el estudio. En las hembras progenitoras (P), la administración se iniciará después de 5 días de aclimatación, por lo menos, y continuará durante al menos 2 semanas antes del apareamiento. El tratamiento diario de las hembras P proseguirá durante todo el período de apareamiento de 3 semanas, en la gestación y hasta el destete de la generación F1. Cabe considerar la introducción de modificaciones del esquema posológico si se dispone de otros datos sobre la sustancia estudiada, como la inducción del metabolismo o la bioacumulación.
Método de apareamiento
En los estudios de toxicidad para la reproducción puede utilizarse apareamiento 1:1 (1 macho con 1 hembra) o 1:2 (1 macho con 2 hembras).
Si el apareamiento es 1:1, cada hembra se colocará con el mismo macho hasta que exista gestación o hayan transcurrido 3 semanas. Se examinará todas las mañanas a las hembras para determinar la presencia de esperma o tapones vaginales. Se considera día 0 de la gestación aquel en que se encuentre un tapón vaginal o esperma.
Las parejas que no se apareen se examinarán para determinar la causa de la infertilidad aparente.
Para ello, cabe recurrir a métodos como nuevas oportunidades de aparearse con machos o hembras que ya hayan procreado, examen microscópico de los órganos reproductores y examen del ciclo estral o de la espermatogénesis.
Tamaño de la camada
Se permitirá a los animales tratados durante el estudio de fertilidad parir naturalmente y criar a su camada libremente hasta el destete.
Cuando se recurra a un método de homogeneización de las camadas, se sugiere la técnica siguiente. Entre los días 1 y 4 tras el nacimiento, puede adaptarse el tamaño de cada camada mediante la eliminación por selección de las crías sobrantes para obtener, en la medida de lo posible, 4 machos y 4 hembras por camada.
Cuando el número de crías machos y hembras impida lograr que cada camada cuente con 4 de cada sexo, es aceptable una adaptación parcial (por ejemplo, 5 machos y 3 hembras). Los ajustes no serán posibles con camadas de menos de 8 crías.
Observaciones
Se observará a cada animal al menos una vez al día durante la totalidad del período de prueba. Se anotarán los cambios de conducta pertinentes, los signos de parto difícil o prolongado y todos los signos de toxicidad, incluida la mortalidad. En los períodos de preapareamiento y de apareamiento, puede determinarse a diario el consumo de alimento. Tras el parto y durante la lactancia, se determinará el consumo de alimento (o de agua cuando la sustancia en estudio se administre en el agua de bebida) en los mismos días en que se pesen las camadas. Los machos y hembras P se pesarán el primer día de tratamiento, y luego semanalmente. Estas observaciones se anotarán por separado para cada animal adulto.
La duración de la gestación se calculará a partir del día 0 de gestación. Cada camada se examinará lo antes posible tras el alumbramiento para establecer el número y sexo de las crías, las nacidas muertas, las vivas y la presencia de anomalías macroscópicas.
Las crías muertas y las sacrificadas en el día 4 se conservarán y estudiarán en busca de posibles defectos. Se contarán las crías vivas y se pesarán las camadas la mañana siguiente al nacimiento, los días 4 y 7 siguientes y, por fin, semanalmente hasta la conclusión del estudio, momento en que debe pesarse por separado a los animales.
Se registrarán las anomalías físicas o de conducta observadas en las madres o su progenie.
Patología
Autopsia
Cuando los animales de la generación P se sacrifiquen, o si han muerto a lo largo del estudio, se examinarán al microscopio en busca de anomalías estructurales o alteraciones patológicas, prestando una atención especial a los órganos del sistema reproductor. Las crías muertas o moribundas se examinarán por si sufrieran malformaciones.
Examen histopatológico
Se conservarán para examen microscópico ovarios, útero, cérvix, vagina, testículos, epidídimos, vesículas seminales, próstata, glándula coagulante, hipófisis y órganos efectores de todos los animales P. En caso de que estos órganos no se hayan examinado en otros estudios con varias dosis, se estudiarán al microscopio en todos los animales con dosis máxima y en los controles y en los animales que mueran durante el estudio, siempre que sea posible.
Se examinarán entonces, en todos los demás animales P, los órganos que muestren anomalías en aquellos. En estos casos, se practicará examen microscópico de todos los tejidos que muestren alteraciones patológicas macroscópicas. Como se ha indicado al exponer los métodos de apareamiento, pueden someterse a examen microscópico los órganos reproductores de los animales que se sospeche sufren esterilidad.
2. RESULTADOS
Los resultados se resumirán en forma de tabla y mostrarán, para cada grupo de prueba, el número de animales al comienzo del ensayo, el de machos fértiles, el de hembras grávidas, los tipos de alteraciones y el porcentaje de animales que mostraban cada alteración,
Cuando sea posible, los resultados numéricos se enumerarán por un método estadístico apropiado. Puede usarse cualquier método estadístico reconocido.
3. INFORME
3.1. DATOS DEL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
especie y cepa utilizada, |
|
— |
datos de respuesta tóxica en función del sexo y la dosis, incluidos los índices de fertilidad, gestación y viabilidad, |
|
— |
momento de la muerte durante el estudio, o indicación de que los animales sobrevivieron hasta el día previstopara el sacrificio al final del estudio, |
|
— |
tabla en la que aparezcan los pesos de cada camada, los pesos medios de las crías y los pesos de las distintas crías tras concluir el estudio, |
|
— |
efectos tóxicos o de otro tipo sobre la reproducción, la progenie y el crecimiento postnatal, |
|
— |
día de observación de cada signo anómalo y su evolución, |
|
— |
datos de peso de los animales P, |
|
— |
hallazgos de autopsia, |
|
— |
descripción detallada de todos los hallazgos microscópicos, |
|
— |
tratamiento estadístico de los resultados, cuando proceda, |
|
— |
comentario de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
B.35. ESTUDIO DE TOXICIDAD PARA LA REPRODUCCIÓN EN DOS GENERACIONES
1. MÉTODO
El presente método reproduce las directrices de ensayo de la OCDE TG 416 (2001).
1.1. INTRODUCCIÓN
El presente método de ensayo de toxicidad para la reproducción en dos generaciones tiene por objeto proporcionar información general sobre los efectos de una sustancia de ensayo en la integridad y funcionamiento del aparato reproductor del macho y la hembra, en particular, la función gonadal, ciclo estrual, comportamiento relativo al apareamiento, concepción, gestación, parto, lactancia y destete, así como el desarrollo y crecimiento de la progenie. El estudio también puede mostrar los efectos de la sustancia de ensayo en la morbilidad y mortalidad neonatales, proporcionar datos preliminares sobre la toxicidad para el desarrollo prenatal y postnatal y servir de orientación para ensayos posteriores. Además de estudiar el desarrollo y crecimiento de la generación F1, el ensayo tiene por objeto evaluar la integridad y el funcionamiento del aparato reproductor del macho y la hembra, así como el desarrollo y crecimiento de la generación F2. Puede obtenerse mayor información sobre la toxicidad para el desarrollo y las deficiencias funcionales completando el presente protocolo mediante estudios descritos en los ensayos de toxicidad para el desarrollo y/o neurotoxicidad para el desarrollo, según proceda, o estudiando dichos aspectos de forma independiente empleando métodos de ensayo apropiados.
1.2. PRINCIPIO DEL MÉTODO DE ENSAYO
Se administra la sustancia de ensayo en dosis escalonadas a varios lotes de machos y hembras. En el caso de los machos de la generación P (generación parental), se administra a lo largo del crecimiento y al menos durante un ciclo de espermatogénesis completo (unos 56 días en el ratón y 70 en la rata) para poder observar los posibles efectos adversos sobre la espermatogénesis. Los efectos en el esperma se valoran mediante diversos parámetros (morfología y motilidad de los espermatozoides, etc.), preparaciones tisulares y análisis histopatológico exhaustivo. Si se dispone de datos relativos a la espermatogénesis obtenidos en estudios previos con dosis repetidas de duración suficiente (por ejemplo, 90 días), no será preciso incluir los machos de la generación P en la evaluación. No obstante, se recomienda conservar muestras o grabaciones digitales del esperma de esta generación para su posterior evaluación. La sustancia de ensayo se administra a las hembras de la generación P durante el crecimiento y a lo largo de varios ciclos estruales completos para detectar todos los efectos adversos en el ciclo estrual. Asimismo, se administra a los animales de la generación P durante el período de apareamiento y las gravideces resultantes hasta el destete de su descendencia (generación F1). Tras el destete, se sigue administrando la sustancia a la generación F1 durante todo el crecimiento, el período de apareamiento y el nacimiento de la generación F2, hasta el destete de esta última.
Se someten todos los animales a observación clínica y examen patológico para detectar signos de toxicidad. El examen tendrá por objeto principal evaluar los efectos sobre la integridad y el funcionamiento del aparato reproductor de las hembras y los machos y sobre el desarrollo y crecimiento de la descendencia.
1.3. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
1.3.1. Selección de la especie animal
La especie idónea para el ensayo es la rata. Si se emplea otra distinta, debe justificarse y procederse a las adaptaciones pertinentes. Debe evitarse la utilización de cepas que presenten un bajo índice de fertilidad o una incidencia notoriamente elevada de anomalías del desarrollo. Al principio del experimento, la variación ponderal de los animales empleados ha de ser mínima y no superar el ± 20 % del peso medio de cada sexo.
1.3.2. Condiciones de alojamiento y alimentación
El cuarto de experimentación ha de estar a una temperatura de 22 oC (± 3o) y con una humedad relativa mínima del 30 % y preferiblemente inferior al 70 %, salvo durante la limpieza del local, si bien lo ideal es que esté comprendida entre el 50 y el 60 %. Se aplica una iluminación artificial en una secuencia de 12 horas de luz y 12 horas de oscuridad. Puede proporcionarse una dieta alimentaria corriente para animales de laboratorio y agua potable a voluntad. Si la sustancia de ensayo se administra con los alimentos, es preciso obtener una mezcla adecuada, lo cual puede influir en la elección de la dieta.
Los animales pueden enjaularse por separado o en pequeños grupos del mismo sexo. El apareamiento debe llevarse a cabo en jaulas adecuadas al efecto. Una vez comprobado el apareamiento, las hembras que se hayan apareado se colocarán en jaulas individuales preparadas para el parto y la maternidad. También pueden enjaularse en grupos reducidos y separarse uno o dos días antes del parto. Cuando se acerque la fecha de este, se les proporcionará material adecuado y determinado de nidificación.
1.3.3. Preparación de los animales
Deben emplearse animales jóvenes sanos, que se hayan mantenido al menos 5 días en las condiciones de laboratorio para su aclimatación y que no hayan sido sometidos a experimentos previos. Se caracteriza la especie, cepa, procedencia, sexo, peso y/o edad de los animales de experimentación. Es preciso conocer las relaciones de consanguinidad entre los animales para evitar el apareamiento entre hermanos. Los animales se reparten al azar entre los lotes tratados y los de control (se recomienda agruparlos por niveles ponderales). Las jaulas se disponen de forma que se reduzcan al mínimo los posibles efectos debidos al enjaulamiento. Se asigna a cada animal un número de identificación distinto. Los animales de la generación P deben numerarse antes de iniciar el tratamiento y los de la generación F1 que se seleccionen para el apareamiento, en el momento del destete. Debe registrarse la camada de origen de todos los animales de la generación F1 seleccionados. Además de ello, se recomienda identificar cada cría tan pronto como sea posible tras el nacimiento si se tiene intención de pesarlas por separado o someterlas a ensayos funcionales.
Los animales de la generación P han de tener entre 5 y 9 semanas de edad cuando se inicie el tratamiento. Los lotes de ensayo han de ser tan homogéneos como sea posible por lo que respecta al peso y edad de los animales.
1.4. PROCEDIMIENTO
1.4.1. Número y sexo de los animales
Cada lote de ensayo y de control debe estar integrado por una cantidad de animales que permita disponer de un mínimo de 20 hembras grávidas a término o casi. En el caso de las sustancias que provocan efectos indeseables (por ejemplo, esterilidad o toxicidad excesiva a la dosis más alta), puede resultar imposible. Se trata de obtener un número suficiente de hembras grávidas para realizar una evaluación significativa de los efectos de la sustancia de ensayo sobre la fertilidad, la gestación y el comportamiento materno, así como sobre la lactancia, desarrollo y crecimiento dé la descendencia F1 desde la concepción hasta la madurez, y sobre el desarrollo de la generación F2 hasta el destete. Por consiguiente, el hecho de no conseguir el número deseado de hembras grávidas (es decir, 20) no invalida necesariamente el estudio y debe valorarse caso por caso.
1.4.2. Preparación de las dosis
Se recomienda administrar la sustancia de ensayo por vía oral (con el alimento o el agua de bebida o por sonda), salvo que se considere más apropiado emplear otra vía (cutánea o inhalatoria).
En caso necesario, la sustancia de ensayo se disuelve o suspende en un vehículo adecuado. Se recomienda considerar en primer lugar, siempre que sea posible, el uso de una solución o suspensión acuosa, después el uso de una solución o emulsión oleosa (por ejemplo, en aceite de maíz) y, por último, la posible disolución en otros vehículos. Si se emplean vehículos distintos del agua, deben conocerse sus características tóxicas. Debe determinarse la estabilidad de la sustancia de ensayo en el vehículo.
1.4.3. Posología
Se emplean al menos tres dosis de ensayo y un lote de control en paralelo. A menos que las características fisicoquímicas o las propiedades biológicas de la sustancia de ensayo impongan restricciones, la dosis superior debe seleccionarse con el propósito de inducir efectos tóxicos, pero sin llegar a provocar la muerte ni un sufrimiento intenso. En caso de que se produzca un índice de mortalidad inesperado, si el correspondiente a la generación P es inferior al 10 % aproximadamente, el estudio sigue siendo aceptable. Se selecciona una serie de dosis decrecientes para poner de manifiesto las reacciones en función de la dosis y la dosis sin efectos adversos observados (NOAEL). Los intervalos del doble al cuádruple suelen ser óptimos para establecer las dosis decrecientes y a menudo es preferible añadir un cuarto lote de ensayo en lugar de utilizar intervalos muy amplios (por ejemplo, con un factor superior a 10) entre dosis. Si la sustancia de ensayo se añade a los alimentos, el intervalo entre las dosis no debe superar un factor de 3. Las dosis deben establecerse tomando en consideración todos los datos disponibles sobre la toxicidad, en particular los resultados de los estudios de dosis repetidas, y la información complementaria relativa al metabolismo y la cinética de la sustancia de ensayo o productos afines. Dicha información servirá asimismo para justificar la pertinencia de la gama de dosis.
Se emplea un lote de control en paralelo, que no recibe la sustancia de ensayo, pero sí el vehículo en caso de que este se utilice para la sustancia. Salvo por lo que respecta a la administración de sustancia de ensayo, los animales del lote de control deben tratarse de la misma manera que los de los lotes de ensayo. En su caso, los lotes de control han de recibir el mayor volumen de vehículo que se haya utilizado. Si la sustancia de ensayo se administra con los alimentos y provoca una disminución de la ingesta o asimilación, puede ser útil utilizar un lote de control alimentado en paralelo. En lugar de ello, pueden emplearse los resultados de estudios de control destinados a evaluar los efectos de la disminución del consumo de alimentos sobre los parámetros de la reproducción.
Debe prestarse atención a las siguientes características del vehículo u otros aditivos, según proceda: efectos sobre la absorción, distribución, metabolismo o retención de la sustancia de ensayo, efectos sobre las propiedades químicas de la sustancia de ensayo que puedan modificar su toxicidad y efectos sobre el consumo de alimentos y agua o el estado nutricional de los animales.
1.4.4. Ensayo límite
Si en un ensayo con una sola dosis equivalente al menos a 1 000 mg/kg peso corporal/día, administrada por vía oral, o un porcentaje equivalente si se incorpora en los alimentos o el agua, y siguiendo el procedimiento descrito en el presente estudio, no se produce ningún efecto tóxico observable en los padres ni en la progenie y si, a la luz de los datos disponibles (por ejemplo, sobre sustancias con características estructurales y/o metabólicas similares), no cabe esperar efectos tóxicos, puede considerarse innecesario realizar un estudio completo con varias dosis. El ensayo límite es válido excepto cuando la exposición humana indique la necesidad de utilizar una dosis oral superior. Si se trata de otras vías de administración, como la inhalación o la aplicación cutánea, suelen ser las características fisicoquímicas de la sustancia de ensayo (solubilidad, etc.) las que determinan y limitan el grado máximo de exposición que puede alcanzarse.
1.4.5. Administración de las dosis
La sustancia de ensayo se administra diariamente a los animales, los 7 días de la semana y preferiblemente por vía oral (con los alimentos o el agua de bebida o por sonda). En caso de emplearse otra vía, debe justificarse y efectuarse las modificaciones oportunas. Se utilizará la misma forma de administración para todos los animales durante el período experimental apropiado. Si la sustancia de ensayo se administra por sonda, debe hacerse con una sonda gástrica. El volumen máximo de líquido administrado de una sola vez no debe superar 1 ml/100 g de peso corporal (0,4 ml/100 g de peso corporal en caso de que se emplee aceite de maíz como vehículo), salvo en el caso de las soluciones acuosas, en que puede llegarse a 2 ml/100 g de peso corporal. Excepto en el caso de sustancias irritantes o corrosivas que provoquen normalmente efectos exacerbados a concentraciones superiores, la variabilidad del volumen de ensayo debe reducirse al mínimo ajustando la concentración para que el volumen sea constante en todas las dosis. En los ensayos en que se emplee sonda, en principio las crías reciben la sustancia de ensayo indirectamente a través de la leche materna hasta que se inicia la administración directa en el destete. En los estudios en que la sustancia de ensayo se mezcla con los alimentos o el agua de bebida, las crías también la reciben directamente cuando empiezan a alimentarse solas en la última semana de lactancia.
Si la sustancia se administra con los alimentos o el agua de bebida, es importante cerciorarse de que las cantidades de sustancia de ensayo administradas no interfieren con la nutrición normal ni el equilibrio hídrico. Cuando la sustancia de ensayo se administre con los alimentos, puede utilizarse una concentración constante en la dieta (ppm) o bien una dosis constante respecto al peso corporal de los animales, pero debe indicarse qué método se ha elegido. Si la sustancia se administra por sonda, la dosis debe darse todos los días a la misma hora y ajustarse al menos una vez por semana para mantener una dosis constante respecto al peso corporal del animal. El ajuste deberá tener presente la difusión placentaria.
1.4.6. Programa experimental
La administración diaria de la sustancia de ensayo a los machos y las hembras de la generación P se inicia cuando tienen entre 5 y 9 semanas de edad y a los de la generación F1, en el destete. Debe tenerse presente que cuando la sustancia se administra con los alimentos o el agua de bebida, las crías de la generación F1 ya han estado expuestas directamente durante la lactancia. Se sigue administrando la sustancia a los animales de ambos sexos de las generaciones P y F1 al menos durante 10 semanas antes del período de apareamiento y durante las dos semanas de dicho período. Los machos que no vayan a emplearse para evaluar los efectos sobre la reproducción se sacrifican por métodos compasivos y se estudian. La sustancia de ensayo sigue administrándose a las hembras de la generación P durante la gestación y hasta el destete de su progenie F1. Puede ser oportuno modificar el programa de administración de las dosis a la luz de la información disponible sobre la sustancia de ensayo, en particular sobre la toxicidad, la inducción metabólica o la bioacumulación. Por lo general, la dosis que se administra a cada animal se calcula con arreglo a la última determinación del peso corporal, si bien conviene ser prudente al adaptar la dosis en el último período de la gestación.
Los machos y hembras de las generaciones P y F1 siguen recibiendo sustancia de ensayo hasta que sean sacrificados por métodos compasivos, cuando ya no sean necesarios para evaluar los efectos sobre la reproducción. Los descendientes F1 que no se seleccionen para el apareamiento y todos los descendientes F2 se sacrificarán por métodos compasivos después del destete.
1.4.7. Apareamiento
1.4.7.1. Apareamiento de la generación parental (P)
Para cada apareamiento, se juntan una hembra y un macho tratados con la misma dosis (apareamiento 1:1) hasta que se produzca la cópula o durante 2 semanas. Las hembras se observan todos los días para detectar la presencia de esperma o tapón vaginal. El día 0 de la gestación es el día en que se observa un tapón vaginal o la presencia de esperma. Si no se produce apareamiento, puede plantearse otro intento con hembras y machos de comprobada capacidad reproductora del mismo lote. Las parejas deben quedar claramente identificadas en los resultados. Debe evitarse el apareamiento entre animales hermanos.
1.4.7.2. Apareamiento de la generación F1
Se seleccionan al menos un macho y una hembra de cada camada de la generación F1 en el momento del destete con objeto de aparearlos con otras crías tratadas con la misma dosis, pero pertenecientes a otra camada, para obtener la generación F2. La selección de las crías de la misma camada debe hacerse al azar si no presentan diferencias significativas de peso corporal ni de aspecto. Si se observan diferencias, se seleccionan los mejores ejemplares de cada camada. Desde el punto de vista pragmático, resulta más fácil seleccionarlos en función del peso corporal, si bien puede estar más indicado basarse en el aspecto. Los descendientes F1 no deben aparearse antes de alcanzar la plena madurez sexual.
Las parejas sin descendencia deben estudiarse con objeto de determinar la causa aparente de infertilidad. Para ello, puede dárseles otras oportunidades de aparearse con machos o hembras de comprobada capacidad reproductora, efectuarse el examen microscópico de los órganos reproductores o estudiarse los ciclos estruales o la espermatogénesis.
1.4.7.3. Segundo apareamiento
En algunos casos, en particular cuando las dimensiones de la camada se ven modificadas por el tratamiento o cuando se observa un efecto equívoco en el primer apareamiento, se recomienda volver a aparear los adultos P o F1 para obtener una segunda camada. Conviene volver a aparear las hembras o machos que no hayan engendrado con un reproductor o reproductora de capacidad comprobada. Si se considera necesaria una segunda camada de alguna de las generaciones, los animales deben aparearse más o menos una semana después del destete de la primera camada.
1.4.7.4. Dimensión de la camada
Se deja que los animales paran y críen con normalidad a su progenie hasta el destete. La normalización de la dimensión de las camadas es facultativa. Si se lleva a cabo, debe describirse detalladamente el método empleado.
1.5. OBSERVACIONES
1.5.1. Observaciones clínicas
Debe hacerse una observación clínica general una vez al día y, en caso de administración por sonda, teniendo en cuenta el período más agudo de los efectos previstos tras la administración. Se registran los cambios en el comportamiento, los signos de parto difícil o prolongado y todos los signos de toxicidad. Además de ello, al menos una vez por semana se examinan todos los animales de forma más exhaustiva; puede hacerse coincidiendo con los días en que se pesen los animales. Se examinan todos los animales dos veces al día y una vez al día durante los fines de semana, según proceda, para detectar signos de morbilidad y mortalidad.
1.5.2. Peso corporal y consumo de alimentos y agua de los animales parentales
Los animales de las generaciones P y F1 deben pesarse el día de la primera administración y a continuación al menos una vez por semana. Las madres (P y F1) se pesan, al menos, los días 0, 7, 14 y 20 o 21 de la gestación; durante la lactancia, los mismos días en que se pesen las crías y, por último, el día del sacrificio. Los resultados se registran para cada animal adulto por separado. Durante el período previo al apareamiento y durante la gestación, se mide al menos una vez por semana el consumo de alimentos. Si la sustancia de ensayo se administra con el agua, el consumo de esta se mide al menos una vez por semana.
1.5.3. Ciclo estrual
Se estudia la duración y normalidad del ciclo estrual en las hembras P y F1 mediante frotis vaginales antes del apareamiento y, con carácter facultativo, durante este, hasta que se compruebe que ha habido apareamiento. La toma de células vaginales o cervicales ha de hacerse con cuidado para no dañar la mucosa y evitar una pseudogestación (1).
1.5.4. Parámetros de evaluación del esperma
Se registra el peso de los testículos y epidídimos de todos los machos P y F1 tras el sacrificio y se conserva un ejemplar de cada órgano para el examen histopatológico (véanse los apartados 1.5.7 y 1.5.8.1). Se conservan los testículos y epidídimos restantes de un sublote integrado al menos por 10 machos de cada grupo de machos P y F1, para contar las espermátides resistentes a la homogeneización y los espermatozoos almacenados en la cola del epidídimo, respectivamente. En ese mismo sublote de machos, se recoge el contenido de la cola del epidídimo o el conducto deferente para evaluar la motilidad y morfología de los espermatozoides. Si se observan efectos relacionados con el tratamiento o los resultados de otros estudios ponen de manifiesto que la sustancia de ensayo puede afectar a la espermatogénesis, debe efectuarse la evaluación del esperma en todos los machos de cada lote tratado; en caso contrario, el recuento puede limitarse a los machos P y F1 del lote de control y los lotes tratados con la dosis superior.
Debe contarse la totalidad de las espermátides testiculares resistentes a la homogeneización y los espermatozoos de la cola del epidídimo (2) (3). La reserva caudal de espermatozoos puede deducirse de la concentración y el volumen presentes en la suspensión utilizada para completar las evaluaciones cualitativas y de la cantidad de espermatozoos recuperados tras triturar u homogeneizar el tejido caudal restante. El recuento debe efectuarse inmediatamente después del sacrificio, en el sublote de machos seleccionados en los grupos tratados con las distintas dosis, salvo que se realicen grabaciones digitales o de vídeo, o se congelen las muestras para su posterior análisis. En estos casos pueden analizarse en primer lugar el lote de control y el tratado con la dosis más alta y, si no se observan efectos relacionados con el tratamiento (por ejemplo, en la cantidad, motilidad o morfología de los espermatozoides), no es preciso analizar los demás lotes. Si se observan efectos relacionados con el tratamiento en el lote tratado con la dosis más alta, deberán someterse a análisis los lotes tratados con dosis inferiores.
Debe evaluarse o grabarse en soporte de vídeo inmediatamente después del sacrificio la motilidad de los espermatozoides en el epidídimo o el conducto deferente. Se toma una muestra de esperma evitando al máximo dañar los tejidos y se diluye para estudiar la motilidad mediante métodos aceptables (4). El porcentaje de espermatozoides progresivamente móviles se determina de forma objetiva o subjetiva. Si se realiza un análisis de la motilidad asistido por ordenador (5) (6) (7) (8) (9) (10), la motilidad progresiva se evalúa con arreglo a umbrales de velocidad media de trayectoria y movimiento en línea recta o índice lineal, definidos por el usuario. Si las muestras se graban en vídeo (11) o se graban las imágenes de otra manera en el momento de la autopsia, el análisis posterior puede limitarse a los machos P y F1 del lote de control y del tratado con la dosis más alta, salvo que se hayan observado efectos relacionados con el tratamiento, en cuyo caso habrá que evaluar asimismo los lotes tratados con dosis inferiores. Si no se dispone de imágenes de vídeo ni digitales, se analizarán en la autopsia todas las muestras de todos los lotes tratados.
Se llevará a cabo el análisis morfológico de una muestra de esperma del epidídimo o el conducto deferente. Los espermatozoides (al menos 200 por muestra) se fijan y analizan en preparaciones húmedas (12), y se clasifican entre normales o anómalos. Las anomalías morfológicas de los espermatozoides incluyen las fusiones, las cabezas aisladas y las cabezas o las colas deformadas. La evaluación ha de efectuarse en el sublote de machos seleccionados de los grupos tratados con las distintas dosis, inmediatamente después del sacrificio o posteriormente a partir de grabaciones de vídeo o digitales. Una vez fijados, los frotis también pueden analizarse en un momento posterior. En estos casos pueden analizarse en primer lugar el lote de control y el tratado con la dosis más alta y, si no se observan efectos relacionados con el tratamiento (sobre la morfología de los espermatozoides, por ejemplo), no es preciso analizar los demás lotes. Si se observan efectos relacionados con el tratamiento en el lote tratado con la dosis más alta, deberán someterse a análisis los lotes tratados con dosis inferiores.
Si alguno de los parámetros mencionados de evaluación del esperma ya ha sido analizado en un estudio de toxicidad sistémica de 90 días al menos, no es necesario repetir el análisis en el estudio de la toxicidad en dos generaciones. Se recomienda, no obstante, conservar muestras o grabaciones digitales del esperma de la generación P para poder repetir la evaluación, si fuera necesario.
1.5.5. Descendencia
Se examinan todas las camadas lo antes posible después del parto (día 0 de la lactancia) para determinar el número y sexo de las crías, la mortinatalidad, el número de nacidos vivos y la presencia de anomalías macroscópicas. Es preferible examinar las crías halladas muertas el día 0, si no están maceradas, para detectar posibles anomalías y determinar la causa de la muerte y, a continuación, conservarlas. Las crías vivas se cuentan y pesan por separado al nacer (día 0 de la lactancia) o el día 1 y luego de forma periódica (por ejemplo, los días 4, 7, 14 y 21 de la lactancia). Se registran las anomalías físicas y las alteraciones del comportamiento de las madres y las crías.
Se registra el desarrollo físico de la progenie, anotando la ganancia de peso corporal. Otros parámetros físicos (apertura de las orejas y los ojos, erupción de dientes, crecimiento del pelo, etc.) pueden proporcionar información adicional, si bien es preferible emplear esos datos para evaluar la madurez sexual (edad y peso corporal en el momento de la apertura de la vagina o la separación balanoprepucial, etc.) (13). Si no forma parte de otros estudios, se recomienda efectuar la valoración funcional (actividad motriz, funciones sensoriales, ontogenia de los reflejos, por ejemplo) de la progenie F1 antes y/o después del destete, sobre todo por lo que respecta a la maduración sexual. En los descendientes F1 recién destetados y seleccionados para el apareamiento debe determinarse la edad a la que se produzca la apertura de la vagina y la separación balanoprepucial. Si se observa una alteración de la proporción de machos y hembras o de la edad de maduración sexual en la generación F1, debe medirse la distancia anogenital en las crías F2 el día del nacimiento.
Las observaciones funcionales no son obligatorias en los lotes que presenten signos evidentes de toxicidad (disminución significativa de la ganancia de peso, etc.). Si se efectúan exploraciones funcionales, se harán en las crías que no se hayan seleccionado para el apareamiento.
1.5.6. Autopsia macroscópica
Justo después del sacrificio o la muerte ocurrida durante el estudio, se someten a examen macroscópico para detectar toda anomalía estructural o cambio patológico todos los animales parentales (P y F1), todas las crías que presenten anomalías externas o signos clínicos y una cría seleccionada al azar de cada sexo y camada de las generaciones F1 y F2. Se prestará especial atención a los órganos reproductores. Deben examinarse y conservarse las crías moribundas que se sacrifiquen por métodos compasivos y las crías muertas, si no están maceradas, para detectar posibles anomalías y establecer la causa de la muerte.
Debe procederse al examen de los úteros de todas las hembras primíparas, sin comprometer la evaluación histopatológica, para detectar la presencia de puntos de implantación y contarlos.
1.5.7. Pesaje de los órganos
Tras el sacrificio, se determina el peso corporal de todos los animales parentales P y F1, así como el de los órganos siguientes (los órganos pares se pesarán por separado):
|
— |
utero y ovarios, |
|
— |
testículos y epidídimos (enteros y colas), |
|
— |
próstata, |
|
— |
vesículas seminales con glándulas coagulantes y sus líquidos, y próstata (conjunto), |
|
— |
cerebro, hígado, riñones, bazo, hipófisis, glándula tiroides, glándulas suprarrenales y órganos diana conocidos. |
Se determina el peso corporal de las crías F1 y F2 sacrificadas que se hayan seleccionado para la autopsia, así como el peso del cerebro, bazo y timo de una cría seleccionada al azar de cada sexo y camada (véase el apartado 1.5.6).
Si es posible, los resultados de la autopsia macroscópica y del pesaje de órganos se interpretan a la luz de las observaciones realizadas en otros estudios con dosis repetidas.
1.5.8. Histopatología
1.5.8.1. Animales parentales
Los siguientes órganos y tejidos de los animales parentales (P y F1), o muestras representativas de estos, se fijan y conservan en un medio apropiado con vistas al examen histopatológico:
|
— |
vagina, útero con cuello, y ovarios (conservados en un fijador apropiado), |
|
— |
un testículo (conservado en líquido de Bouin o un fijador comparable), un epidídimo, las vesículas seminales, la próstata y una glándula coagulante, |
|
— |
órgano u órganos diana previamente identificados de todos los animales P y F1 seleccionados para el apareamiento. |
Debe realizarse un examen histopatológico completo de los órganos y tejidos antes mencionados de todos los animales P y F1 del lote de control y el lote tratado con la dosis más alta, que se hayan seleccionado para el apareamiento. El examen de los ovarios de las hembras P es facultativo. Deben examinarse, asimismo, los órganos que presenten alteraciones relacionadas con el tratamiento en los lotes tratados con las dosis inferior y media, con objeto de facilitar la determinación de la NOAEL. Además de ello, se someterán a evaluación histopatológica los órganos reproductores de los animales tratados con las dosis inferior y media, en los que se sospeche una disminución de la fertilidad, por ejemplo, los que no se hayan apareado, no hayan concebido, no hayan engendrado o no hayan tenido una progenie sana, o en los que se hayan observado alteraciones del ciclo estrual o de la cantidad, motilidad o morfología de los espermatozoides. Se examinarán todas las lesiones macroscópicas como atrofias o tumores.
Debe efectuarse un examen histopatológico pormenorizado de los testículos (por ejemplo, con líquido de Bouin, inclusión en parafina y cortes transversales de 4-5μm de espesor) para poner de manifiesto los efectos relacionados con el tratamiento como la retención de espermátides, la ausencia de algunas capas o tipos de células germinales, la presencia de células gigantes plurinucleadas o el desprendimiento de células espermatogénicas a la luz de los túbulos seminíferos (14). El examen del epidídimo intacto ha de abarcar la cabeza, el cuerpo y la cola, y puede efectuarse en una sección longitudinal. Se analiza el epidídimo para ver si hay infiltración leucocitaria, cambios en la frecuencia de los tipos celulares, células aberrantes y fagocitosis de espermatozoides. El análisis de los órganos reproductores de los machos puede realizarse mediante coloración con PAS o hematoxilina.
Tras la lactancia, el ovario debe contener folículos primordiales, folículos en crecimiento y grandes cuerpos amarillos de la lactancia. El examen histopatológico debe poner de manifiesto la depleción cualitativa de la población de folículos primordiales. Debe efectuarse una evaluación cuantitativa de los folículos primordiales de las hembras F1; el número de animales, la elección de la sección ovárica y el tamaño de las muestras de secciones han de ser válidas desde el punto de vista estadístico para el método de evaluación empleado. En el examen debe hacerse el recuento de los folículos primordiales, que pueden estar combinados con pequeños folículos en crecimiento, para efectuar la comparación de los ovarios de las hembras de los lotes tratados y del de control (15) (16) (17) (18) (19).
1.5.8.2. Crías destetadas
Se fijan y conservan en un medio adecuado para el posterior examen histopatológico los tejidos que presenten anomalías macroscópicas y los órganos diana de todas las crías que presenten anomalías externas o signos clínicos, así como de la cría seleccionada al azar de cada sexo y camada de las generaciones F1 y F2, que no haya sido seleccionada para el apareamiento. La descripción histopatológica completa de los tejidos conservados se centrará sobre todo en los órganos reproductores.
2. RESULTADOS
2.1. TRATAMIENTO DE LOS RESULTADOS
Deben proporcionarse datos de cada animal por separado y resumirse en un cuadro que recoja, para cada lote de ensayo y cada generación, el número de animales al inicio del ensayo, el número de animales hallados muertos durante el mismo o sacrificados por razones compasivas, el momento de la muerte o sacrificio compasivo, el número de animales fértiles, el número de hembras grávidas, el número de animales que presenten signos de toxicidad, una descripción de dichos signos (con inclusión del momento de su aparición, duración y gravedad), los tipos de observaciones de los animales parentales y las crías, los tipos de cambios histopatológicos y todos los datos pertinentes sobre la camada.
Los resultados numéricos deben evaluarse mediante un método estadístico adecuado y comúnmente aceptado. La elección de los métodos estadísticos debe efectuarse en la fase de diseño del estudio y justificarse. Los modelos estadísticos aplicables a las relaciones dosis-respuesta pueden resultar útiles para analizar los resultados. El informe debe recoger información suficiente sobre el método y el programa informático empleados, de manera que un revisor o estadístico independiente pueda reevaluar y reconstruir el análisis.
2.2. EVALUACIÓN DE LOS RESULTADOS
Los resultados del estudio de toxicidad para la reproducción en dos generaciones deben evaluarse respecto a los efectos observados, en particular en la autopsia y los exámenes microscópicos. La evaluación debe referirse a la relación, o la ausencia de relación, entre la dosis de sustancia de ensayo y la presencia o ausencia, incidencia y gravedad de las anomalías, incluyendo lesiones macroscópicas, órganos diana identificados, alteración de la fertilidad, anomalías clínicas, alteración de la capacidad de reproducción y del rendimiento de la camada, cambios del peso corporal, efectos sobre la mortalidad y cualquier otro efecto tóxico. Los resultados del estudio han de interpretarse teniendo presentes las propiedades fisicoquímicas de la sustancia de ensayo y, en su caso, los datos toxicocinéticos.
Un ensayo de toxicidad para la reproducción correctamente realizado debe proporcionar una estimación satisfactoria de la dosis sin efecto y poner de manifiesto los efectos adversos sobre la reproducción, el parto, la lactancia y el desarrollo postnatal, en particular el crecimiento y la maduración sexual.
2.3. INTERPRETACIÓN DE LOS RESULTADOS
El estudio de toxicidad para la reproducción en dos generaciones proporciona información sobre los efectos de la exposición repetida a una sustancia durante todas las fases del ciclo de reproducción y, en particular, sobre los parámetros de la reproducción y el desarrollo, crecimiento, maduración y supervivencia de la descendencia. Los resultados del estudio han de interpretarse a la luz de los de los estudios subcrónicos, de desarrollo prenatal, toxicocinéticos y de otro tipo. Los resultados del presente estudio pueden servir para valorar la necesidad de realizar más ensayos con una sustancia química. La validez de la extrapolación de los resultados del estudio al hombre es limitada. Resultan más útiles para determinar las dosis sin efecto y el grado de exposición humana aceptable (20) (21) (22) (23).
3. INFORME
3.1. INFORME DEL ENSAYO
El informe del ensayo debe incluir la información siguiente:
|
|
Sustancia de ensayo:
|
|
|
Vehículo (si procede):
|
|
|
Animales sometidos a ensayo:
|
|
|
Condiciones de ensayo:
|
|
|
Resultados:
|
|
|
Discusión de los resultados. . |
|
|
Conclusiones, incluida la NOAEL en la madre y la progenie. |
4. REFERENCIAS
|
(1) |
Sadleir, R.M.F.S. (1979). Cycles and Seasons, En: Reproduction in Mammals: I. Germ Cells and Fertilization, C.R. Auston and R.V. Short (eds.), Cambridge, New York. |
|
(2) |
Gray, L.E. et al., (1989). A Dose-Response Analysis of Methoxychlor-lnduced Alterations of Reproductive Development and Function in the Rat. Fundamental and Applied Toxicology 12:92-108. |
|
(3) |
Robb, G.W. et al., (1978). Daily Sperm Production and Epididymal Sperm Reserves of Pubertal and Adult Rats. Journal of Reproduction and Fertility 54:103-107. |
|
(4) |
Klinefelter, G.R. et al., (1991). The Method of Sperm Collection Signifícantly Influences Sperm Motion Parameters Following Ethane Dimethanesulfonate Administration in the Rat. Reproductive Toxicology 5:39 44. |
|
(5) |
Seed, J. et al. (1996). Methods for Assessing Sperm Motility, Morphology, and Counts in the Rat, Rabbit, and Dog: a Consensus Report. Reproductive Toxicology 10(3):237- 244. |
|
(6) |
Chapin, R.E. et al., (1992). Methods for Assessing Rat Sperm Motility. Reproductive Toxicology 6:267-273. |
|
(7) |
Klinefelter, G.R. et al., (1992). Direct Effects of Ethane Dimethanesulphonate on Epididymal Function in Adult Rats: an In Vitro Demonstration. Journal of Andrology 13:409-421. |
|
(8) |
Slott, V.L. et al., (1991). Rat Sperm Motility Analysis: Methodologic Considerations. Reproductive Toxicology 5:449-458. |
|
(9) |
Slott, V.L. y Perreault, S.D. (1993). Computer-Assisted Sperm Analysis of Rodent Epididymal Sperm Motility Using the Hamilton-Thorn Motility Analyzer. En: Methods in Toxicology, Part A., Academic, Orlando, Florida, pp. 319-333. |
|
(10) |
Toth, G.P. et al. (1989). The Automated Analysis of Rat Sperm Motility Following Subchronic Epichlorhydrin Administration: Methodologic and Statistical Considerations. Journal of Andrology 10: 401-415. |
|
(11) |
Working, P.K. y M. Hurtt, (1987). Computerized Videomicrographic Analysis of Rat Sperm Motility. Journal of Andrology 8:330-337. |
|
(12) |
Linder, R.E. et al., (1992). Endpoints of Spermatoxicity in the Rat After Short Duration Exposures to Fourteen Reproductive Toxicants. Reproductive Toxicology 6:491-505. |
|
(13) |
Korenbrot, C.C. et al., (1977). Preputial Separation as an External Sign of Pubertal Development in the Male Rat. Biological Reproduction 17:298303. |
|
(14) |
Russell, L.D. et al., (1990). Histological and Histopathological Evaluation of the Testis, Cache River Press, Clearwater, Florida. |
|
(15) |
Heindel, J.J. y R.E. Chapin, (eds.) (1993). Part B. Female Reproductive Systems, Methods in Toxicology, Academic, Orlando, Florida. |
|
(16) |
Heindel, J.J. et al., (1989) Histological Assessment of Ovarían Follicle Number in Mice As a Screen of Ovarían Toxicity. En: Growth Factors and the Ovary, A.N. Hirshfield (ed.), Plenum, New York, pp. 421-426. |
|
(17) |
Manson, J.M. y Y.J. Kang, (1989). Test Methods for Assessing Female Reproductive and Developmental Toxicology. En: Principies and Methods of Toxicology, A.W. Hayes (ed.), Raven, New York. |
|
(18) |
Smith, BJ. et al. (1991). Comparison of Random and Serial Sections in Assessment of Ovarian Toxicity. Reproductive Toxicology 5:379-383. |
|
(19) |
Heindel, J.J. (1999). Oocyte Quantitation and Ovarian Histology. En: An Evaluation and Interpretation of Reproductive Endpoints for Human Health Risk Assessment, G. Daston, and C.A. Kimmel, (eds.), ILSI Press, Washington, DC. |
|
(20) |
Thomas, J.A. (1991). Toxic Responses of the Reproductive System. En: Casarett and Doull's Toxicology, M.O. Amdur, J. Doull, and C.D. Klaassen (eds.), Pergamon, New York. |
|
(21) |
Zenick, H. y E.D. Clegg, (1989). Assessment of Male Reproductive Toxicity: A Risk Assessment Approach. En: Principies and Methods of Toxicology, A.W. Hayes (ed.), Raven Press, New York. |
|
(22) |
Palmer, A.K. (1981). En: Developmental Toxicology, Kimmel, C.A. and J. Buelke-Sam (eds.), Raven Press, New York. |
|
(23) |
Palmer, A.K. (1978). In Handbook of Teratology, Vol. 4, J.G. Wilson and F.C. Fraser (eds.), Plenum Press, New York. |
B.36. TOXICOCINÉTICA
1. MÉTODO
1.1. INTRODUCCIÓN
Véase la Introducción general, Parte B.
1.2. DEFINICIONES
Véase la Introducción general, Parte B.
1.3. SUSTANCIAS DE REFERENCIA
Ninguna.
1.4. PRINCIPIO DEL MÉTODO
La sustancia objeto de estudio se administra por una vía apropiada. Según el objetivo del estudio, puede administrarse en dosis única o repetida, durante períodos determinados, a uno o varios grupos de animales de experimentación. Con posterioridad, y en función del tipo de estudio, se determinan la sustancia, o sus metabolitos, o ambos. en los líquidos, los tejidos o las excreciones corporales.
Pueden realizarse estudios con formas «marcadas» o «no marcadas» de la sustancia probada. Cuando se utilice el marcado, su posición en la sustancia debe suministrar la mayor información posible sobre el destino del compuesto.
1.5. CRITERIOS CUALITATIVOS
Ninguno.
1.6. DESCRIPCIÓN DEL MÉTODO
Preparativos
Se aclimatan animales jóvenes y sanos a las condiciones del laboratorio durante, al menos, 5 días previos a la prueba. Antes de ella, se distribuye al azar a los animales para formar grupos de tratamiento. En casos especiales pueden emplearse animales muy jóvenes, grávidos o tratados previamente.
Condiciones del ensayo
Animales de experimentación
Los estudios toxicocinéticos pueden efectuarse en una o varias especies animales apropiadas; se tendrán en cuenta las especies utilizadas o que esté previsto utilizar en otros estudios toxicológicos con la misma sustancia. Cuando se empleen roedores en una prueba, la variación ponderal no será superior al ± 20 % del peso medio.
Número y sexo
Para estudios de absorción y excreción, cada grupo contendrá inicialmente 4 animales. No es necesaria la elección de un sexo determinado, pero en ciertas circunstancias puede ser preciso estudiar ambos sexos. Si se apreciaran respuestas diferentes según el sexo, se estudiarán 4 animales de cada uno. Cuando los estudios se hagan con no roedores, pueden utilizarse menos animales. Si se estudia la distribución en los tejidos, al considerar el tamaño del grupo inicial se tendrán en cuenta el número de animales que se sacrificarán en cada fecha de examen establecida y el número de exámenes.
Cuando se estudie el metabolismo, el tamaño del grupo estará acorde con las necesidades del estudio. En los estudios con varias dosis y exámenes intermedios, al considerar el tamaño del grupo se tendrá en cuenta el número de exámenes y de sacrificios previstos; no obstante, estará formado por al menos dos animales. El tamaño del grupo será suficiente para permitir una evaluación aceptable del aumento, la estabilización y la reducción (según convenga) de las concentraciones de la sustancia estudiada, de sus metabolitos o de ambos.
Dosis
Cuando se administre una sola dosis, se utilizarán al menos dos niveles posológicos: una dosis baja con la que no se observen efectos tóxicos, y otra alta capaz de producir modificaciones de los parámetros toxicocinéticos o efectos tóxicos.
Si se administran dosis reiteradas, suele bastar la dosis baja, aunque en circunstancias determinadas quizá sea necesaria también una dosis alta.
Vía de administración
Los estudios toxicocinéticos se efectuarán por medio de la misma vía y, cuando convenga, del mismo vehículo empleado o que esté previsto emplear en los demás estudios de toxicidad. La sustancia analizada suele administrarse oralmente por alimentación forzada o en el alimento, aplicarse a la piel o administrarse por inhalación durante períodos definidos a grupos de animales de experimentación. La administración intravenosa de la sustancia puede ser útil para determinar la absorción relativa por otras vías. Además, es posible obtener información útil sobre el patrón de distribución poco después de la administración intravenosa de una sustancia.
Se tendrá presente la posibilidad de una interferencia del vehículo con la sustancia estudiada. Se prestará atención a las diferencias de absorción entre la administración de las sustancias analizadas por alimentación forzada y en el alimento y la necesidad de una determinación exacta de la dosis, sobre todo cuando se administre el compuesto en el alimento.
Período de observación
Se observará a diario a todos los animales, se registrarán los signos de toxicidad y otros rasgos clínicos relevantes, incluidos el momento de comienzo, el grado y la duración de los mismos.
Procedimiento
Después de pesar a los animales, se administra la sustancia objeto de estudio por una vía apropiada. Si se considera oportuno, puede mantenerse a los animales en ayunas antes de la administración de la sustancia.
Absorción
El índice y el grado de absorción de la sustancia administrada pueden valorarse por métodos diversos, con grupos de referencia (8) o sin ellos, por ejemplo mediante:
|
— |
determinación de la cantidad de la sustancia estudiada, de sus metabolitos o de ambos en las excretas, como orina, bilis, heces, aire espirado y el contenido en el caparazón, |
|
— |
comparación de la respuesta biológica (por ejemplo, estudios de toxicidad aguda) entre los grupos de experimentación y de control o referencia (o ambos), |
|
— |
comparación de la cantidad de sustancia, metabolitos o ambos, excretada por vía renal en los grupos de prueba y de referencia, |
|
— |
determinación de la zona bajo la curva, concentración plasmática/tiempo de la sustancia analizada, sus metabolitos o ambos, y comparación con datos de un grupo de referencia. |
Distribución
Existen actualmente dos métodos, de los que uno o ambos pueden utilizarse para analizar los patrones de distribución:
|
— |
se obtiene información cualitativa útil, mediante técnicas autorradiográficas del cuerpo entero, |
|
— |
se obtiene información cuantitativa, por sacrificio de los animales en momentos diferentes tras la exposición para determinar la concentración y la cantidad de la sustancia estudiada, sus metabolitos o ambos en tejidos y órganos. |
Excreción
En los estudios de excreción, se recogen orina, heces y aire espirado, y, en determinadas circunstancias, bilis. La cantidad de la sustancia estudiada, sus metabolitos o ambos en estas excretas se determinarán, en varios momentos tras la exposición, hasta que se haya excretado alrededor del 95 % de la dosis administrada o durante 7 días, si tal porcentaje no se alcanza antes.
En casos especiales, tal vez deba tenerse en cuenta la excreción de la sustancia en la leche de animales lactantes de experimentación.
Metabolismo
Para determinar el modo y la intensidad del metabolismo, se analizarán muestras biológicas por técnicas apropiadas. Se estudiarán las estructuras de los metabolitos y se propondrán vías metabólicas apropiadas cuando haya necesidad de responder a interrogantes planteados por estudios toxicológicos previos. Quizá sea útil realizar estudios in vitro para obtener información sobre las vías metabólicas.
Es posible obtener información complementaria sobre la relación del metabolismo con la toxicidad mediante estudios bioquímicos, como la determinación de los efectos sobre sistemas enzimáticos metabolizantes, la depleción de compuestos endógenos con grupos sulfhidrilos no proteicos y la unión de la sustancia a macromoléculas.
2. RESULTADOS
Según el tipo de estudio realizado, se resumirán los resultados en forma de tablas, complementadas por gráficos cuando proceda. Se mostrarán para cada grupo, cuando convenga, las variaciones medias y estadísticas de las determinaciones en relación con el tiempo, la posología, los tejidos y los órganos. Se establecerán, por métodos apropiados, el grado de absorción y la cantidad e índices de excreción. Cuando se realicen estudios de metabolismo, se indicará la estructura de los metabolitos identificados, y se presentarán las vías metabólicas posibles.
3. INFORME
3.1. DATOS DEL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
especie, cepa, origen, condiciones ambientales, alimentación, |
|
— |
caracterización de los materiales marcados, cuando se utilicen, |
|
— |
niveles posológicos e intervalos utilizados, |
|
— |
vía o vías de administración y vehículos utilizados, |
|
— |
efectos tóxicos y de otro tipo observados, |
|
— |
métodos de determinación de la sustancia objeto de ensayo, sus metabolitos o ambos en muestras biológicas, incluido el aire espirado, |
|
— |
presentación en tablas de las determinaciones en función de sexo, dosis, régimen, tiempo, tejidos y órganos, |
|
— |
indicación del grado de absorción y excreción con el tiempo, |
|
— |
métodos de caracterización e identificación de los metabolitos en muestras biológicas, |
|
— |
métodos de las determinaciones bioquímicas relacionadas con el metabolismo, |
|
— |
vías de metabolismo propuestas, |
|
— |
comentario de los resultados, |
|
— |
interpretación de los resultados. |
3.2. EVALUACIÓN E INTERPRETACIÓN
Véase la Introducción general, Parte B.
4. REFERENCIAS
Véase la Introducción general, Parte B.
«B.37. NEUROTOXICIDAD RETARDADA DE SUSTANCIAS ORGANOFOSFORADAS POR ADMINISTRACIÓN ÚNICA
1. MÉTODO
1.1. INTRODUCCIÓN
En la evaluación de los efectos tóxicos de las sustancias, es importante considerar el potencial de ciertas clases de sustancias para causar tipos específicos de neurotoxicidad que pueden no detectarse con otros estudios de toxicidad. Se ha visto que ciertas sustancias organofosforadas producen neurotoxicidad retardada, por lo que debe considerarse su evaluación.
Pueden emplearse ensayos de cribado in vitro para detectar las sustancias capaces de producir polineuropatía retardada; sin embargo, unos resultados negativos en los estudios in vitro no excluyen que la sustancia estudiada sea neurotóxica.
Véase la Introducción general, Parte B.
1.2. DEFINICIONES
Sustancias organofosforadas: ésteres organofosforados sin carga, tioésteres o anhídridos de ácidos organofosfóricos, organofosfónicos u organofosforamídicos, o de ácidos fosforotioicos, fosfono-tioicos o fosforotioamídicos afines, u otras sustancias que puedan producir la neurotoxicidad retardada que a veces se aprecia en esta clase se sustancias.
Neurotoxicidad retardada: síndrome asociado con la aparición retardada y prolongada de ataxia, axonopatías distales en la médula espinal y en los nervios periféricos, e inhibición y envejecimiento de la esterasa diana de la neuropatía (NTE) en el tejido nervioso.
1.3. SUSTANCIAS DE REFERENCIA
Puede someterse a ensayo una sustancia de referencia con un lote de control positivo como medio para demostrar que, en las condiciones de ensayo del laboratorio, no cambia significativamente la respuesta de las especies utilizadas.
Como ejemplo de neurotóxico de amplia utilización está el folfato de tri-o-tolilo [no CAS 78-30-8, no EINECS 201-103-5, denominación CAS: tris(2-metilfenil) éster del ácido fosfórico], conocido también como tri-o-cresilfosfato.
1.4. PRINCIPIO DEL MÉTODO
Se da una sola dosis oral de la sustancia estudiada a gallinas domésticas, protegidas de los efectos colinérgicos agudos, cuando convenga. Se observa durante 21 días la aparición en los animales de anomalías de comportamiento, ataxia y parálisis. Se realizan medidas bioquímicas, especialmente la inhibición de la esterasa diana de la neuropatía (NTE), en gallinas seleccionadas aleatoriamente de cada lote, normalmente a las 24 y 48 horas de la administración. A los 21 días de la exposición, se sacrifican las gallinas restantes y se procede al examen histopatológico de tejidos nerviosos seleccionados.
1.5. DESCRIPCIÓN DEL MÉTODO
1.5.1. Preparación
Se seleccionan al azar gallinas jóvenes sanas, libres de enfermedades víricas que puedan interferir y sin tratamiento médico, que no presenten anomalías de la marcha, se asignan a los lotes de tratamiento y de control y se aclimatan a las condiciones del laboratorio durante un mínimo de 5 días antes del inicio del estudio.
Deben utilizarse jaulas o recintos de la capacidad suficiente para permitir la libre movilidad de las gallinas y la fácil observación de su marcha.
La sustancia estudiada se administrará normalmente por vía oral, utilizando sondas gástricas, cápsulas de gelatina o un método similar. Los líquidos pueden administrarse sin diluir o disueltos en un vehículo apropiado, como el aceite de maíz; los sólidos deben disolverse siempre que sea posible, ya que las grandes dosis de productos sólidos en cápsulas de gelatina pueden no absorberse de forma eficaz. Respecto a los vehículos no acuosos, deben conocerse sus características tóxicas; en caso de no conocerse, se determinarán antes de la prueba.
1.5.2. Condiciones del ensayo
1.5.2.1. Animales de experimentación
Se recomienda la gallina ponedora doméstica adulta (Gallus gallus domesticus), de una edad comprendida entre 8 y 12 meses. Deben emplearse razas y variedades de tamaño normal y las gallinas se habrán criado normalmente en condiciones que permitan su movilidad libre.
1.5.2.2. Número y sexo
Además del lote de tratamiento, se utilizarán tanto un lote de control del vehículo como un lote de control positivo. El lote de control del vehículo debe tratarse de la misma forma que el lote de tratamiento, salvo que se omitirá la administración de la sustancia estudiada.
En cada lote de aves debe utilizarse un número suficiente de gallinas de forma que puedan matarse al menos 6 aves para hacer las determinaciones bioquímicas (3 en cada uno de los dos momentos seleccionados) y otras 6 puedan sobrevivir durante el período de 21 días de observación de las patologías.
El lote de control positivo puede estudiarse a la vez o bien ser un lote de control estudiado recientemente. Debe incluir al menos 6 gallinas, tratadas con un neurotóxico conocido de efecto retardado, de las que 3 se destinarán a las pruebas bioquímicas y otras 3 a la observación de la patología. Se recomienda la actualización periódica de los datos de los antecedentes. Deben obtenerse nuevos datos de lotes de control positivo cuando el laboratorio que realice las pruebas haya cambiado algún elemento fundamental de estas (por ejemplo, raza, alimentación, condiciones de alojamiento).
1.5.2.3. Dosis
Debe realizarse un estudio preliminar con un número adecuado de gallinas y lotes de distintas dosis, a fin de determinar la dosis que debe utilizarse en el estudio principal. En el estudio preliminar es necesario que se produzca letalidad, a fin de definir una dosis adecuada para el estudio principal. No obstante, para evitar la muerte debida a los efectos colinérgicos agudos, puede utilizarse atropina u otro agente protector que no interfiera con las respuestas neurotóxicas retardadas. Pueden utilizarse diversos métodos de ensayo para evaluar la dosis máxima no letal de las sustancias estudiadas (véase el método B.1 bis). También puede ayudar a seleccionar la dosis la consideración de datos de antecedentes de las gallinas u otra información toxicológica.
La dosis de la sustancia en el estudio principal debe ser lo más elevada posible, teniendo en cuenta los resultados del estudio preliminar de selección de la dosis y el límite superior a 2 000 mg/kg de peso corporal. La mortalidad que pueda darse no debe interferir con la supervivencia de animales suficientes para las pruebas bioquímicas (6) e histológicas (6) hasta el día 21. Debe utilizarse atropina u otro agente protector que no interfiera con las respuestas neurotóxicas retardadas, para evitar la muerte por efectos colinérgicos agudos.
1.5.2.4. Ensayo límite
Si un ensayo con una dosis de al menos 2 000 mg/kg de peso corporal/día, siguiendo los procedimientos descritos para el presente estudio, no produce ningún efecto tóxico observable y si, a partir de los datos procedentes de sustancias relacionadas estructuralmente, no cabe esperar la aparición de fenómenos tóxicos, puede considerarse innecesario un estudio con una dosis superior. El ensayo límite es aplicable excepto cuando la exposición humana indique la necesidad de utilizar una dosis superior.
1.5.2.5. Período de observación
El período de observación debe ser de 21 días.
1.5.3. Procedimiento
Tras administrar un protector para evitar la muerte por efectos colinérgicos agudos, se administra una sola dosis de la sustancia estudiada.
1.5.3.1. Observación general
La observación debe iniciarse inmediatamente tras la administración. Todas las gallinas deben observarse cuidadosamente varías veces durante los 2 primeros días y posteriormente al menos una vez al día durante un período de 21 días o hasta el sacrificio programado. Deben registrarse todos los signos de toxicidad, con inclusión del momento de aparición, tipo, gravedad y duración de las anomalías de comportamiento. La ataxia debe medirse con una escala de clasificación ordinal consistente en un mínimo de cuatro niveles, y debe registrarse la eventual parálisis. Al menos dos veces por semana deben sacarse de las jaulas las gallinas seleccionadas para la observación de las patologías y someterse a un período de actividad motriz forzada (como subida de una escalera) a fin de facilitar la observación de efectos tóxicos mínimos. Los animales moribundos o que presenten dolor o sufrimiento intenso deben sacarse en cuanto se observen, sacrificarse de forma compasiva y someterse a autopsia.
1.5.3.2. Peso corporal
Deben pesarse todas las gallinas justo antes de la administración de la sustancia estudiada y al menos una vez por semana posteriormente.
1.5.3.3. Bioquímica
En el plazo de unos días después de la administración, deben sacrificarse 6 gallinas seleccionadas aleatoriamente de cada uno de los lotes de tratamiento y de control del vehículo, y tres del lote de control positivo (cuando este lote se utilice en paralelo). El cerebro y la médula lumbar se prepararán y estudiarán para detectar la actividad de inhibición de la esterasa diana de la neuropatía. Además, también puede ser útil preparar y estudiar tejido del nervio ciático para medir la actividad de inhibición de la esterasa diana de la neuropatía. Normalmente, se sacrificarán 3 aves del lote de control y de cada lote de tratamiento a las 24 horas, y otras tres a las 48 horas, mientras que las tres gallinas de los controles positivos se sacrificarán a las 24 horas. Si la observación de signos clínicos de intoxicación (esta puede evaluarse frecuentemente observando el momento de aparición de signos colinérgicos) indica que el agente tóxico se elimina muy despacio, podrá ser preferible tomar dos veces tejidos de 3 aves entre las 24 y las 72 horas a partir de la administración.
También pueden hacerse análisis de la acetilcolinesterasa (ACE) con estas muestras, si se considera conveniente. No obstante, puede darse in vivo una reactivación espontánea de la ACE, llevando así a una subestimación de la potencia de la sustancia como inhibidora de la ACE.
1.5.3.4. Autopsia macroscópica
La autopsia macroscópica de todos los animales (sacrificados de forma programada y sacrificados por encontrarse moribundos) debe incluir la observación del aspecto del cerebro y de la médula espinal.
1.5.3.5. Examen histopatológico
Se someterán a examen microscópico muestras de tejido nervioso de los animales supervivientes al final del período de observación que no se hayan utilizado en los estudios bioquímicos. Los tejidos se fijarán in situ, utilizando técnicas de perfusión. Las secciones serán del cerebelo (nivel medio longitudinal), bulbo raquídeo, médula espinal y nervios periféricos. Las secciones de la médula espinal deben tomarse del segmento cervical superior y de las regiones media-dorsal y lumbo-sacra. Deben tomarse también secciones de la región distal del nervio tibial y sus ramificaciones al músculo gastrocnemio y del nervio ciático. Las secciones se someterán a tinción con colorantes adecuados específicos de la mielina y del axón.
2. RESULTADOS
La obtención de resultados negativos respecto a los parámetros seleccionados en este método (bioquímica, histopatología y observación del comportamiento) no requiere normalmente la realización de más ensayos de neurotoxicidad retardada. La obtención de resultados dudosos o ambiguos respecto a estos parámetros sí puede hacer necesaria otra evaluación.
Deben proporcionarse datos individuales. Además, todos los datos deben resumirse en forma tabular, indicando para cada lote de ensayo el número de animales al inicio de la prueba, el número de animales que presenten lesiones o efectos bioquímicos o de comportamiento, los tipos y gravedad de estas alteraciones, y el porcentaje de animales que presenten cada tipo y grado de alteración.
Las observaciones del presente estudio deben evaluarse en términos de incidencia, gravedad y correlación de los efectos bioquímicos, histopatológicos y de comportamiento, así como cualquier otro efecto observado en los lotes tratados y de control.
Los resultados numéricos deben evaluarse mediante métodos estadísticos adecuados de aceptación general. Los métodos estadísticos deben seleccionarse durante el diseño del estudio.
3. INFORME
INFORME DEL ENSAYO
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
|
Animales de ensayo:
|
|
|
Condiciones del ensayo:
|
|
|
Resultados:
|
|
|
Discusión de tos resultados. |
|
|
Conclusiones. |
4. REFERENCIAS
El presente método es análogo a la TG 418 de la OCDE
B.38. NEUROTOXICIDAD RETARDADA DE SUSTANCIAS ORGANOFOSFORADAS. ESTUDIO POR ADMINISTRACIÓN CONTINUADA DE 28 DÍAS
1. MÉTODO
1.1. INTRODUCCIÓN
En la evaluación de los efectos tóxicos de las sustancias, es importante considerar el potencial de ciertas clases de sustancias para causar tipos específicos de neurotoxicidad que pueden no detectarse con otros estudios de toxicidad. Se ha visto que ciertas sustancias organofosforadas producen neurotoxicidad retardada, por lo que debe considerarse su evaluación.
Pueden emplearse ensayos de cribado in vitro para detectar las sustancias capaces de producir polineuropatía retardada; sin embargo, unos resultados negativos en los estudios in vitro no excluyen que la sustancia estudiada sea neurotóxica.
Este ensayo de 28 días de neurotoxicidad retardada proporciona información sobre los riesgos sanitarios que puede provocar una exposición repetida a lo largo de un período limitado de tiempo. Da información sobre la respuesta a las dosis y proporciona una estimación del nivel sin efectos adversos observados que puede ser útil para establecer criterios de seguridad de la exposición.
Véase la Introducción general, Parte B.
1.2. DEFINICIONES
Sustancias organofosforadas: ésteres organofosforados sin carga, tioésteres o anhídridos de ácidos organofosfóricos, organofosfóricos u organofosforamídicos, o de ácidos fosforotioicos, fosfono-tioicos o fosforotioamídicos afines, u otras sustancias que puedan producir la neurotoxicidad retardada que a veces se aprecia en esta clase de sustancias.
Neurotoxicidad retardada: síndrome asociado con la aparición retardada y prolongada de ataxia, axonopatías distales en la médula espinal y en los nervios periféricos, e inhibición y envejecimiento de la esterasa diana de la neuropatía (NTE) en el tejido nervioso.
1.3. PRINCIPIO DEL MÉTODO
Se dan dosis orales diarias de la sustancia estudiada a gallinas domésticas durante 28 días. Se observa al menos diariamente la aparición en los animales de anomalías de comportamiento, ataxia y parálisis, hasta que pasen 14 días desde la última administración. Se realizan medidas bioquímicas, especialmente la inhibición de la esterasa diana de la neuropatía (NTE), en gallinas seleccionadas aleatoriamente de cada lote, normalmente a las 24 y 48 horas de la última administración. A las dos semanas de la última dosis, se sacrifican las gallinas restantes y se procede al examen histopatológico de tejidos nerviosos seleccionados.
1.4. DESCRIPCIÓN DEL MÉTODO
1.4.1. Preparación
Se seleccionan al azar gallinas jóvenes sanas, libres de enfermedades víricas que puedan interferir y sin tratamiento médico, que no presenten anomalías de la marcha, se asignan a los lotes de tratamiento y de control y se aclimatan a las condiciones del laboratorio durante un mínimo de 5 días antes del inicio del estudio.
Deben utilizarse jaulas o recintos de la capacidad suficiente para permitir la libre movilidad de las gallinas y la fácil observación de su marcha.
La sustancia estudiada se administrará todos y cada uno de los días por vía oral, de preferencia utilizando sondas gástrica o cápsulas de gelatina. Los líquidos pueden administrarse sin diluir o disueltos en un vehículo apropiado, como el aceite de maíz; los sólidos deben disolverse siempre que sea posible, ya que las grandes dosis de productos sólidos en cápsulas de gelatina pueden no absorberse de forma eficaz. Respecto a los vehículos no acuosos, deben conocerse sus características tóxicas; en caso de no conocerse, se determinarán antes de la prueba.
1.4.2. Condiciones del ensayo
1.4.2.1. Animales de experimentación
Se recomienda la gallina ponedora doméstica adulta (Gallus gallus domesticus), de una edad comprendida entre 8 y 12 meses. Deben emplearse razas y variedades de tamaño normal y las gallinas se habrán criado normalmente en condiciones que permitan su movilidad libre.
1.4.2.2. Número y sexo
Se utilizarán generalmente al menos tres lotes de tratamiento y un lote de control del vehículo. El lote de control del vehículo debe tratarse de la misma forma que el lote de tratamiento, salvo que se omitirá la administración de la sustancia estudiada.
En cada lote de aves debe utilizarse un número suficiente de gallinas de forma que puedan matarse al menos 6 aves para hacer las determinaciones bioquímicas (3 en cada uno de los dos momentos seleccionados) y otras 6 puedan sobrevivir durante el período de 14 días tras la exposición destinado a la observación.
1.4.2.3. Dosis
Las dosis deben seleccionarse teniendo en cuenta los resultados de un ensayo de neurotoxicidad aguda retardada y cualesquiera otros datos existentes sobre toxicidad o cinética del compuesto estudiado. La dosis superior debe elegirse con el fin de inducir efectos tóxicos, de preferencia neurotoxicidad retardada, pero sin producir la muerte ni sufrimiento patente. Posteriormente, debe seleccionarse una secuencia descendente de dosis destinadas a demostrar la eventual relación dosis-respuesta y la ausencia de efectos adversos observados con la dosis inferior.
1.4.2.4. Ensayo límite
Si un ensayo con una dosis de al menos 1 000 mg/kg de peso corporal/día, siguiendo los procedimientos descritos para el presente estudio, no produce ningún efecto tóxico observable y si, a partir de los datos procedentes de sustancias relacionadas estructuralmente, no cabe esperar la aparición de fenómenos tóxicos, puede considerarse innecesario un estudio con una dosis superior. El ensayo límite es aplicable excepto cuando la exposición humana prevista indique la necesidad de utilizar una dosis superior
1.4.2.5. Período de observación
Todos los animales se observarán al menos una vez al día durante el período de exposición y durante 14 días después, salvo en los casos de autopsia prevista.
1.4.3. Procedimiento
Los animales reciben la sustancia estudiada todos y cada uno de los días durante un período de 28 días.
1.4.3.1. Observación general
La observación debe iniciarse inmediatamente tras el inicio del tratamiento. Todas las gallinas deben observarse cuidadosamente al menos una vez al día durante cada uno de los 28 días del tratamiento y durante 14 días tras la última administración o hasta el sacrificio programado. Deben registrarse todos los signos de toxicidad, con inclusión del momento de aparición, tipo, gravedad y duración. Las observaciones incluirán las anomalías del comportamiento, sin limitarse a ellas. La ataxia debe medirse con una escala de clasificación ordinal consistente en un mínimo de cuatro niveles, y debe registrarse la eventual parálisis. Al menos dos veces por semana deben sacarse de las jaulas las gallinas y someterse a un período de actividad motriz forzada (como subida de una escalera) a fin de facilitar la observación de efectos tóxicos mínimos. Los animales moribundos o que presenten dolor o sufrimiento intenso deben sacarse en cuanto se observen, sacrificarse de forma compasiva y someterse a autopsia.
1.4.3.2. Peso corporal
Deben pesarse todas las gallinas justo antes de la primera administración de la sustancia estudiada y al menos una vez por semana posteriormente.
1.4.3.3. Bioquímica
En el plazo de unos días después de la última administración, deben sacrificarse 6 gallinas seleccionadas aleatoriamente de cada uno de los lotes de tratamiento y de control del vehículo. El cerebro y la médula lumbar se prepararán y estudiarán para detectar la actividad de inhibición de la esterasa diana de la neuropatía. Además, también puede ser útil preparar y estudiar tejido del nervio ciático para medir la actividad de inhibición de la esterasa diana de la neuropatía. Normalmente, se sacrificarán 3 aves del lote de control y de cada lote de tratamiento a las 24 horas, y otras 3 a las 48 horas de la última administración. Si de datos procedentes del estudio de toxicidad aguda o de otros estudios (por ejemplo, de toxicocinética), se deduce que es preferible utilizar otros tiempos de sacrificio tras la última administración, entonces deberán aplicarse estos tiempos aportando la justificación pertinente.
También pueden hacerse análisis de la acetilcolinesterasa (ACE) con estas muestras, si se considera conveniente. No obstante, puede darse in vivo una reactivación espontánea de la ACE, llevando así a una subestimación de la potencia de la sustancia como inhibidora de la ACE.
1.4.3.4. Autopsia macroscópica
La autopsia macroscópica de todos los animales (sacrificados de forma programada y sacrificados por encontrarse moribundos) debe incluir la observación del aspecto del cerebro y de la médula espinal.
1.4.3.5. Examen histopatológico
Se someterán a examen microscópico muestras de tejido nervioso de los animales supervivientes al final del período de observación que no se hayan utilizado en los estudios bioquímicos. Los tejidos se fijarán in situ, utilizando técnicas de perfusión. Las secciones serán del cerebelo (nivel medio longitudinal), bulbo raquídeo, médula espinal y nervios periféricos. Las secciones de la médula espinal deben tomarse del segmento cervical superior y de las regiones media-dorsal y lumbo-sacra. Deben tomarse también secciones de la región distal del nervio tibial y sus ramificaciones al músculo gastrocnemio y del nervio ciático. Las secciones se someterán a tinción con colorantes adecuados específicos de la mielina y del axón. En principio, debe hacerse el examen microscópico de los tejidos conservados de todos los animales de los lotes de control y de dosis alta. Cuando haya pruebas de efectos en el lote de dosis alta, deberá hacerse también el examen microscópico de las gallinas de los lotes de dosis intermedia y baja.
2. RESULTADOS
La obtención de resultados negativos respecto a los parámetros seleccionados en este método (bioquímica, histopatología y observación del comportamiento) no requiere normalmente la realización de más ensayos de neurotoxicidad retardada. La obtención de resultados dudosos o ambiguos respecto a estos parámetros sí puede hacer necesaria otra evaluación.
Deben proporcionarse datos individuales. Además, todos los datos deben resumirse en forma tabular, indicando para cada lote de ensayo el número de animales al inicio de la prueba, el número de animales que presenten lesiones o efectos bioquímicos o de comportamiento, los tipos y gravedad de estas alteraciones, y el porcentaje de animales que presenten cada tipo y grado de alteración.
Las observaciones del presente estudio deben evaluarse en términos de incidencia, gravedad y correlación de los efectos bioquímicos, histopatológicos y de comportamiento, así como cualquier otro efecto observado en los lotes tratados y de control.
Los resultados numéricos deben evaluarse mediante métodos estadísticos adecuados de aceptación general. Los métodos estadísticos deben seleccionarse durante el diseño del estudio.
3. INFORME
INFORME DEL ENSAYO
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
|
Animales de ensayo:
|
|
|
Condiciones del ensayo:
|
|
|
Resultados:
|
|
|
Discusión de los resultados |
|
|
Conclusiones |
4. REFERENCIAS
El presente método es análogo a la TG 419 de la OCDE.»
«B.39. ENSAYO DE SÍNTESIS DE ADN NO PROGRAMADA (UDS) EN HEPATOCITOS DE MAMÍFERO IN VIVO
1. MÉTODO
El presente método reproduce las directrices del documento OCDE TG 486 sobre el ensayo de síntesis de ADN no programada (UDS) en hepatocitos de mamífero in vivo (1997).
1.1. INTRODUCCIÓN
El ensayo de síntesis de ADN no programada (UDS) en hepatocitos de mamífero in vivo tiene por objeto determinar sustancias de ensayo que inducen la reparación del ADN en los hepatocitos de animales tratados (1) (2) (3) (4).
El presente ensayo in vivo constituye un método de investigación de los efectos genotóxicos de los productos químicos en el hígado y sirve para detectar lesiones en el ADN y su posterior reparación en las células hepáticas. El hígado suele ser el lugar donde más se metabolizan los compuestos absorbidos y por ello constituye el órgano más apropiado para la detección de lesiones del ADN in vivo.
Si hay pruebas de que la sustancia de ensayo no llega al tejido diana, no procede realizar el presente ensayo.
La síntesis de ADN no programada (UDS) se determina midiendo la incorporación de nucleósidos marcados en células que no se encuentran en la fase S de síntesis programada de ADN. La técnica más empleada consiste en determinar la captación de timidina tritiada (3H-TdR) por autorradiografía. Para los ensayos de UDS in vivo es preferible utilizar hígados de rata. También pueden emplearse otros tejidos, si bien no se tratan en las presentes directrices.
La detección de una respuesta de UDS depende del número de bases del ADN eliminadas y repuestas en el sitio de la lesión. De ahí que el ensayo de UDS sea particularmente útil para detectar la reparación de secuencias largas (20 a 30 bases; secuencias largas de reparación) inducida por una sustancia. Sin embargo, la sensibilidad del ensayo para detectar la reparación de secuencias cortas (1 a 3 bases; secuencias cortas de reparación) es mucho menor. Por otra parte, las alteraciones mutagénicas pueden deberse a una falta de reparación o a una reparación o replicación erróneas de las lesiones del ADN. La magnitud de la respuesta de UDS no es indicativa de la fidelidad del proceso de reparación. Además de ello, es posible que un mutágeno reaccione con el ADN, pero que la lesión de este no se repare por un mecanismo de reparación de escisión. La falta de información específica sobre la actividad mutagénica que caracteriza al ensayo UDS se ve compensada por su sensibilidad potencial, ya que detecta los efectos en todo el genoma.
Véase asimismo la Introducción general de la parte B.
1.2. DEFINICIONES
Células en reparación: células con un número neto de granulaciones nucleares (NGN) superior a un número preestablecido que el laboratorio donde se realiza el ensayo ha de fundamentar.
Número neto de granulaciones nucleares (NGN): medida cuantitativa de la síntesis de ADN no programada en las células por autorradiografía: se calcula restando el número medio de granulaciones citoplasmáti-cas (GC) en las zonas citoplasmáticas equivalentes al núcleo del número de granulaciones nucleares (GN): NGN = GN - GC. Primero se calcula el NGN de cada célula y después el correspondiente a todo un cultivo o a cultivos paralelos, etc.
Síntesis de ADN no programada (UDS): síntesis de reparación de ADN tras la escisión y eliminación de un segmento de ADN que contiene una región dañada por sustancias químicas o agentes físicos.
1.3. PRINCIPIO DEL MÉTODO
El ensayo de síntesis de ADN no programada en hepatocitos de mamífero in vivo detecta la síntesis de reparación del ADN tras la escisión y eliminación de un segmento de ADN que contiene una región dañada por sustancias químicas o agentes físicos. El ensayo suele fundarse en la incorporación de 3H-TdR al ADN de hepatocitos de los cuales una escasa proporción se encuentra en la fase S del ciclo celular. La incorporación de 3H-TdR suele determinarse por autorradiografía, ya que esta técnica no es tan sensible a las interferencias debidas a las células en fase S como lo es, por ejemplo, el recuento por centelleo de líquidos.
1.4. DESCRIPCIÓN DEL MÉTODO
1.4.1. Preparación
1.4.1.1. Selección de la especie animal
Si bien suelen emplearse ratas, pueden utilizarse otras especies de mamíferos apropiadas. Deben usarse animales adultos jóvenes y sanos de cepas utilizadas habitualmente en laboratorio. La variación del peso de los animales al empezar el estudio ha de ser mínima y no debe exceder de ± 20 % del peso medio de cada sexo.
1.4.1.2. Alojamiento y alimentación de los animales
Se aplicarán las condiciones generales recogidas en la Introducción general de la parte B. La humedad debe ser del 50 al 60 %.
1.4.1.3. Preparación de los animales
Se reparten al azar animales adultos jóvenes y sanos entre los lotes tratados y los lotes de control. Las jaulas se colocan de manera que los posibles efectos debidos a la posición de las mismas sean mínimos. Se identifica a los animales de forma unívoca y se los mantiene en las jaulas al menos 5 días antes de empezar el estudio para que se acostumbren a las condiciones de laboratorio.
1.4.1.4. Preparación de la sustancia de ensayo
Las sustancias de ensayo sólidas deben disolverse o suspenderse en disolventes o vehículos adecuados y, si es preciso, diluirse antes de su administración a los animales. Las sustancias de ensayo líquidas pueden administrarse directamente o diluirse antes de la administración. Han de emplearse soluciones de la sustancia de ensayo recién preparadas, salvo que los datos relativos a la estabilidad demuestren que es posible conservarlas.
1.4.2. Condiciones del ensayo
1.4.2.1. Disolvente o vehículo
El disolvente o vehículo no deberá producir efectos tóxicos a las dosis empleadas. Es preciso cerciorarse de que el disolvente o vehículo no produce reacciones químicas con la sustancia de ensayo. Si se emplean disolventes o vehículos poco conocidos, debe disponerse de información que avale su compatibilidad. Siempre que sea posible, se recomienda considerar en primer lugar la utilización de un disolvente o vehículo acuoso.
1.4.2.2. Controles
Deberán realizarse controles positivos y negativos (disolvente o vehículo) en paralelo en cada parte independiente del experimento. Salvo por lo que respecta a la exposición a la sustancia de ensayo, los animales de los lotes de control deben tratarse exactamente igual que los de los lotes tratados.
En los controles positivos se emplearán sustancias de las que se sepa que inducen la síntesis de ADN no programada cuando se administran en dosis con las que se prevé un aumento detectable respecto a la frecuencia espontánea. Los controles positivos que requieran activación metabólica se realizarán con dosis que provoquen una respuesta moderada (4). Las dosis del control positivo pueden ser tales que los efectos sean claros, pero no revelen inmediatamente al lector la identidad de los portaobjetos codificados. A continuación figuran algunos ejemplos de sustancias para los controles positivos:
|
Período de muestreo |
Sustancia |
No CAS |
No EINECS |
|
Corto (2 a 4 horas) |
N-nitrosodimetilamina |
62-75-9 |
200-249-8 |
|
Largo (12 a 16 horas) |
N-2-fluorenilacetamida (2-AAF) |
53-96-3 |
200-188-6 |
Pueden emplearse otras sustancias adecuadas para los controles positivos. La sustancia del control positivo puede administrarse por una vía distinta de la de la sustancia de ensayo.
1.5. PROCEDIMIENTO
1.5.1. Número y sexo de los animales
El número de animales deberá decidirse tomando en consideración la variación biológica natural de la respuesta al ensayo. Cada lote estará constituido al menos por 3 animales analizables. Si se dispone de una base de datos de controles históricos importante, bastarán 1 o 2 animales para los lotes de los controles negativos y positivos realizados en paralelo.
Si en el momento en que se efectúe el estudio se dispone de datos sobre otros estudios realizados con la misma especie y la misma vía de exposición, que demuestren que no hay diferencia significativa entre sexos en cuanto a la toxicidad, será suficiente el ensayo en un solo sexo, preferiblemente en machos. En caso de que la exposición humana a sustancias químicas pueda ser específica de un sexo, como sucede con algunos productos farmacéuticos, el ensayo se realizará con animales del sexo correspondiente.
1.5.2. Pauta de tratamiento
Las sustancias de ensayo suelen administrarse en una sola vez.
1.5.3. Dosis
En principio, se administran al menos dos dosis distintas. Se entiende por “dosis máxima” la que produce tales signos de toxicidad que, si se administrara una dosis superior según el mismo protocolo, resultaría probablemente letal. Por lo general, la dosis inferior es del 50 % al 25 % de la dosis máxima.
Las sustancias con actividades biológicas específicas a dosis bajas no tóxicas (como las hormonas y los mitó-genos) pueden constituir excepciones en cuanto a los criterios de establecimiento de la dosis y han de evaluarse caso por caso. Si se lleva a cabo un estudio previo de determinación de la gama de dosis por falta de datos adecuados, ha de hacerse en el mismo laboratorio, con la misma especie, cepa, sexo y protocolo de tratamiento que el estudio principal.
La dosis máxima también puede definirse como la dosis que produce algún signo de toxicidad hepática (por ejemplo, núcleos picnóticos).
1.5.4. Ensayo límite
Si en un ensayo realizado con una dosis de al menos 2 000 mg/kg de peso corporal administrada en una sola vez, o dos veces en el mismo día, no se observan efectos tóxicos y si, sobre la base de datos relativos a sustancias de estructura afín, no cabe esperar genotoxicidad, puede no ser necesario llevar a cabo un estudio completo. Si se prevé la exposición humana a la sustancia, puede ser preciso administrar una dosis mayor en el ensayo límite.
1.5.5. Administración de las dosis
La sustancia de ensayo suele administrarse por sonda gástrica o con una cánula de intubación adecuada. Pueden emplearse otras vías de administración, siempre que se justifiquen. La vía intraperitoneal no está indicada, ya que podría exponer el hígado a la sustancia de ensayo directamente en lugar de a través del sistema circulatorio. El volumen máximo de líquido que puede administrarse de una sola vez por sonda o inyección depende del tamaño del animal de ensayo, pero no excederá de 2 ml/100 g de peso corporal. Deberá justificarse la administración de volúmenes superiores a este. Salvo en el caso de sustancias irritantes o corrosivas, que por lo general producen efectos exacerbados con concentraciones mayores, deberá reducirse al mínimo la variabilidad del volumen de ensayo ajustando la concentración de manera que el volumen sea el mismo en todas las dosis.
1.5.6. Preparación de los hepatocitos
Se extraen hepatocitos de los animales tratados, por lo general de 12 a 16 horas después de la administración de la sustancia de ensayo. Suele ser necesaria otra toma previa (generalmente de 2 a 4 horas después del tratamiento), salvo que a las 12-16 horas haya una respuesta positiva clara. No obstante, pueden aplicarse otros períodos de muestreo, si así lo justifican los datos toxicocinéticos.
Los cultivos de hepatocitos de mamífero a corto plazo se realizan perfundiendo el hígado in situ con colage-nasa y dejando que los hepatocitos recién disociados se adhieran a una superficie adecuada. La viabilidad de los hepatocitos de los animales del control negativo ha de ser del 50 % como mínimo (5).
1.5.7. Determinación de la síntesis de ADN no programada
Los hepatocitos de mamífero recién aislados se incuban en un medio con 3H-TdR durante un tiempo adecuado, por ejemplo, de 3 a 8 horas. Al término de la incubación, se retiran las células del medio y pueden entonces incubarse en otro medio con un exceso de timidina sin tratar con el fin de reducir la radiactividad no incorporada (cold chase). A continuación, se aclaran las células, se fijan y se tiñen. Esta segunda incubación puede no ser necesaria si la primera ha sido más larga. Se bañan los portaobjetos en una emulsión auto-rradiográfica, se exponen en la oscuridad (por ejemplo, refrigerados, de 7 a 17 días), se revelan y tiñen y se cuentan los granos de plata expuestos. Se preparan dos o tres portaobjetos por animal.
1.5.8. Análisis
Las preparaciones han de contener un número suficiente de células de morfología normal para que la evaluación de la síntesis de ADN no programada sea significativa.
Se observan las preparaciones al microscopio en busca de signos de citotoxicidad manifiesta (por ejemplo, picnosis, índice reducido de marcado radiactivo, etc.).
Se codifican los portaobjetos antes del recuento de granulaciones. Por lo general, se cuentan 100 células por animal, en dos portaobjetos como mínimo. El recuento de menos de 100 células por animal deberá estar debidamente justificado. Si bien los núcleos en fase S no se consideran en el recuento de granulaciones, puede registrarse la proporción de células en fase S.
Se determinará con métodos apropiados la cantidad de 3H-TdR incorporada en los núcleos y el citoplasma de las células con morfología normal, revelada por el depósito de granos de plata.
2. RESULTADOS
2.1. TRATAMIENTO DE LOS RESULTADOS
Se proporcionarán los datos de cada portaobjetos y de cada animal. Se resumirán todos los datos en forma de tabla. Se calculará el número neto de granulaciones nucleares (NGN) por célula, por animal y por dosis y tiempo restando el número de GC del de GN. Si se cuentan las células en reparación, se motivará el criterio aplicado para definir las “células en reparación”, que deberá fundarse en datos relativos a controles históricos negativos o realizados en paralelo al ensayo. Los resultados numéricos pueden evaluarse por medio de métodos estadísticos, que deberán seleccionarse y justificarse antes de llevar a cabo el estudio.
2.2. EVALUACIÓN E INTERPRETACIÓN DE LOS RESULTADOS
Entre los criterios para determinar si una respuesta es positiva o negativa se encuentran los siguientes:
|
respuesta positiva: |
i) |
el NGN es superior a un valor preestablecido fundado en datos de controles históricos obtenidos en el laboratorio, o |
|
|
ii) |
el NGN supera de forma significativa al de los controles realizados en paralelo; |
|
respuesta negativa: |
i) |
el NGN no supera el umbral correspondiente a controles históricos, o |
|
|
ii) |
el NGN no supera de forma significativa al de los controles realizados en paralelo. |
Debe considerarse la importancia biológica de los resultados. Se tomarán en consideración, por ejemplo, la variabilidad entre animales, la relación dosis-respuesta y la citotoxicidad. Pueden emplearse métodos estadísticos como apoyo para evaluar los resultados del ensayo. El hecho de que los datos estadísticos sean significativos no ha de ser el único factor para determinar que una respuesta es positiva.
Si bien en la mayoría de los experimentos los resultados serán claramente positivos o negativos, en algunos casos el conjunto de datos no permitirá emitir un juicio definitivo sobre la actividad de la sustancia de ensayo. Con independencia del número de veces que se repita el experimento, los resultados pueden seguir siendo ambiguos o dudosos.
Un resultado positivo en el ensayo de síntesis de ADN no programada en hepatocitos de mamífero in vivo indica que la sustancia de ensayo provoca lesiones del ADN en los hepatocitos de mamífero in vivo que pueden repararse por síntesis de ADN no programada in vitro. Un resultado negativo indica que, en las condiciones del ensayo, la sustancia de ensayo no provoca lesiones del ADN detectables mediante este ensayo.
Deberá estudiarse la probabilidad de que la sustancia de ensayo pase al torrente circulatorio o llegue específicamente al tejido diana (toxicidad sistémica, etc.).
3. INFORME
INFORME DEL ENSAYO
El informe del ensayo incluirá la información siguiente:
|
|
Disolvente o vehículo:
|
|
|
Animales de ensayo:
|
|
|
Condiciones del ensayo:
|
|
|
Resultados:
|
|
|
Discusión de los resultados. |
|
|
Conclusiones. |
4. REFERENCIAS
|
(1) |
Ashby, J., Lefevre, F. A., Burlinson, B. and Penman, M. G. (1985), An Assessment of the In Vivo Rat Hepatocyte DNA Repair Assay, Miuation Res., 156, pp. 1-18. |
|
(2) |
Butterworth, B. E., Ashby, J., Bermúdez, E., Casciano, D., Mirsalis, J., Probst G. and Williams, G. (1987), A Protocol and Guide for the In Vivo Rat Hepatocyte DNA-Repair Assay, Mutation Res., 189, pp. 123-133. |
|
(3) |
Kennelly, J. C, Waters, R., Ashby, J., Lefevre, P. A., Burlinson B., Benford, D. J., Dean, S. W. and Mitchell I. de G. (1993), In Vivo Rat Liver UDS Assay, in: Kirkland D. J. and Fox M., (eds.), Suppkmentary Mutage-nicity Tests; UKEM Recommended Procedures. UKEMS Subcommittee on Guidelines for Mutagenicity Testing Repon. Part II revised, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 52-77. |
|
(4) |
Madle, S., Dean, S. W., Andrae, U., Brambilla, G., Burlinson, B., Doolittle, D. J., Furihata, C, Hertner, T., McQueen, C. A. and Mori, H. (1993), Recommendations for the Performance of UDS Tests In Vitro and In Vivo, Mutations Res, 312, pp. 263-285. |
|
(5) |
Fautz, R., Hussain, B., Efstathiou, E. and Hechenberger-Freudl, C. (1993), Assessment of the Relation Bet-ween the Initital Viability and the Attachment of Freshly Isolated Rat Hepatocytes Used for the In Vivo/In Vitro DNA Repair Assay (UDS), Mutation Res., 291, pp. 21-27. |
|
(6) |
Mirsalis, J. C, Tyson, C. K. and Butterworth, B. E. (1982), Detection of Genotoxic Carcinogens in the In Vivo/In Vitro Hepatocyte DNA Repair Assay, Environ Mutagen, 4, pp. 55 3-562.» |
B.40. CORROSIÓN CUTÁNEA IN VITRO: ENSAYO DE RESISTENCIA ELÉCTRICA TRANSCUTÁNEA (RET)
1. MÉTODO
El presente método de ensayo es equivalente a las directrices de ensayo de la OCDE TG 430 (2004).
1.1. INTRODUCCIÓN
La corrosión cutánea se refiere a la producción de daños tisulares irreversibles en la piel tras la aplicación de una sustancia [según la definición del Sistema Armonizado Mundial (Globally Harmonized System, GHS) de clasificación y etiquetado de productos químicos] (1). El presente método contempla un procedimiento para evaluar la corrosividad sin recurrir a animales vivos.
Tradicionalmente, la evaluación de la corrosividad cutánea se hacía con animales de laboratorio (2). La preocupación por el dolor y el sufrimiento de los animales utilizados en esa evaluación se ha tenido en cuenta en la revisión del método de ensayo B.4, para permitir la determinación de la corrosión cutánea mediante métodos alternativos, in vitro, que eviten el dolor y el sufrimiento.
Un primer paso hacia la definición de métodos alternativos que se pudieran utilizar en los ensayos de corrosividad cutánea con fines normativos fue la realización de estudios de prevalidación (3). Posteriormente se llevó a cabo un estudio oficial de validación de métodos in vitro para evaluar la corrosión cutánea (4) (5) (6) (7) (8). Como consecuencia de estos estudios y de otros datos publicados, se llegó a la recomendación de que los siguientes métodos podían utilizarse para predecir el efecto corrosivo in vivo de la piel (9) (10) (11): el ensayo con un modelo de piel humana (véase el método de ensayo 40 bis) y el ensayo de resistencia eléctrica transcutánea (el presente método).
Mediante un estudio de validación y otros estudios publicados se ha sabido que el ensayo de resistencia eléctrica transcutánea (RET) (12) (13) puede discriminar de forma fiable entre agentes corrosivos y no corrosivos conocidos de la piel (5) (9).
El ensayo descrito en el presente método permite la identificación de sustancias químicas y mezclas corrosivas. Asimismo permite la identificación de sustancias y mezclas no corrosivas con el apoyo de pruebas sopesadas derivadas de otro tipo de información disponible (por ejemplo, pH, relaciones estructura-actividad, datos con hombres o animales) (1) (2) (11) (14). No da información sobre la irritación cutánea ni permite la subcategorización de sustancias corrosivas que se contempla en el Sistema Armonizado Mundial de clasificación (1).
Para una evaluación completa de los efectos cutáneos locales tras una única exposición de la piel, se recomienda seguir la estrategia de ensayos secuenciales que figura adjunta al método de ensayo B.4 (2) y que proporciona el Sistema Armonizado Mundial (1). Esta estrategia de ensayos incluye la realización de pruebas in vitro de corrosión cutánea (como las descritas en el presente método) y de irritación cutánea antes de considerar las pruebas con animales vivos.
1.2. DEFINICIÓN
Se entenderá por: corrosión cutánea in vivo la producción de lesiones cutáneas irreversibles consistentes en necrosis visible en todo el espesor de la epidermis y hasta en la dermis, debida a la aplicación de una sustancia durante hasta 4 horas; las reacciones de corrosión se tipifican como úlceras, hemorragia, costras hemorrágicas y, hacia el final del período de observación de 14 días, como decoloración de la piel, zonas de alopecia completa y cicatrices; debe recurrirse a la histopatología para evaluar las lesiones dudosas;
resistencia eléctrica transcutánea (RET) la medida de la impedancia eléctrica de la piel, como valor de resistencia en kiloohmios; se trata de un método simple y sólido para evaluar la función de barrera, consistente en registrar el paso de iones a través de la piel mediante el uso de un puente de Wheatstone.
1.3. SUSTANCIAS DE REFERENCIA
Cuadro 1
Sustancias de referencia
|
Denominación |
No EINECS |
No CAS |
|
|
1,2-diaminopropano |
201-155-9 |
78-90-0 |
Fuertemente corrosivo |
|
Ácido acrílico |
201-177-9 |
79-10-7 |
Fuertemente corrosivo |
|
2-terc-butilfenol |
201-807-2 |
88-18-6 |
Corrosivos |
|
Hidróxido de potasio (10 %) |
215-181-3 |
1310-58-3 |
Corrosivos |
|
Ácido sulfúrico (10 %) |
231-639-5 |
7664-93-9 |
Corrosivos |
|
Ácido octanoico (ácido caprílico) |
204-677-5 |
124-07-02 |
Corrosivos |
|
4-amino-1,2,4-triazol |
209-533-5 |
584-13-4 |
No corrosivo |
|
Eugenol |
202-589-1 |
97-53-0 |
No corrosivo |
|
Bromuro de fenetilo |
203-130-8 |
103-63-9 |
No corrosivo |
|
Tetracloroetileno |
204-825-9 |
27-18-4 |
No corrosivo |
|
Ácido isoesteárico |
250-178-0 |
30399-84-9 |
No corrosivo |
|
4-(metiltio)-benzaldehído |
222-365-7 |
3446-89-7 |
No corrosivo |
La mayoría de estas sustancias proceden de la lista de sustancias seleccionadas para el estudio internacional de validación del ECVAM (4). Su elección se basa en los criterios siguientes:
|
i) |
igual número de sustancias corrosivas y no corrosivas, |
|
ii) |
sustancias disponibles en el comercio y representativas de la mayoría de las clases químicas pertinentes, |
|
iii) |
inclusión de sustancias fuertemente corrosivas y de sustancias menos corrosivas para permitir la discriminación según la potencia corrosiva, |
|
iv) |
elección de sustancias que puedan manipularse en un laboratorio sin plantear peligros graves aparte de la corrosividad. |
1.4. PRINCIPIO DEL MÉTODO DE ENSAYO
La sustancia problema se aplica durante un máximo de 24 horas en la superficie epidérmica de discos de piel en un sistema de ensayo bicompartimental, donde los discos hacen de separación entre los compartimentos. Los discos de piel se toman de ratas, sacrificadas de forma compasiva a los 28 o 30 días de edad. Las sustancias corrosivas se caracterizan por su capacidad de producir una pérdida de la integridad de la capa córnea normal y de la función de barrera. Esta pérdida se mide como reducción de la RET por debajo de un umbral (12). En relación con la RET de ratas, se ha elegido un valor límite de 5 kΩ basándose en muchos datos sobre una amplia gama de sustancias, ya que la gran mayoría de valores estaba claramente muy por encima (a menudo > 10 kΩ), o muy por debajo (a menudo < 3 kΩ) de este valor (12). Generalmente, el valor de la RET reducida por los materiales que no son corrosivos para los animales, tanto si son irritantes como si no, queda por encima de este límite. Por otra parte, el uso de otros preparados de piel o de otros equipos puede alterar el valor límite, lo que hace necesaria otra validación.
Se incorpora al procedimiento una fase de fijación de colorante para confirmar los resultados positivos con la RET, incluidos los casos de valores próximos a 5 kΩ. La fijación de colorante determina si el aumento de la permeabilidad iónica se debe a la destrucción física de la capa córnea. Se ha visto que el método de RET con piel de rata sirve para predecir el valor de la corrosividad in vivo del conejo, evaluada según el método de ensayo B.4 (2). Debe señalarse que el ensayo in vivo con conejos da unos valores muy moderados de corrosividad cutánea y de irritación cutánea si se compara con el ensayo del parche de piel humana (15).
1.5. DESCRIPCIÓN DEL MÉTODO
1.5.1. Animales
La rata es la especie elegida porque ya se ha demostrado previamente la sensibilidad de su piel a las sustancias de esta prueba (10). La edad (en el momento de tomar la piel) y la cepa de la rata son especialmente importantes para conseguir que los folículos pilosos estén en fase latente antes de que se inicie el crecimiento del pelo de los adultos.
Se quita cuidadosamente con maquinilla pequeña el pelo dorsal y lateral de ratas jóvenes, de unos 22 días de edad, machos o hembras, de una cepa derivada de la Wistar o de otra comparable. A continuación se lavan los animales frotándolos cuidadosamente al mismo tiempo que se cubre la zona pelada con una solución antibiótica (que contenga, por ejemplo, estreptomicina, penicilina, cloranfenicol y anfotericina en concentraciones que sean eficaces para inhibir el crecimiento bacteriano). Los animales se vuelven a lavar con antibióticos al tercer o cuarto día después del primer lavado y se utilizan en el plazo de 3 días desde el segundo lavado, cuando la capa córnea se ha recuperado de la eliminación del pelo.
1.5.2. Preparación de los discos de piel
Los animales se sacrifican compasivamente cuando tienen de 28 a 30 días (esta edad es de importancia fundamental). A continuación se retira la piel dorsolateral de cada animal y se le quita el exceso de grasa subcutánea, separando esta cuidadosamente de la piel. Se toman discos de piel de un diámetro aproximado de 20 mm. Puede guardarse la piel antes de utilizar los discos si se demuestra que los datos obtenidos con controles positivos y negativos son equivalentes a los obtenidos con piel fresca.
Cada disco de piel se coloca sobre uno de los extremos de un tubo de PTFE (politetrafluoretileno), comprobando que la superficie epidérmica esté en contacto con el mismo. Se coloca a presión un anillo de goma en el extremo del tubo para mantener la piel en su sitio y se recorta el tejido sobrante. En la figura 2 se dan las dimensiones del anillo y del tubo. Después se sella con cuidado el anillo de goma al extremo del tubo de PTFE con vaselina. El tubo se sujeta mediante una pinza de muelle dentro de una cámara receptora que contiene una solución de MgSO4 (154 mM) (figura 1). El disco de piel debe quedar totalmente sumergido en la solución de MgSO4. De la piel de una sola rata pueden obtenerse hasta 10 o 15 discos de piel.
Antes de iniciar el ensayo, se mide la resistencia eléctrica de dos discos de piel, para controlar la calidad de la piel de cada animal. El valor de la resistencia de ambos discos debe ser superior a 10 kΩ para que los discos restantes puedan utilizarse en el ensayo. Si el valor de la resistencia es inferior a 10 kΩ, hay que desechar los discos restantes de la misma piel.
1.5.3. Aplicación de las sustancias problema y de control
En cada estudio deben utilizarse a la vez controles positivos y negativos para garantizar el funcionamiento adecuado del modelo experimental. Han de utilizarse discos de piel procedentes de un solo animal. Las sustancias de control positivo y negativo que se proponen son ácido clorhídrico 10 M y agua destilada, respectivamente.
Las sustancias problema líquidas (150 μL) se aplican uniformemente a la superficie epidérmica dentro del tubo. Cuando se prueban materias sólidas se aplica uniformemente al disco una cantidad suficiente del sólido de tal manera que quede cubierta toda la superficie de la epidermis. A continuación se vierte encima del sólido agua desionizada (150 μL) y se agita suavemente el tubo. A fin de conseguir el contacto máximo con la piel, es posible que los sólidos deban calentarse a 30 oC para fundir o reblandecer la sustancia problema, o bien molerse para obtener polvo o material granuloso.
Para cada sustancia problema y de control se utilizan tres discos de piel. Las sustancias problema se aplican durante 24 horas a 20-23 oC; luego se eliminan lavando con un chorro de agua del grifo a una temperatura máxima de 30 oC, hasta que no pueda eliminarse más material.
1.5.4. Mediciones de la RET
La impedancia de la piel, tomada como RET, se mide con un puente de Wheatstone de corriente alterna y baja tensión (13). Las especificaciones generales del puente son una tensión de funcionamiento de 1-3 V, una corriente alterna de forma sinusoidal o rectangular de 50-1 000 Hz, y un intervalo de medición como mínimo de 0,1 a 30 kΩ. El puente utilizado en el estudio de validación medía valores de inductancia, capacitancia y resistencia de hasta 2 000 H, 2 000 μF, y 2 MΩ, respectivamente, a frecuencias de 100 Hz o 1 kHz, utilizando montajes en serie o en paralelo. A efectos de la RET, las mediciones del ensayo de corrosividad se registran como resistencia, a la frecuencia de 100 Hz y utilizando montajes en serie. Antes de medir la resistencia eléctrica, se reduce la tensión superficial de la piel añadiendo el volumen suficiente de etanol al 70 % para cubrir la epidermis. Tras unos segundos se retira el etanol y después se hidrata el tejido aplicando 3 mL de una solución de MgSO4 (154 mM). Los electrodos del puente se colocan a ambos lados del disco de piel para medir la resistencia en kΩ/disco de piel (figura 1). En la figura 2 se dan las dimensiones de los electrodos y la longitud del electrodo expuesta por debajo de las pinzas de cocodrilo. La pinza que sujeta el electrodo interior se apoya encima del tubo de PTFE durante la medición de la resistencia para asegurarse de que es constante la longitud del electrodo que queda sumergida en la solución de MgSO4. El electrodo exterior se coloca dentro de la cámara receptora de manera que se apoye en el fondo de la misma. La distancia entre la pinza de muelle y el fondo del tubo de PTFE se ha de mantener constante (figura 2), ya que afecta al valor de la resistencia obtenido. Por tanto, la distancia entre el electrodo interior y el disco de piel debe ser constante y mínima (1-2 mm).
Si el valor medido de la resistencia es superior a 20 kΩ, puede deberse a que haya restos de la sustancia problema recubriendo la superficie epidérmica del disco de piel. Puede intentarse la eliminación de este recubrimiento, por ejemplo, sellando el tubo de PTFE con el pulgar cubierto de un guante y agitándolo aproximadamente 10 segundos; se tira la solución de MgSO4 y se repite la medición de la resistencia con solución nueva de MgSO4.
Las propiedades y dimensiones del instrumental y el procedimiento experimental utilizado pueden influir en los valores obtenidos de RET. El umbral de corrosión de 5 kΩ se ha determinado a partir de datos obtenidos con el instrumental y el procedimiento concretos descritos en el presente método. Si se modifican las condiciones del ensayo o se utiliza un instrumental diferente, es posible que deban aplicarse diferentes umbrales y valores de control. Por tanto, es necesario calibrar el método y los valores umbral de resistencia sometiendo a ensayo una serie de de patrones de referencia elegidos de entre las sustancias utilizadas en el estudio de validación (4) (5) o de clases químicas similares a las sustancias problema. En el cuadro 1 se ofrece un conjunto de sustancias de referencia adecuadas.
1.5.5. Métodos de fijación de colorante
La exposición de determinados materiales no corrosivos puede dar lugar a una reducción de la resistencia por debajo del umbral de 5 kΩ por permitir el paso de iones a través de la capa córnea, reduciendo así la resistencia eléctrica (5). Por ejemplo, los productos orgánicos neutros y las sustancias químicas que tienen propiedades tensoactivas (incluidos los detergentes, emulgentes y otros agentes tensoactivos) pueden eliminar lípidos cutáneos, lo que aumenta la permeabilidad de la barrera a los iones. Así pues, si los valores de RET de una sustancia problema están por debajo o cerca de 5 kΩ, sin que visualmente se aprecien lesiones, deberá efectuarse una evaluación de la penetración de colorante en los tejidos tratado y de control, a fin de determinar si los valores de RET obtenidos son resultado de un aumento de la permeabilidad de la piel o bien de la corrosión de esta (3) (5). En este último caso, si la capa córnea está destruida, el colorante sulforrodamina B aplicado a la superficie de la piel penetra rápidamente en el tejido subyacente y lo tiñe. Este colorante en particular es estable ante una amplia gama de sustancias y no se ve afectado por el procedimiento de extracción que se describe más abajo.
1.5.5.1. Aplicación y eliminación del colorante sulforrodamina B
Tras la evaluación de la RET, se retira el sulfato de magnesio del tubo y se examina cuidadosamente la piel para detectar la presencia de lesiones evidentes. Si no hay ninguna lesión evidente importante, se aplican durante 2 horas a la superficie epidérmica de cada disco de piel 150 μL de una dilución al 10 % (p/v) del colorante sulforrodamina B (Acid Red 52; C.I. 45100; no EINECS 222-529-8; no CAS 3520-42-1) en agua destilada. Después se lavan estos discos de piel con agua del grifo, durante unos 10 segundos y a temperatura no superior a la ambiente, para eliminar el exceso de colorante no fijado. Cada disco de piel se retira cuidadosamente del tubo de PTFE y se coloca en un frasco (por ejemplo, un frasco de cristal de centelleo de 20 mL) con agua desionizada (8 mL). Los frascos se agitan suavemente durante 5 minutos para eliminar los eventuales restos de colorante no fijado. Este procedimiento de aclarado se repite a continuación, después de lo cual se retiran los discos cutáneos y se colocan en frascos que contengan 5 ml de dodecilsulfato sódico (SDS) al 30 % (p/v) en agua destilada y se dejan incubar una noche a 60 oC.
Después de la incubación, se retiran y desechan los discos de piel y la solución restante se centrifuga durante 8 minutos a 21 oC (fuerza centrífuga relativa ~175 × g). Luego, se diluye de 1 a 5 (v/v) [es decir, 1 mL + 4 mL] una alícuota de 1 mL del sobrenadante con dodecilsulfato sódico al 30 % (p/v) en agua destilada. Se mide la densidad óptica (DO) de la solución a 565 nm.
1.5.5.2. Cálculo del contenido de colorante
El contenido del colorante sulforrodamina B por disco se calcula a partir de los valores de DO (5) (coeficiente de extinción molar del colorante sulforrodamina B a 565 nm = 8,7 × 104; peso molecular = 580). Se determina el contenido de colorante de cada disco de piel utilizando una curva de calibración adecuada, y después se calcula la media del contenido de colorante de las muestras paralelas.
2. DATOS
Deben indicarse en un cuadro los valores de resistencia (kΩ) y los valores medios de contenido de colorante (μg/disco), en su caso, correspondientes al material problema y a los controles positivos y negativos (todos los datos del ensayo y las medias ± D.T.), incluidos los datos de las muestras paralelas o de experimentos repetidos, las medias y cada uno de los valores.
2.1. INTERPRETACIÓN DE LOS RESULTADOS
Los resultados medios de la RET se aceptan a condición de que los valores de los controles positivos y negativos correspondientes queden dentro de los intervalos aceptables para el método en el laboratorio de ensayo. Los intervalos aceptables de resistencia del método y del instrumental antes descritos se indican en el cuadro siguiente:
|
Control |
Sustancia |
Intervalo de resistencia (kΩ) |
|
Positivo |
Ácido clorhídrico 10 M |
0,5-1,0 |
|
Negativo |
Agua destilada |
10-25 |
Los resultados medios de fijación del colorante se aceptan a condición de que los valores de los controles correspondientes queden dentro de los intervalos aceptables para el método. A continuación se dan los intervalos de contenido de colorante aceptables que se proponen para las sustancias de control con el método y el instrumental antes descritos:
|
Control |
Sustancia |
Intervalo del contenido de colorante (μg/disco) |
|
Positivo |
Ácido clorhídrico 10 M |
40-100 |
|
Negativo |
Agua destilada |
15-35 |
Se considera que la sustancia problema no es corrosiva para la piel:
|
i) |
si el valor medio de RET de la sustancia que se ha obtenido es superior a 5 kΩ, o |
|
ii) |
si el valor medio de RET es inferior o igual a 5 kΩ, y
|
Se considera que la sustancia problema es corrosiva para la piel:
|
i) |
si el valor medio de RET es inferior o igual a 5 kΩ y el disco de piel muestra lesiones evidentes, o |
|
ii) |
si el valor medio de RET es inferior o igual a 5 kΩ, y
|
3. INFORMES
3.1. INFORME DE ENSAYO
El informe del ensayo debe incluir la información siguiente:
|
|
Sustancias problema y de control:
|
|
|
Animales de ensayo:
|
|
|
Condiciones de los ensayos:
|
|
|
Resultados:
|
|
|
Discusión de los resultados. |
|
|
Conclusiones. |
4. REFERENCIAS
|
1 |
OECD (2001) Harmonised Integrated Classification System for Human Health and Environmental Hazards of Chemical Substances and Mixtures. OECD Series on Testing and Assessment Number 33. ENV/JM/MONO(2001)6, Paris. http://www.olis.oecd.org/olis/2001 doc.nsf/LinkTo/env-jm-mono(2001)6. |
|
2 |
Método de ensayo B.4. Toxicidad aguda: irritación/corrosión cutánea. |
|
3 |
Botham, P.A., Chamberlain, M., Barratt, M.D., Curren, R.D., Esdaile, D.J., Gardner, J.R., Gordon, V.C., Hildebrand, B., Lewis, R.W., Liebsch, M., Logemann, P., Osborne, R., Ponec, M., Regnier, J.F., Steiling, W., Walker, A.P., and Balls, M. (1995). A prevalidation study on in vitro skin corrosivity testing. The report and recommendations of ECVAM Workshop 6. ATLA 23, 219-255. |
|
4. |
Barratt, M.D., Brantom, P.G., Fentem, J.H., Gerner, I., Walker, A.P., and Worth, A.P. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 1. Selection and distribution of the test chemicals. Toxic. in Vitro 12, 471-482. |
|
5. |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhütter, H.-G., and Liebsch, M. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxic. In Vitro 12, 483-524. |
|
6. |
OECD (1996). Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, 62 pp. |
|
7. |
Balls, M., Blaauboer, B.J., Fentem. J.H., Bruner. L., Combes, R.D., Ekwall, B., Fielder. R.J., Guillouzo, A., Lewis, R.W., Lovell, D.P., Reinhardt, C.A., Repetto, G., Sladowski. D., Spielmann, H., and Zucco, F. (1995). Practical aspects of the validation of toxicity test procedures. The report and recommendations of ECVAM workshops. ATLA 23, 129-147. |
|
8. |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (1997). Validation and Regulatory Acceptance of Toxicological Test Methods. NIH Publication No. 97-3981. National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/docs/guidelines/validate.pdf |
|
9. |
ECVAM (1998). ECVAM News & Views. ATLA 26, 275-280. |
|
10. |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (2002). ICCVAM evaluation of EpiDermTM, EPISKINTM (EPI-200), and the Rat Skin Transcutaneous Electrical Resistance (TER) assay: In Vitro test methods for assessing dermal corrosivity potential of chemicals. NIH Publication No. 02-4502. National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/methods/epiddocs/epis_brd.pdf. |
|
11. |
OECD (2002) Extended Expert Consultation Meeting on The In Vitro Skin Corrosion Test Guideline Proposal, Berlin, 1st-2nd November 2001, Secretariat's Final Summary Report, 27th March 2002, OECD ENV/EHS, available upon request from the Secretariat |
|
12. |
Oliver, G.J.A., Pemberton, M.A., and Rhodes, C. (1986). An in vitro skin corrosivity test -modificationsand validation. Fd. Chem. Toxicol. 24, 507-512. |
|
13. |
Botham, P.A., Hall, T.J., Dennett, R., McCall, J.C., Basketter, D.A., Whittle, E., Cheeseman, M., Esdaile, D.J., and Gardner, J. (1992). The skin corrosivity test in vitro: results of an interlaboratory trial. Toxic. in Vitro 6, 191-194. |
|
14. |
Worth AP, Fentem JH, Balls M, Botham PA, Curren RD, Earl LK, Esdaile DJ, Liebsch M (1998). An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26: 709-720. |
|
15. |
Basketter, D.A., Chamberlain, M., Griffiths, H.A., Rowson, M., Whittle, E., York, M. (1997). The classification of skin irritants by human patch test. Fd. Chem. Toxicol. 35, 845-852. |
|
16. |
Oliver G.J.A, Pemberton M.A and Rhodes C. (1988). An In Vitro model for identifying skin-corrosive chemicals. I. Inicial Validation. Toxic. in Vitro. 2, 7-17. |
Figura 1
Instrumental para el ensayo de la RET en piel de rata

Figura 2
Dimensiones del tubo de politetrafluoretileno (PTFE), de la cámara receptora y de los electrodos usados

Factores críticos del instrumental mostrado arriba:
|
— |
el diámetro interior del tubo de PTFE, |
|
— |
la longitud de los electrodos respecto al tubo de PTFE y a la cámara receptora, de forma que el disco de piel no toque los electrodos y que haya en contacto con la solución de MgSO4 una longitud fijada de electrodo, |
|
— |
la cantidad de solución de MgSO4 presente en la cámara receptora debe hacer que el líquido tenga cierta profundidad respecto al nivel dentro del tubo de PTFE, como se indica en la figura 1, |
|
— |
el disco de piel debe fijarse bien al tubo de PTFE, de forma que la resistencia eléctrica sea una medida fiel de las propiedades de la piel. |
B.40 BIS. CORROSIÓN CUTÁNEA IN VITRO: ENSAYO CON MODELO DE PIEL HUMANA
1. MÉTODO
El presente método de ensayo es equivalente a las directrices de ensayo de la OCDE TG 431(2004).
1.1. INTRODUCCIÓN
La corrosión cutánea se refiere a la producción de daños tisulares irreversibles en la piel tras la aplicación de una sustancia [según la definición del Sistema Armonizado Mundial (Globally Harmonized System, GHS) de clasificación y etiquetado de productos químicos] (1). El presente método de ensayo no exige el uso de animales vivos ni de tejidos animales para la evaluación de la corrosividad cutánea.
Tradicionalmente, la evaluación de la corrosividad cutánea se hacía con animales de laboratorio (2). La preocupación por el dolor y el sufrimiento que implicaba esa evaluación se ha tenido en cuenta en la revisión del método de ensayo B.4, para permitir la determinación de la corrosión cutánea mediante métodos alternativos, in vitro, que eviten el dolor y el sufrimiento de los animales.
Un primer paso hacia la definición de métodos alternativos que se pudieran utilizar en los ensayos de corrosividad cutánea con fines normativos fue la realización de estudios de prevalidación (3). Posteriormente se llevó a cabo un estudio oficial de validación de métodos in vitro para evaluar la corrosión cutánea (4) (5) (6) (7) (8). Como consecuencia de estos estudios y de otros datos publicados (9), se llegó a la recomendación de que los siguientes métodos podían utilizarse para predecir el efecto corrosivo in vivo de la piel (10) (11) (12) (13): el ensayo con un modelo de piel humana (el presente método) y el ensayo de resistencia eléctrica transcutánea (véase el método de ensayo B.40).
Los estudios de validación han señalado que los ensayos que utilizan modelos de piel humana (3) (4) (5) (9) pueden discriminar de manera fiable entre agentes corrosivos y no corrosivos cutáneos conocidos. El protocolo del ensayo puede distinguir asimismo entre agentes corrosivos cutáneos fuertes y menos fuertes.
El ensayo descrito en el presente método permite la identificación de sustancias químicas y mezclas corrosivas. Asimismo, permite la identificación de sustancias y mezclas no corrosivas con el apoyo de pruebas sopesadas derivadas de otro tipo de información disponible (por ejemplo, pH, relaciones estructura-actividad, datos con hombres o animales) (1) (2) (13) (14). En principio, no da información adecuada sobre la irritación cutánea ni permite la subcategorización de sustancias corrosivas que se contempla en el Sistema Armonizado Mundial de clasificación (1).
Para una evaluación completa de los efectos cutáneos locales tras una única exposición de la piel, se recomienda seguir la estrategia de ensayos secuenciales que figura adjunta al método de ensayo B.4 (2) y que proporciona el Sistema Armonizado Mundial (1). Esta estrategia de ensayos incluye la realización de pruebas in vitro de corrosión cutánea (como las descritas en el presente método) y de irritación cutánea antes de considerar las pruebas con animales vivos.
1.2. DEFINICIONES
Se entenderá por:
corrosión cutánea in vivo la producción de lesiones cutáneas irreversibles consistentes en necrosis visible en todo el espesor de la epidermis y hasta en la dermis, debida a la aplicación de una sustancia durante hasta 4 horas; las reacciones de corrosión se tipifican como úlceras, hemorragia, costras hemorrágicas y, hacia el final del período de observación de 14 días, como decoloración de la piel, zonas de alopecia completa y cicatrices; debe recurrirse a la histopatología para evaluar las lesiones dudosas;
viabilidad celular el parámetro que mide la actividad total de una población celular (por ejemplo, capacidad de las deshidrogenasas mitocondriales celulares para reducir el colorante vital MTT), que, según el efecto que se mida y el tipo de ensayo realizado, se corresponde con el número total o con la vitalidad de las células.
1.3. SUSTANCIAS DE REFERENCIA
Cuadro 1
Sustancias de referencia
|
Denominación |
No EINECS |
No CAS |
|
|
1,2-diaminopropano |
201-155-9 |
78-90-0 |
Fuertemente corrosivo |
|
Ácido acrílico |
201-177-9 |
79-10-7 |
Fuertemente corrosivo |
|
2-terc-butilfenol |
201-807-2 |
88-18-6 |
Corrosivos |
|
Hidróxido de potasio (10 %) |
215-181-3 |
1310-58-3 |
Corrosivos |
|
Ácido sulfúrico (10 %) |
231-639-5 |
7664-93-9 |
Corrosivos |
|
Ácido octanoico (ácido caprílico) |
204-677-5 |
124-07-02 |
Corrosivos |
|
4-amino-1,2,4-triazol |
209-533-5 |
584-13-4 |
No corrosivo |
|
Eugenol |
202-589-1 |
97-53-0 |
No corrosivo |
|
Bromuro de fenetilo |
203-130-8 |
103-63-9 |
No corrosivo |
|
Tetracloroetileno |
204-825-9 |
27-18-4 |
No corrosivo |
|
Ácido isoesteárico |
250-178-0 |
30399-84-9 |
No corrosivo |
|
4-(metiltio)-benzaldehído |
222-365-7 |
3446-89-7 |
No corrosivo |
La mayoría de estas sustancias proceden de la lista de sustancias seleccionadas para el estudio internacional de validación del ECVAM (4). Su elección se basa en los criterios siguientes:
|
i) |
igual número de sustancias corrosivas y no corrosivas, |
|
ii) |
sustancias disponibles en el comercio y representativas de la mayoría de las clases químicas pertinentes, |
|
iii) |
inclusión de sustancias fuertemente corrosivas y de sustancias menos corrosivas para permitir la discriminación según la potencia corrosiva, |
|
iv) |
elección de sustancias que puedan manipularse en un laboratorio sin plantear peligros graves aparte de la corrosividad. |
1.4. PRINCIPIO DEL MÉTODO DE ENSAYO
El material problema se aplica tópicamente a un modelo tridimensional de piel humana que comprende al menos una epidermis reconstruida con una capa córnea funcional. Los materiales corrosivos se caracterizan por su capacidad de producir una disminución de la viabilidad celular [determinada, por ejemplo, utilizando el ensayo de reducción del MTT (15)] por debajo de determinados umbrales en períodos de exposición especificados. El principio del ensayo con modelo de piel humana se basa en la hipótesis de que las sustancias corrosivas pueden penetrar en la capa córnea por difusión o erosión, y son citotóxicas para las células de las capas subyacentes.
1.4.1. Procedimiento
1.4.1.1. Modelos de piel humana
Los modelos de piel humana pueden construirse u obtenerse en el comercio (por ejemplo, los modelos EpiDermMR y EPISKINMR) (16) (17) (18) (19), o bien desarrollarse o construirse en el laboratorio de ensayo (20) (21). Se sabe que el uso de piel humana es objeto de condiciones y consideraciones éticas tanto nacionales como internacionales. Todo nuevo modelo debe validarse (al menos en la medida descrita en el punto 1.4.1.1.2). Los modelos de piel humana utilizados para este ensayo deben cumplir las siguientes condiciones:
1.4.1.1.1. Condiciones generales del modelo
Para construir el epitelio deben utilizarse queratinocitos humanos. Bajo una capa córnea funcional, debe haber presentes múltiples capas de células epiteliales viables. El modelo de piel puede tener asimismo una capa de componentes del estroma. La capa córnea debe constar de múltiples estratos con el perfil lipídico necesario para constituir una barrera funcional con la suficiente resistencia contra la penetración rápida de marcadores citotóxicos. Las propiedades de aislamiento del modelo deben evitar que pase material rodeando la capa córnea hasta el tejido viable. El paso de sustancias problema rodeando la capa córnea reduce la calidad del modelo en cuanto a la exposición de la piel. El modelo de piel debe estar exento de contaminación por bacterias (incluidos micoplasmas) y hongos.
1.4.1.1.2. Condiciones funcionales del modelo
La magnitud de la viabilidad se cuantifica normalmente utilizando MTT o los otros colorantes vitales que se transforman en el metabolismo. En estos casos, la densidad óptica (DO) del colorante extraído (solubilizado) del tejido de control negativo debe ser al menos 20 veces mayor que la DO del disolvente de extracción solo [para una visión general, consúltese (22)]. El tejido de control negativo debe ser estable en cultivo (proporcionar unas mediciones similares de la viabilidad) durante la duración del tiempo de exposición del ensayo. La capa córnea debe tener la suficiente resistencia contra la penetración rápida de determinados marcadores citotóxicos (por ejemplo, tritón X-100 al 1 %). Esta propiedad puede estimarse mediante el tiempo de exposición necesario para reducir la viabilidad celular en un 50 % (TE50) (por ejemplo, en el caso de los modelos EpiDermMR y EPISKINMR, es > 2 horas). Los resultados obtenidos con el tejido han de poder reproducirse posteriormente y, de preferencia, en otros laboratorios. Por otra parte, el tejido ha de ser tal que permita prever el potencial corrosivo de las sustancias de referencia (véase el cuadro 1) cuando se utiliza en el protocolo de ensayo seleccionado.
1.4.1.2. Aplicación de las sustancias problema y de control
Por cada tratamiento (tiempo de exposición), incluidos los controles, se utilizan dos muestras de tejido. En caso de materiales líquidos, debe aplicarse una cantidad de sustancia problema suficiente para cubrir uniformemente la superficie cutánea (un mínimo de 25 μL/cm2. En caso de materiales sólidos, tiene que aplicarse uniformemente una cantidad suficiente de sustancia problema para cubrir la piel y debe humedecerse con agua desionizada o destilada para asegurar un buen contacto con la piel. Cuando convenga, deberán triturarse los sólidos para transformarlos en polvo antes de su aplicación. El método de aplicación debe ser adecuado para la sustancia problema (véase, por ejemplo, la cita bibliográfica 5). Al final del período de exposición, el material problema debe retirarse cuidadosamente de la superficie de la piel lavando con una solución amortiguadora adecuada o con NaCl al 0,9 %.
En cada estudio deben utilizarse a la vez controles positivos y negativos para garantizar el funcionamiento adecuado del modelo experimental. Las sustancias de control positivo que se proponen son ácido acético glacial o KOH 8 N. Los controles negativos propuestos son NaCl al 0,9 % o agua.
1.4.1.3. Medición de la viabilidad celular
Para medir la viabilidad celular solo pueden emplearse métodos cuantitativos validados. Por otra parte, la medición de la viabilidad debe ser compatible con el uso de un modelo de tejido tridimensional. La medición de la viabilidad no debe sufrir ninguna interferencia con una fijación inespecífica del colorante. Por tanto, no son adecuados los colorantes que se fijan a proteínas ni los que no se transforman en el metabolismo (por ejemplo, el rojo neutro). El ensayo más utilizado es la reducción del MTT [bromuro de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazolio, azul de tiazolilo: número EINECS 206-069-5, número CAS 298-93-1)], que, según se ha demostrado, da resultados exactos y reproducibles (5), pero también pueden utilizarse otros. La muestra de piel se coloca en una solución de MTT de la concentración adecuada (por ejemplo, 0,3-1 mg/mL), a una temperatura adecuada de incubación, durante 3 horas. A continuación, utilizando un disolvente (isopropanol), se extrae el precipitado que se produce de formazano azul y se mide la concentración de formazano determinando la DO a una longitud de onda entre 540 y 595 nm.
Es posible que la acción química de la sustancia problema sobre el colorante vital imite a la del metabolismo celular, provocando una estimación falsa de la viabilidad. Se ha visto que esto sucede cuando una sustancia problema de este tipo no se elimina completamente de la piel mediante lavado (9). Si la sustancia problema actúa directamente sobre el colorante vital, deben utilizarse controles adicionales para detectar y corregir la interferencia de la sustancia problema con la medición de la viabilidad (9) (23).
2. DATOS
Respecto a cada tejido, deben recogerse en un cuadro los valores de DO y los porcentajes calculados de viabilidad celular de la sustancia problema y de los controles positivo y negativo, incluidos los datos de los experimentos repetidos paralelos en su caso, con cada uno de los valores obtenidos y la media.
2.1. INTERPRETACIÓN DE LOS RESULTADOS
Los valores de DO obtenidos con cada muestra problema pueden utilizarse para calcular un porcentaje de viabilidad respecto al control negativo, que se fija arbitrariamente en el 100 %. Es necesario definir claramente y documentar, así como demostrar que es adecuado, el porcentaje límite de viabilidad celular que permite distinguir entre sustancias problema corrosivas y no corrosivas (o discriminar entre diferentes tipos de sustancias corrosivas), o el método o métodos estadísticos utilizados para evaluar los resultados y señalar las sustancias corrosivas. En general, estos valores límite se establecen durante la fase de optimización del ensayo, se prueban durante una fase de prevalidación y se confirman en un estudio de validación. Como ejemplo, la predicción de corrosividad asociada con el modelo EpiDermMR es la siguiente (9):
Se considera que la sustancia problema es corrosiva para la piel:
|
i) |
si la viabilidad tras 3 minutos de exposición es inferior al 50 %, o |
|
ii) |
si la viabilidad tras 3 minutos de exposición es superior o igual al 50 % y la viabilidad tras una hora de exposición es inferior al 15 %. |
Se considera que la sustancia problema no es corrosiva para la piel:
|
i) |
si la viabilidad tras 3 minutos de exposición es superior o igual al 50 % y la viabilidad tras una hora de exposición es superior o igual al 15 %. |
3. INFORMES
3.1. INFORME DE ENSAYO
El informe del ensayo debe incluir la información siguiente:
|
|
Sustancias problema y de control:
|
|
|
Justificación del modelo de piel y del protocolo utilizados. |
|
|
Condiciones de ensayo:
|
|
|
Resultados:
|
|
|
Discusión de los resultados. |
|
|
Conclusión. |
4. REFERENCIAS
|
1. |
OECD (2001) Harmonised Integrated Classification System for Human Health and Environmental Hazards of Chemical Substances and Mixtures. OECD Series on Testing and Assessment Number 33. ENV/JM/MONO(2001)6, Paris. http://www.olis.oecd.org/olis/200 1doc.nsf/LinkTo/env-jm-mono(2001)6. |
|
2. |
Testing Method B.4. Acute Toxicity: Dermal Irritation/Corrosion. |
|
3. |
Botham, P.A., Chamberlain, M., Barratt, M.D., Curren, R.D., Esdaile, D.J., Gardner, J.R., Gordon, V.C., Hildebrand, B., Lewis, R.W., Liebsch, M., Logemann, P., Osborne, R., Ponec, M., Regnier, J.F., Steiling, W., Walker, A.P., and Balls, M. (1995). A prevalidation study on in vitro skin corrosivity testing. The report recommendations of ECVAM Workshop 6 ATLA 23,219- 255. |
|
4. |
Barratt, M.D., Brantom, P.G., Fentem, J.H., Gerner, I., Walker, A.P., and Worth, A.P. (1998). The ECVAM international validation study on in Vitro tests for skin corrosivity. 1. Selection and distribution of the test chemicals. Toxic. In Vitro 12, 471-482. |
|
5. |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.G. and Liebsch, M. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxic. In Vitro 12, 483-524. |
|
6. |
OECD (1996). Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, 62pp. |
|
7. |
Balls, M., Blaauboer, B.J., Fentem, J.H., Bruner, L., Combes, R.D., Ekwall, B., Fielder, R.J., Guillouzo, A., Lewis, R.W., Lovell, D.P., Reinhardt, C.A., Repetto, G., Sladowski, D., Spielmann, H., and Zucco, F. (1995). Practical aspects of the validation of toxicity test procedures. Test report and recommendations of ECVAM workshops. ATLA 23, 129-147. |
|
8. |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (1997). Validation and Regulatory Acceptance of Toxicological Test Methods. NIH Publication No. 97-3981. National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/docs/guidelines/validate.pdf. |
|
9. |
Liebsch, M., Traue, D., Barrabas, C., Spielmann, H., Uphill, P., Wilkins, S., McPherson, J.P., Wiemann, C., Kaufmann, T., Remmele, M. and Holzhutter, H.G. (2000). The ECVAM prevalidation study on the use of EpiDerm for skin Corrosivity testing. ATLA 28, pp.371-401. |
|
10. |
ECVAM (1998). ECVAM News & Views. ATLA 26, 275-280. |
|
11. |
ECVAM (2000). ECVAM News & Views. ATLA 28, 365-67. |
|
12. |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (2002). ICCVAM evaluation of EpiDermTM, EPISKINTM (EPI-200), and the Rat Skin Transcutaneous Electrical Resistance (TER) assay: In Vitro test methods for assessing dermal corrosivity potential of chemicals. NIH Publication No. 02-4502. National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/methods/epiddocs/epis_brd.pdf. |
|
13. |
OECD (2002) Extended Expert Consultation Meeting on The In Vitro Skin Corrosion Test Guideline Proposal, Berlin, 1st-2nd November 2001, Secretariat's Final Summary Report, 27th March 2002, OECD ENV/EHS, available upon request from the Secretariat |
|
14. |
Worth AP, Fentem JH, Balls M, Botham PA, Curren RD, Earl LK, Esdaile DJ, Liebsch M (1998). An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26: 709-720. |
|
15. |
Mosmann, T. (1983). Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity asssays. J.Immunol. Meth. 65, 55-63. |
|
16. |
Cannon, C.L., Neal, P.J., Southee, J.A., Kubilus, J., and Klausner, M., 1994. New epidermal model for dermal irritancy testing. Toxic. In Vitro 8, 889-891. |
|
17. |
Ponec, M., Boelsma, E., Weerheim, A., Mulder, A., Boutwstra, J., and Mommaas, M., 2000. Lipid and ultrastructural characterization of reconstructed skin models. International Journal of Pharmaceutics. 203, 211-225. |
|
18. |
Tinois E, Gaetani Q, Gayraud B, Dupont D, Rougier A, Pouradier DX (1994). The Episkin model: Successful reconstruction of human epidermis in vitro. In vitro Skin Toxicology. Edited by A Rougier, AM Goldberg and HI Maibach: 133-140 |
|
19. |
Tinois E, Tiollier J, Gaucherand M, Dumas H, Tardy M, Thivolet J (1991). In vitro and post -transplantation differentiation of human keratinocytes grown on the human type IV collagen film of a bilayered dermal substitute. Experimental Cell Research 193:310-319 |
|
20. |
Parentau, N.L., Bilbo, P., Molte, C.J., Mason, V.S., and Rosenberg, H. (1992). The organotypic culture of human skin keratinocytes and fibroblasts to achieve form and function. Cytotechnology 9, 163-171. |
|
21. |
Wilkins, L.M., Watson, S.R., Prosky, S.J., Meunier, S.F., Parentau, N.L. (1994). Development of a bilayered living skin construct for clinical applications. Biotechnology and Bioengineering 43/8, 747-756. |
|
22. |
Marshall, N.J., Goodwin, C.J., Holt, S.J. (1995). A critical assessment of the use of microculture tetrazolium assays to measure cell growth and function. Growth Regulation 5, 69-84. |
|
23. |
Fentem, J.H., Briggs, D., Chesne', C., Elliot, G.R., Harbell, J.W., Heylings, J.R., Portes, P., Rouget, R., and van de Sandt, J.J.M., and Botham, P.A. (2001). A prevalidation study on in vitro tests for acute skin irritation: results and evaluation by the Management Team. Toxic. In Vitro 15, 57-93. |
B.41. ENSAYO DE FOTOTOXICIDAD IN VITRO 3T3 ARN (ABSORCIÓN DE ROJO NEUTRO)
1. MÉTODO
El presente método es equivalente a las directrices de ensayo de la OCDE TG 432 (2004).
1.1. INTRODUCCIÓN
La fototoxicidad se define como una respuesta tóxica ante una sustancia aplicada al cuerpo que es provocada o aumentada (se hace perceptible a dosis menores) tras una exposición posterior a la luz, o que es inducida por irradiación de la piel tras la administración sistémica de una sustancia.
El ensayo de fototoxicidad in vitro 3T3 ARN se emplea para identificar el potencial fototóxico de una sustancia problema inducido por la sustancia excitada tras su exposición a la luz. El ensayo evalúa la fototoxicidad mediante la reducción relativa de la viabilidad de las células expuestas a la sustancia en presencia de luz, respecto a las expuestas en la oscuridad. Es probable que las sustancias identificadas mediante este ensayo sean fototóxicas in vivo, tras su aplicación sistémica seguida de su distribución a la piel, o tras una aplicación tópica.
Se han señalado numerosos tipos de sustancias químicas que producen efectos fototóxicos (1) (2) (3) (4). Tienen en común la capacidad de absorber energía luminosa del espectro de la luz solar. Con arreglo a la primera ley de la fotoquímica (ley de Grotthaus-Draper), para que se produzca una fotorreacción es necesaria la absorción suficiente de cuantos de luz. Por tanto, antes de plantearse la realización de ensayos biológicos, debe determinarse el espectro de absorción de luz UV/visible de la sustancia problema según las directrices de ensayo TG 101 de la OCDE. Se ha indicado la escasa probabilidad de que una sustancia sea fotorreactiva si su coeficiente de absorción/extinción molar es inferior a 10 litros × mol-1 × cm-1. Es posible que las sustancias de este tipo no tengan que someterse al ensayo de fototoxicidad in vitro 3T3 ARN ni a ningún otro ensayo biológico de efectos fotoquímicos adversos (1) (5). Véase también a este respecto el anexo 1.
La fiabilidad y la pertinencia del ensayo de fototoxicidad in vitro 3T3 ARN se han evaluado recientemente (6) (7) (8) (9). Se ha comprobado que este ensayo sirve para predecir los efectos de fototoxicidad aguda in vivo en animales y en el hombre. Por el contrario, no está concebido para predecir otros efectos adversos que puedan deberse a la acción combinada de una sustancia y de la luz; así, por ejemplo, no se refiere a la fotogenotoxicidad, a la fotoalergia ni a la fotocarcinogénesis, y tampoco permite la evaluación de la potencia fototóxica. Tampoco se ha designado el ensayo para estudiar mecanismos indirectos de fototoxicidad, efectos de metabolitos de la sustancia problema ni efectos de mezclas.
Si bien en general es necesario utilizar sistemas de metabolización en todos los ensayos in vitro de predicción del potencial genotóxico o carcinogénico, en el caso de la fototoxicología hasta la fecha solo se conocen escasos ejemplos de que una sustancia química requiera una transformación metabólica para actuar como fototoxina in vivo o in vitro. Por consiguiente, no resulta necesario ni está fundado científicamente realizar el presente ensayo con un sistema de activación metabólica.
1.2. DEFINICIONES
Se entenderá por:
irradiancia la intensidad de luz ultravioleta (UV) o visible que incide en una superficie, medida en W/m2 o mW/cm2;
dosis de luz la cantidad (= intensidad × tiempo) de radiación ultravioleta (UV) o visible que incide en una superficie, expresada en julios (= W × s) por unidad de superficie, por ejemplo J/m2 o J/cm2;
bandas de luz UV las siguientes designaciones recomendadas por la CIE (Commission Internationale de l'Eclairage): UVA (315-400 nm), UVB (280-315 nm) y UVC (100-280 nm); también se emplean otras designaciones: la separación entre UVB y UVA se sitúa con frecuencia en 320 nm y las radiaciones UVA pueden subdividirse en UV-A1 y UV-A2, con una separación a 340 nm aproximadamente;
viabilidad celular el parámetro que mide la actividad total de una población celular (por ejemplo, absorción del colorante vital rojo neutro por los lisosomas celulares), que, según el efecto que se mida y el tipo de ensayo realizado, se corresponde con el número total o con la vitalidad de las células;
viabilidad celular relativa la viabilidad celular respecto a la de los controles negativos (disolvente) que se someten a todo el procedimiento de ensayo (+ Irr o - Irr), pero no se tratan con la sustancia problema;
PIF (Photo Irritation Factor — factor de fotoirritación) el factor que se obtiene comparando dos concentraciones citotóxicas igualmente eficaces (CI50) de la sustancia problema con irradiación no citotóxica de luz UVA/visible (+ Irr) y sin dicha irradiación (- Irr);
CI50 la concentración de la sustancia problema a la que la viabilidad celular se reduce en un 50 %;
MPE (Mean Photo Effect — fotoefecto medio) la medida derivada del análisis matemático de las curvas respuesta-concentración obtenidas con una irradiación no citotóxica de luz UVA/visible (+ Irr) y sin dicha irradiación (- Irr);
fototoxicidad la reacción tóxica aguda provocada tras una primera exposición de la piel a determinadas sustancias químicas y su posterior exposición a la luz, o inducida análogamente por irradiación de la piel tras la administración sistémica de una sustancia química.
1.3. PRINCIPIO DEL MÉTODO DE ENSAYO
El ensayo de fototoxicidad in vitro 3T3 ARN se basa en la comparación de la citotoxicidad de una sustancia cuando se somete a ensayo con exposición y sin exposición a una dosis no citotóxica de luz solar simulada. La citotoxicidad en este ensayo se expresa como la disminución, ligada a la concentración, de la absorción del colorante vital rojo neutro a las 24 horas de la administración de la sustancia problema y la irradiación (10). El rojo neutro es un colorante catiónico débil que penetra fácilmente a través de las membranas celulares por un mecanismo distinto de la difusión, y se acumula intracelularmente en los lisosomas. Las alteraciones de la superficie de la sensible membrana de los lisosomas provocan un aumento de la fragilidad de estos, junto con otros cambios que gradualmente se van haciendo irreversibles. Tales cambios inducidos por la acción de agentes xenobióticos ocasionan un descenso de la absorción y fijación del rojo neutro, con lo que es posible distinguir entre células viables, lesionadas y muertas, lo que constituye la base del ensayo.
Se mantienen en cultivo células Balb/c 3T3 durante 24 horas para que se formen monocapas. Se preincuban durante 1 hora dos placas de 96 pocillos por sustancia problema, con 8 concentraciones diferentes de esta sustancia. Después se expone una de las dos placas a la dosis máxima de irradiación no citotóxica, mientras que la otra placa se mantiene a oscuras. A continuación se sustituye en ambas placas el medio de tratamiento por medio de cultivo y, tras otras 24 horas de incubación, se determina la viabilidad celular mediante la absorción de rojo neutro. La viabilidad celular se expresa en porcentaje respecto a los controles de disolvente sin tratar y se calcula para cada una de las concentraciones del ensayo. Para predecir el potencial fototóxico, se comparan las respuestas a las distintas concentraciones obtenidas con y sin irradiación, por lo general al nivel CI50, es decir, a la concentración que reduce la viabilidad celular en un 50 % respecto a los controles sin tratar.
1.4. DESCRIPCIÓN DEL MÉTODO
1.4.1. Preparación
1.4.1.1. Células
En el estudio de validación se utilizó una línea celular de fibroblastos permanentes de ratón, la Balb/c 3T3, clon 31, bien de la Colección Americana de Cultivos Tipo (ATCC), Manassas, VA, EEUU, o bien de la Colección Europea de Cultivos Celulares (ECACC), Salisbury, Wiltshire, RU, por lo que se recomienda obtenerla de una colección reconocida. También pueden utilizarse con el mismo protocolo de ensayo otras células o líneas celulares, siempre y cuando las condiciones de cultivo se adapten a las necesidades particulares de las células, si bien en tal caso debe demostrarse la equivalencia de los resultados.
Debe comprobarse periódicamente que las células están libres de contaminación por micoplasmas y solo se utilizarán si se confirma la ausencia de estos microorganismos (11).
Es importante comprobar periódicamente la sensibilidad de las células a las radiaciones UV conforme al procedimiento de control de calidad descrito en el presente método. Dado que la sensibilidad de las células a las radiaciones UVA puede aumentar con el número de pases, deben emplearse células Balb/c 3T3 sometidas al menor número posible de pases, preferentemente menos de 100. (Véanse el punto 1.4.2.2.2 y el anexo 2).
1.4.1.2. Medios y condiciones de cultivo
Deben emplearse medios de cultivo y condiciones de incubación apropiados para los pases celulares corrientes y durante el ensayo; por ejemplo, en el caso de células Balb/c 3T3, el medio de cultivo es DMEM (Dulbecco's Modified Eagle's Medium) enriquecido con un 10 % de suero de ternera recién nacida, glutamina 4 mM, penicilina (100 UI) y estreptomicina (100 μg/ml), y la incubación ha de hacerse en atmósfera humidificada, a 37 oC, con 5-7,5 % de CO2 según el amortiguador (véase el segundo párrafo del punto 1.4.1.4). Es particularmente importante que las condiciones del cultivo celular permitan mantener la duración del ciclo celular dentro de los límites normales conocidos de la línea celular o las células utilizadas.
1.4.1.3. Preparación de los cultivos
Se siembran en medio de cultivo, a una densidad apropiada, células procedentes de cultivos madre congelados y se subcultivan al menos una vez antes de utilizarlas en el ensayo de fototoxicidad in vitro 3T3 ARN.
Para el ensayo de fototoxicidad, las células se siembran en el medio de cultivo a una densidad apropiada de manera que los cultivos no confluyan antes de que finalice el ensayo, es decir, cuando se determine la viabilidad celular 48 horas después de la siembra celular. Para las células Balb/c 3T3 cultivadas en placas de 96 pocillos, se recomienda una densidad de siembra de 1 × 104 células por pocillo.
Por cada sustancia problema se siembran las células de igual modo en dos placas distintas de 96 pocillos, que se mantendrán en paralelo en condiciones de cultivo idénticas a lo largo de todo el ensayo, excepto durante el tiempo en que una de ellas se irradia (+ Irr) y la otra se mantiene en la oscuridad (- Irr).
1.4.1.4. Preparación de la sustancia problema
Las sustancias problema deben prepararse justo antes de su empleo, salvo que se disponga de datos que demuestren que pueden conservarse estables. Se recomienda efectuar todas las manipulaciones químicas y el tratamiento inicial de las células en unas condiciones luminosas que eviten la fotoactivación o la degradación de la sustancia problema antes de la irradiación.
Las sustancias problema se disuelven en soluciones salinas amortiguadoras, como la solución salina equilibrada de Earle (Earle's Balanced Salt Solution, EBSS) u otras soluciones amortiguadoras equilibradas fisiológicamente, que no deben contener componentes proteínicos ni absorbentes de la luz (por ejemplo, colorantes indicadores de pH y vitaminas), con el fin de evitar interferencias durante la irradiación. Como durante la irradiación las células se mantienen unos 50 minutos fuera de la incubadora de CO2, debe tenerse cuidado para evitar la alcalinización. Si se utilizan amortiguadores débiles, como la EBSS, esto puede conseguirse incubando las células con un 7,5 % de CO2. Si las células se incuban solo con un 5 % de CO2, debe elegirse un amortiguador más fuerte.
Las sustancias problema de hidrosolubilidad limitada deben disolverse en un disolvente adecuado. Si se emplea un disolvente, debe encontrarse a un volumen constante en todos los cultivos, es decir, en los controles negativos (disolvente) y en todas las concentraciones de la sustancia problema, y a esa concentración no debe ser citotóxico. Las concentraciones de la sustancia problema deben elegirse de manera que se evite la aparición de precipitados o de turbidez.
Se recomienda el uso de los disolventes dimetilsulfóxido (DMSO) y etanol (EtOH). También pueden ser útiles otros disolventes de baja citotoxicidad. Antes de utilizar cualquier disolvente, hay que comprobar ciertas propiedades del mismo como, por ejemplo, su capacidad de reacción con la sustancia problema, de atenuación del efecto fototóxico, de captación de radicales, o la estabilidad de la sustancia en el disolvente.
Para facilitar la solubilización puede utilizarse un agitador mecánico, un baño de ultrasonidos o el calentamiento a temperatura adecuada, siempre que no se altere la estabilidad de la sustancia problema.
1.4.1.5. Condiciones de irradiación
1.4.1.5.1. Fuente luminosa
En el ensayo de fototoxicidad, es fundamental la elección de una fuente luminosa y de unos filtros adecuados. La luz de las regiones visible y UVA suele relacionarse con reacciones fototóxicas in vivo (3) (12), mientras que generalmente la UVB reviste menor importancia pero es muy citotóxica; la citotoxicidad se multiplica por 1 000 cuando la longitud de onda pasa de 313 a 280 nm (13). Entre los criterios para elegir una fuente luminosa adecuada debe figurar el requisito de que esta emita longitudes de onda absorbidas por la sustancia problema (espectro de absorción) y que la dosis de luz (conseguida en un tiempo de exposición razonable) sea suficiente para detectar sustancias fotocitotóxicas conocidas. Además, las longitudes de onda y las dosis empleadas no han de ser excesivamente nocivas para el sistema de ensayo, por ejemplo debido a la emisión de calor (región del infrarrojo).
Se considera que la mejor fuente luminosa artificial son los simuladores solares. La distribución de potencia de irradiación del simulador solar filtrado debe parecerse a la de la luz diurna al aire libre que se indica en (14). Se utilizan como simuladores solares tanto los arcos de xenón como los arcos de haluro metálico y mercurio (cebado) (15). Estos últimos presentan la ventaja de emitir menos calor y de ser más baratos, pero no reproducen la luz solar tan bien como los arcos de xenón. Dado que todos los simuladores solares emiten cantidades importantes de rayos UVB, deben emplearse los filtros adecuados para atenuar estas longitudes de onda altamente citotóxicas. Debido a que el material plástico para cultivo celular contiene estabilizadores de UV, el espectro debe medirse a través del mismo tipo de tapa de placa de 96 pocillos que se vaya a utilizar en el ensayo. Independientemente de las medidas tomadas para atenuar partes del espectro mediante filtración o mediante efectos filtrantes inevitables del equipo, el espectro registrado por debajo de estos filtros no debe desviarse de la luz diurna al aire libre normalizada (14). En (8) y (16) se recoge un ejemplo de la distribución de la irradiancia espectral del simulador solar con filtro utilizado en el estudio de validación del ensayo de fototoxicidad in vitro 3T3 ARN. Véase también la figura 1 del anexo 2.
1.4.1.5.2. Dosimetría
La intensidad de la luz (irradiancia) debe comprobarse periódicamente antes de cada ensayo de fototoxicidad utilizando un medidor adecuado de UV de banda ancha. La intensidad debe medirse a través del mismo tipo de tapa de placa de 96 pocillos que se vaya a utilizar en el ensayo. El medidor de UV tiene que haberse calibrado en función de la fuente luminosa. Ha de comprobarse el correcto funcionamiento de dicho aparato, para lo cual se recomienda utilizar otro medidor de UV de referencia, del mismo tipo y calibrado de igual manera. Lo ideal, a intervalos mayores, sería emplear un espectrorradiómetro para medir la irradiancia espectral de la fuente luminosa filtrada y comprobar el calibrado del medidor de UV de banda ancha.
Se ha visto que una dosis de 5 J/cm2 (medida en la región UVA) no es citotóxica para las células Balb/c 3T3 y tiene la potencia suficiente de excitación de sustancias químicas para producir reacciones fototóxicas (6) (17), por ejemplo para conseguir 5 J/cm2 dentro de un plazo de 50 min, se ajustó la irradiancia a 1,7 mW/cm2. Véase la figura 2 del anexo 2. Si se utiliza otra línea celular o una fuente luminosa diferente, es posible que la dosis de irradiación haya de calibrarse de manera que pueda elegirse un régimen de dosis inocuas para las células pero suficientes para excitar fototoxinas de referencia. La duración de la exposición a la luz se calcula de la manera siguiente:
|
|
(1 J = 1 Ws) |
1.4.2. Condiciones del ensayo
1.4.2.1. Concentraciones de la sustancia problema
Las gamas de concentraciones de la sustancia estudiada en presencia (+ Irr) y en ausencia (- Irr) de luz deben determinarse de forma adecuada mediante experimentos destinados a este fin. Puede ser útil evaluar la solubilidad al principio y a los 60 minutos (o el tiempo correspondiente al tratamiento que se vaya a utilizar), ya que la solubilidad puede cambiar a lo largo del tiempo o durante el proceso de exposición. Para evitar la toxicidad debida a unas condiciones de cultivo inadecuadas o a la presencia de sustancias muy ácidas o alcalinas, el pH de los cultivos celulares con la sustancia problema debe situarse entre 6,5 y 7,8.
La concentración máxima de la sustancia problema debe respetar las condiciones fisiológicas de la prueba; por ejemplo, han de evitarse las tensiones osmóticas y de pH. Según la sustancia problema, puede ser necesario estudiar otras propiedades físico-químicas como posibles factores de limitación de la concentración máxima. Cuando se trate de sustancias relativamente insolubles que no sean tóxicas a concentraciones de hasta el punto de saturación, debe utilizarse la concentración máxima que se pueda conseguir. En general, debe evitarse la precipitación de la sustancia problema en cualquiera de las concentraciones del ensayo. La concentración máxima de una sustancia problema no debe superar los 1 000 μg/ml y la osmolaridad no debe pasar de 10 mmolar. Debe utilizarse una serie de diluciones geométricas de 8 concentraciones de la sustancia problema con un factor de dilución constante (véase el punto 2.1, párrafo segundo).
Si se sabe (por un experimento de determinación de la gama de concentraciones) que la sustancia problema no es citotóxica hasta la concentración límite en el experimento sin luz (- Irr), pero es muy citotóxica cuando se ha irradiado (+ Irr), la gama de concentraciones que debe seleccionarse para el experimento (+ Irr) puede ser diferente de la elegida para el experimento (- Irr) a fin de cumplir los requisitos de calidad de los datos.
1.4.2.2. Controles
1.4.2.2.1. Sensibilidad de las células ante la radiación; obtención de datos previos
Debe comprobarse periódicamente (más o menos, cada cinco pases) la sensibilidad de las células ante la fuente luminosa, evaluando su viabilidad tras la exposición a dosis crecientes de radiación. En esta evaluación deben utilizarse varias dosis de radiación, incluyendo dosis sustancialmente más elevadas que las utilizadas en el ensayo de toxicidad 3T3 ARN. Lo más fácil es cuantificar estas dosis por mediciones de la banda UV de la fuente luminosa. Las células se siembran a la densidad utilizada en el ensayo de fototoxicidad in vitro 3T3 ARN y se irradian al día siguiente. La viabilidad de las células se determina un día después mediante la absorción de rojo neutro. Debe demostrarse que la dosis máxima no citotóxica obtenida (por ejemplo, en el estudio de validación: 5 J/cm2 [UVA]) es suficiente para clasificar correctamente las sustancias de referencia (cuadro 1).
1.4.2.2.2. Sensibilidad ante la radiación; comprobación del ensayo en curso
El ensayo cumple los criterios de calidad si los controles negativos/de disolvente irradiados presentan una viabilidad superior al 80 % respecto a los controles negativos/de disolvente no irradiados.
1.4.2.2.3. Viabilidad de los controles de disolvente
La densidad óptica absoluta (DO540 ARN ) del rojo neutro extraído de los controles de disolvente indica si las 1 × 104 células sembradas por pocillo han crecido con un tiempo de generación normal durante los dos días del ensayo. Un ensayo cumple los criterios de aceptación si la media de la DO540 ARN de los controles sin tratar es > 0,4 (es decir, unas 20 veces la absorbancia de fondo del disolvente).
1.4.2.2.4. Control positivo
Por cada ensayo de fototoxicidad in vitro 3T3 ARN, debe someterse a ensayo en paralelo una sustancia fototóxica conocida. Se recomienda el uso de la clorpromazina (CPZ). Se han definido los criterios de aceptación siguientes para el uso de CPZ conforme al protocolo normalizado en el ensayo de fototoxicidad in vitro 3T3 ARN: CPZ irradiada (+ Irr), CI50 = 0,1 a 2,0 μg/ml; CPZ no irradiada (- Irr), CI50 = 7,0 a 90,0 μg/ml. El factor de fotoirritación (PIF) debe ser > 6. Deben supervisarse los datos previos sobre el comportamiento del control positivo.
En lugar de la clorpromazina, pueden emplearse para los controles positivos en paralelo otras sustancias fototóxicas que convengan a la clase química o a las características de solubilidad de la sustancia estudiada.
1.4.3. Procedimiento del ensayo (6) (7) (8) (16) (17)
1.4.3.1. 1er día
Se ponen 100 μl de medio de cultivo en los pocillos periféricos de una placa de microvaloración de cultivo tisular de 96 pocillos (= blancos). En los demás pocillos se vierten 100 μl de una suspensión de 1 × l05 células/ml en medio de cultivo (= 1 × 104 células/pocillo). Deben prepararse dos placas con cada serie de concentraciones de las distintas sustancias problema, así como de los controles positivos y de disolvente.
Se incuban las células durante 24 horas (véase el punto 1.4.1.2) hasta que formen una monocapa semiconfluyente. Este período de incubación permite la recuperación, la adherencia y el crecimiento exponencial de las células.
1.4.3.2. 2o día
Tras la incubación, se decanta el medio de cultivo separándolo de las células y se lava cuidadosamente con 150 μl de la solución amortiguadora utilizada para la dilución. Se añaden 100 μl de la solución amortiguadora con la concentración adecuada de la sustancia problema o del disolvente (control de disolvente). Se utilizan 8 concentraciones distintas de la sustancia problema. Se incuban las células con la sustancia problema en la oscuridad durante 60 minutos (véanse el punto 1.4.1.2 y el punto 1.4.1.4, segundo párrafo).
De las dos placas preparadas con cada serie de concentraciones de la sustancia problema y con los controles, se elige una, generalmente al azar, para la determinación de la citotoxicidad (- Irr) (es decir, la placa de control), y otra (la placa de tratamiento) para la determinación de la fotocitotoxicidad (+ Irr).
Para efectuar la exposición + Irr, se someten las células a la dosis máxima de radiación que no es citotóxica, durante 50 minutos, a través de la tapa de la placa de 96 pocillos, a temperatura ambiente (véase también el anexo 2). Las placas que no se irradian (- Irr) se mantienen a temperatura ambiente en la oscuridad durante 50 minutos (= tiempo de exposición a la luz).
Se decanta la solución de ensayo y se lava cuidadosamente dos veces con 150 μl de la solución amortiguadora utilizada para la incubación, pero sin sustancia problema. Se sustituye la solución amortiguadora por medio de cultivo y se incuban (véase el punto 1.4.1.2.) hasta el día siguiente (18-22 horas).
1.4.3.3. 3er día
1.4.3.3.1. Evaluación microscópica
Deben examinarse las células atendiendo a su crecimiento y morfología, así como a la integridad de la monocapa, utilizando un microscopio de contraste de fase. Deben registrarse los cambios en la morfología y los efectos sobre el crecimiento de las células.
1.4.3.3.2. Ensayo de absorción de rojo neutro
Se lavan las células con 150 μl de solución amortiguadora previamente calentada. Se elimina la solución de lavado con unos golpecitos suaves. Se añaden 100 μl de una solución de 50 μg/ml de rojo neutro (RN) (clorhidrato de 3-amino-7-dimetilamino-2-metilfenazina, número EINECS 209-035-8; número CAS 553-24-2; C.I. 50040) en medio de cultivo sin suero (16) y se incuba durante 3 horas como se indica en el punto 1.4.1.2. Tras la incubación, se elimina el medio de RN y se lavan las células con 15 μL de la solución amortiguadora. Se decanta y se elimina el exceso de solución amortiguadora con material absorbente o por centrifugación.
Se añaden exactamente 150 μl de solución de desorción de RN (recién preparada con 49 partes de agua + 50 partes de etanol + 1 parte de ácido acético).
Se agita suavemente la placa de microvaloración en un agitador de placas durante 10 minutos, hasta que el RN se haya extraído de las células y forme una solución homogénea.
Se mide la densidad óptica del extracto de RN a 540 nm en un espectrofotómetro, utilizando los blancos como referencia. Los datos se conservan en un archivo electrónico adecuado para su análisis posterior.
2. DATOS
2.1. CALIDAD Y CANTIDAD DE LOS RESULTADOS
Los datos del ensayo deben permitir el análisis significativo de la relación respuesta-oncentración con y sin irradiación, y, a ser posible, determinar la concentración de la sustancia problema a la que se reduce al 50 % la viabilidad de las células (CI50). Si se comprueba que hay citotoxicidad, tanto la gama de concentraciones como el intervalo entre las distintas concentraciones deben establecerse de manera que se pueda ajustar una curva a los datos experimentales.
En caso de resultados tanto claramente positivos como claramente negativos (véase el punto 2.3, párrafo primero), puede ser suficiente el experimento primario, con el apoyo de uno o más experimentos previos para determinar la gama de las dosis.
En caso de resultados ambiguos, en el límite o dudosos deben hacerse más ensayos (véase también el punto 2.4, párrafo segundo). En tal caso, debe plantearse la modificación de las condiciones experimentales. Entre las condiciones que podrían modificarse figuran la gama de concentraciones o sus intervalos, el tiempo de preincubación y la duración de la exposición a la radiación. En el caso de las sustancias inestables en el agua puede ser conveniente reducir dicha duración.
2.2. EVALUACIÓN DE LOS RESULTADOS
Para permitir la evaluación de los resultados, puede calcularse el factor de fotoirritación (PIF) o el fotoefecto medio (MPE).
Para calcular las medidas de fotocitotoxicidad (véase más abajo) hay que trazar una curva continua y adecuada respuesta-concentración (modelo) que se aproxime al conjunto de valores discretos de respuestas a las concentraciones. El ajuste de la curva a los datos se suele hacer mediante un método de regresión no lineal (18). Para evaluar la influencia de la variabilidad de los datos sobre la curva ajustada se recomienda un procedimiento bootstrap.
El factor de fotoirritación (PIF) se calcula mediante la siguiente fórmula:
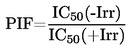
Si no puede calcularse la CI50 en presencia o en ausencia de luz, no es posible determinar el PIF de la sustancia problema. El fotoefecto medio (MPE) se basa en la comparación de las curvas completas respuesta-concentración (19). Se define como la media ponderada de un conjunto representativo de valores de fotoefecto.
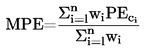
El fotoefecto PEc a una concentración C se define como el producto del efecto de respuesta REc por el efecto de dosis DEc; es decir, PEc = REc × DEc. El efecto de respuesta REc es la diferencia entre las respuestas observadas en ausencia y en presencia de luz, es decir REc = Rc (- Irr) — Rc (+ Irr). El efecto de dosis viene dado por la fórmula siguiente:

donde C* representa la concentración equivalente, es decir, la concentración a la que la respuesta + Irr iguala a la respuesta - Irr a la concentración C. Si C* no puede determinarse porque los valores de respuesta de la curva + Irr son sistemáticamente más altos o más bajos que RC(- Irr), el efecto de dosis se fija en 1. Los factores de ponderación wi vienen dados por el valor máximo de respuesta, es decir, wi = MAX {Ri (+ Irr), Ri (- Irr)}. El cuadro de concentraciones Ci se elige de forma que caiga el mismo número de puntos en cada uno de los intervalos de concentración definidos por los valores de concentración utilizados en el experimento. El cálculo del MPE se limita al valor máximo de concentración al que al menos una de las dos curvas sigue mostrando un valor de respuesta de al menos el 10 %. Si esta concentración máxima es más elevada que la concentración más alta utilizada en el experimento + Irr, la parte residual de la curva + Irr se fija en el valor de respuesta «0». En función de que el valor de MPE sea o no mayor que un valor límite adecuadamente elegido (MPEc = 0,15), la sustancia se clasifica como fototóxica o no.
En la referencia (20) puede obtenerse un programa informático para el cálculo de PIF y MPE.
2.3. INTERPRETACIÓN DE LOS RESULTADOS
Según el estudio de validación (8), una sustancia problema con un PIF < 2 o un MPE < 0,1 no es fototóxica. Un PIF > 2 y < 5 o un MPE > 0,1 y < 0,15 indica fototoxicidad probable, y un PIF > 5 o un MPE > 0,15 indica fototoxicidad.
Cualquier laboratorio que efectúe este ensayo por primera vez debe hacerlo con las sustancias de referencia del cuadro 1 antes de proceder al ensayo de la fototoxicidad de sustancias problema. Los valores de PIF o MPE deben ser similares a los valores indicados en el cuadro 1.
Cuadro 1
|
Nombre químico |
No EINECS |
No CAS |
PIF |
MPE |
Pico de absorción |
Disolvente (9) |
|
Amiodarona HCl |
243-293-2 |
[19774-82-4] |
> 3,25 |
0,27-0,54 |
242 nm 300 nm (hombro) |
etanol |
|
Clorpromazina HCl |
200-701-3 |
[69-09-0] |
> 14,4 |
0,33-0,63 |
309 nm |
etanol |
|
Norfloxacina |
274-614-4 |
[70458-96-7] |
> 71,6 |
0,34-0,90 |
316 nm |
acetonitrilo |
|
Antraceno |
204-371-1 |
[120-12-7] |
> 18,5 |
0,19-0,81 |
356 nm |
acetonitrilo |
|
Protoporfirina IX, disodio |
256-815-9 |
[50865-01-5] |
> 45,3 |
0,54-0,74 |
402 nm |
etanol |
|
L-histidina |
|
[7006-35-1] |
sin PIF |
0,05-0,10 |
211 nm |
agua |
|
Hexaclorofeno |
200-733-8 |
[70-30-4] |
1,1-1,7 |
0,00-0,05 |
299 nm 317 nm (hombro) |
etanol |
|
Laurilsulfato de sodio |
205-788-1 |
[151-21-3] |
1,0-1,9 |
0,00-0,05 |
sin absorción |
agua |
2.4. INTERPRETACIÓN DE LOS DATOS
Si se observan efectos fotocitotóxicos solo a la mayor concentración de ensayo (especialmente en caso de sustancias problema hidrosolubles), para la evaluación del peligro puede ser necesario tener en cuenta otros aspectos, entre los que se pueden incluir datos sobre la absorción cutánea y la acumulación de la sustancia en la piel, así como datos procedentes de otros ensayos, como el del efecto de la sustancia in vitro sobre piel humana o animal, o sobre modelos de piel.
Si no se pone de manifiesto ningún efecto tóxico (+ Irr y - Irr) y si la baja solubilidad limita las concentraciones que se pueden utilizar en el ensayo, puede dudarse de la compatibilidad de la sustancia problema con el ensayo y ha de estudiarse la posibilidad de efectuar un ensayo de confirmación, por ejemplo con otro modelo.
3. INFORMES
INFORME DEL ENSAYO
El informe del ensayo debe dar, como mínimo, la información siguiente:
|
|
Sustancia de ensayo:
|
|
|
Disolvente:
|
|
|
Células:
|
|
|
Condiciones de la prueba (1); incubación antes y después del tratamiento:
|
|
|
Condiciones de la prueba (2); tratamiento con la sustancia:
|
|
|
Condiciones de la prueba (3); irradiación:
|
|
|
Condiciones de la prueba (4); ensayo de viabilidad con rojo neutro:
|
|
|
Resultados:
|
|
|
Discusión de los resultados. |
|
|
Conclusiones. |
4. REFERENCIAS
|
1. |
Lovell W.W. (1993). A scheme for in vitro screening of substances for photoallergenic potential. Toxic. In Vitro 7: 95-102. |
|
2. |
Santamaria, L. and Prino, G. (1972). List of the photodynamic substances. In «Research Progress in Organic, Biological and Medicinal Chemistry» Vol. 3 part 1. North Holland Publishing Co. Amsterdam. p XI-XXXV. |
|
3. |
Spielmann, H., Lovell, W.W., Hölzle, E., Johnson, B.E., Maurer, T., Miranda, M.A., Pape, W.J.W., Sapora, O., and Sladowski, D. (1994). In vitro phototoxicity testing: The report and recommendations of ECVAM Workshop 2. ATLA, 22, 314-348. |
|
4. |
Spikes, J.D. (1989). Photosensitization. In «The science of Photobiology» Edited by K.C. Smith. Plenum Press, New York. 2nd edition, p 79-110. |
|
5. |
OECD (1997) Environmental Health and Safety Publications, Series on Testing and Assessment No.7 «Guidance Document On Direct Phototransformation Of Chemicals In Water» Environment Directorate, OECD, Paris. |
|
6. |
Spielmann, H., Balls, M., Döring, B., Holzhütter, H.G., Kalweit, S., Klecak, G., L'Eplattenier, H., Liebsch, M., Lovell, W.W., Maurer, T., Moldenhauer. F. Moore. L., Pape, W., Pfannbecker, U., Potthast, J., De Silva, O., Steiling, W., and Willshaw, A. (1994). EEC/COLIPA project on in vitro phototoxicity testing: First results obtained with a Balb/c 3T3 cell phototoxicity assay. Toxic. In Vitro 8, 793-796. |
|
7. |
Anon (1998). Statement on the scientific validity of the 3T3 NRU PT test (an in vitro test for phototoxicity), European Commission, Joint Research Centre: ECVAM and DGXI/E/2, 3 November 1997, ATLA, 26, 7-8. |
|
8. |
Spielmann, H., Balls, M., Dupuis, J., Pape, W.J.W., Pechovitch, G. De Silva, O., Holzhütter, H.G., Clothier, R., Desolle, P., Gerberick, F., Liebsch, M., Lovell, W.W., Maurer, T., Pfannenbecker, U., Potthast, J. M., Csato, M., Sladowski, D., Steiling, W., and Brantom, P. (1998). The international EU/COLIPA In vitro phototoxicity validation study: results of phase II (blind trial), part 1: the 3T3 NRU phototoxicity test. Toxic. In Vitro 12, 305-327. |
|
9. |
OECD (2002) Extended Expert Consultation Meeting on The In Vitro 3T3 NRU Phototoxicity Test Guideline Proposal, Berlin, 30th-31th October 2001, Secretariat's Final Summary Report, 15th March 2002, OECD ENV/EHS, available upon request from the Secretariat. |
|
10. |
Borenfreund, E., and Puerner, J.A. (1985). Toxicity determination in vitro by morphological alterations and neutral red absorption. Toxicology Lett., 24,119-124. |
|
11. |
Hay, R.J. (1988) The seed stock concept and quality control for cell lines. Analytical Biochemistry 171, 225-237. |
|
12. |
Lambert L.A, Warner W.G., and Kornhauser A. (1996) Animal models for phototoxicity testing. In «Dermatotoxicology», edited by F.N. Marzulli and H.I. Maibach. Taylor & Francis, Washington DC. 5th Edition, p. 515-530. |
|
13. |
Tyrrell R.M., Pidoux M (1987) Action spectra for human skin cells: estimates of the relative cytotoxicity of the middle ultraviolet, near ultraviolet and violet regions of sunlight on epidermal keratinocytes. Cancer Res., 47, 1825-1829. |
|
14. |
ISO 10977. (1993). Photography — Processed photographic colour films and paper prints — Methods for measuring image stability. |
|
15. |
Sunscreen Testing (UV.B) TECHNICALREPORT, CIE, International Commission on Illumnation, Publication No. 90, Vienna, 1993, ISBN 3900734275 |
|
16. |
ZEBET/ECVAM/COLIPA — Standard Operating Procedure: In Vitro 3T3 NRU Phototoxicity Test. Final Version, 7 September, 1998. 18 pgs. |
|
17. |
Spielmann, H., Balls, M., Dupuis, J., Pape, W.J.W., De Silva, O., Holzhütter, H.G., Gerberick, F., Liebsch, M., Lovell, W.W., and Pfannenbecker, U. (1998) A study on UV filter chemicals from Annex VII of the European Union Directive 76/768/EEC, in the in vitro 3T3 NRU phototoxicity test. ATLA 26, 679-708. |
|
18. |
Holzhütter, H.G., and Quedenau, J. (1995) Mathematical modeling of cellular responses to external signals. J. Biol. Systems 3, 127-138. |
|
19. |
Holzhütter, H.G. (1997). A general measure of in vitro phototoxicity derived from pairs of dose-response curves and its use for predicting the in vivo phototoxicity of chemicals. ATLA, 25, 445-462. |
|
20. |
http://www.oecd.org/document/55/0,2340,en_ 2649 34377 2349687 _1_1_1_1, 00.html |
ANEXO 1
Función del ensayo de fototoxicidad 3T3 ARN en un enfoque secuencial de los ensayos de fototoxicidad de sustancias químicas
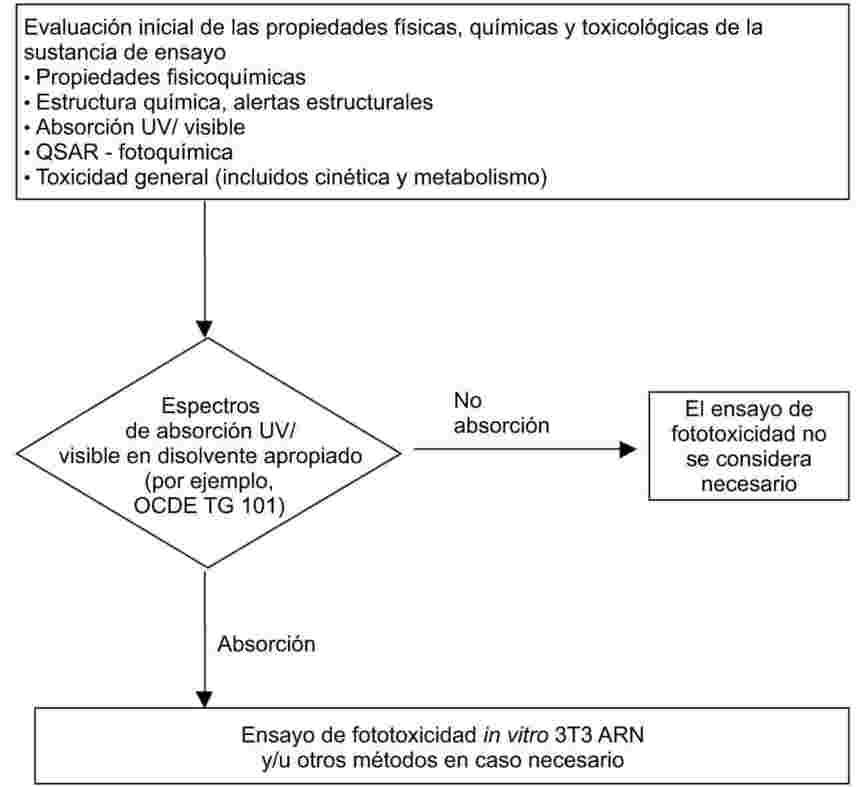
ANEXO 2
Figura 1
Distribución espectral de potencia de un simulador solar con filtro
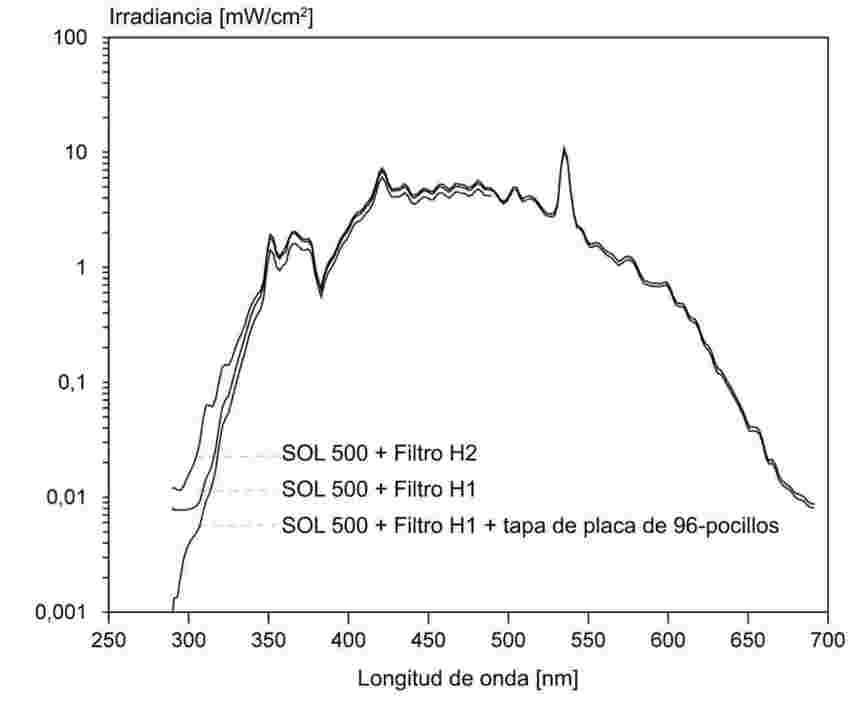
(véase el punto 1.4.1.5, párrafo segundo)
La figura 1 muestra un ejemplo de distribución aceptable de la irradiancia espectral de un simulador solar con filtro. Procede de la fuente de haluro metálico cebado utilizada en la prueba de validación del ensayo de fototoxicidad 3T3 ARN (6) (8) (17). Se indica el efecto de dos filtros distintos y el efecto filtrante adicional de la tapa de una placa de cultivo celular de 96 pocillos. El filtro H2 solo se ha utilizado con sistemas de ensayo que pueden tolerar una cantidad elevada de UVB (ensayo con modelo de piel y ensayo de fotohemólisis de eritrocitos). En el ensayo de fototoxicidad 3T3 ARN se utilizó el filtro H1. La figura muestra que el efecto filtrante adicional de la tapa de la placa se observa sobre todo en la banda de UVB, pero queda en el espectro de irradiación una cantidad suficiente de UVB para excitar sustancias que absorben normalmente en esta banda, como la amiodarona (véase el cuadro 1).
Figura 2
Sensibilidad de las células Balb/c 3T3 ante la irradiación (medida en la banda UVA)
Viabilidad de las células (porcentaje de la absorción de rojo neutro respecto a los controles mantenidos en la oscuridad)

(Véanse el punto 1.4.1.5.2, párrafo segundo, y los puntos 1.4.2.2.1 y 1.4.2.2.2)
Sensibilidad de las células Balb/c 3T3 ante la irradiación con el simulador solar utilizado en la prueba de validación del ensayo de fototoxicidad 3T3 ARN, medida en la banda UVA. La figura muestra los resultados obtenidos en siete laboratorios diferentes en el estudio de prevalidación (1). Las dos curvas con símbolos huecos se obtuvieron con células viejas (gran número de pases), que tuvieron que sustituirse por nuevas poblaciones celulares, mientras que las curvas con símbolos macizos corresponden a células con una tolerancia aceptable a la irradiación.
Con estos datos se determinó que la dosis máxima de irradiación no citotóxica es de 5 J/cm2 (línea discontinua vertical). La línea discontinua horizontal muestra además el efecto aceptable máximo de irradiación contemplado en el punto 1.4.2.2.
B.42. SENSIBILIZACIÓN CUTÁNEA: PRUEBA CON GANGLIOS LINFÁTICOS LOCALES
1. MÉTODO
Este método de evaluación es equivalente al método TG 429 de la OCDE (2002).
1.1. INTRODUCCIÓN
El ensayo con ganglios linfáticos locales (Local Lymph Node Assay, LLNA) ha sido suficientemente validado y aceptado para justificar su adopción como nuevo método (1) (2) (3). Se trata del segundo método para evaluar el poder de sensibilización cutánea de productos químicos en animales. En el otro método (B.6) se utilizan estudios con cobayas, sobre todo el método de ensayo de maximización en cobayas y el método de ensayo de Buehler (4).
El LLNA constituye un método alternativo para la identificación de productos químicos que producen sensibilización cutánea, y para confirmar que los productos químicos carecen de un potencial significativo de producirla. Esto no significa necesariamente que la LLNA deba sustituir en todos los casos a estudios con cobayas, sino que posee la misma capacidad y se puede utilizar como alternativa en que, por lo general, los resultados positivos y negativos ya no necesitan una confirmación posterior.
El LLNA aporta ciertas ventajas en lo que respecta al progreso científico y al bienestar de los animales. Estudia la fase de inducción de la sensibilización cutánea, y aporta datos cuantitativos adecuados para evaluar la respuesta a la dosis. Los detalles de la validación del LLNA y una revisión de los trabajos relacionados ya han sido publicados (5) (6) (7) (8). Hay que señalar, además, que los sensibilizantes leves a moderados, que se recomiendan como sustancias de control positivo adecuadas en los métodos de ensayo con cobayas, también son adecuados para el LLNA (6) (8) (9).
El LLNA es un método in vivo, por lo que no eliminará el uso de animales para la evaluación de la actividad de sensibilización por contacto. No obstante, puede reducir el número de animales necesarios para este fin. Además supone un refinamiento sustancial de la forma de utilizar los animales en los ensayos de sensibilización por contacto. El LLNA se basa en la consideración de acontecimientos inmunológicos estimulados por productos químicos durante la fase de inducción de la sensibilización. Al contrario que los ensayos que utilizan cobayas, el LLNA no necesita provocar reacciones cutáneas de sensibilidad. Tampoco exige el uso de adyuvantes, como ocurre en el método de ensayo de maximización con cobayas. Por consiguiente, el LLNA reduce las molestias para los animales. A pesar de las ventajas que supone con respecto a los ensayos tradicionales con cobayas, hay que reconocer que el LLNA tiene ciertas limitaciones que puede obligar a utilizarlas (por ejemplo, los resultados falsos negativos que se producen en el LLNA con determinados metales, los falsos positivos con algunos irritantes cutáneos) (10).
Véase también la Introducción de la parte B.
1.2. PRINCIPIO DEL MÉTODO DE EVALUACIÓN
El principio básico que subyace en el LLNA es que los sensibilizantes inducen una proliferación primaria de linfocitos en el ganglio linfático que drena la zona de aplicación del producto químico. Esta proliferación es proporcional a la dosis aplicada (y a la potencia del alérgeno) y proporciona un método sencillo para obtener una medida cuantitativa y objetiva de la sensibilización. El LLNA evalúa esta proliferación como si fuera una relación de respuesta a la dosis, en la que se compara la proliferación en los grupos de ensayo con la observada en los controles tratados con un vehículo. Se determina lo que se conoce como índice de estimulación, que es la proporción existente entre la proliferación observada en los grupos tratados y la observada en los controles tratados con vehículo; dicho índice debe alcanzar un valor mínimo de tres para que una sustancia pueda ser sometida a una posterior evaluación como posible sensibilizante cutáneo. Los métodos que se describen aquí se basan en el uso de marcado radiactivo para medir la proliferación celular. Pero se pueden utilizar otros criterios para valorar la proliferación, siempre que se disponga del correspondiente fundamento científico, con toda la bibliografía y la correspondiente descripción de la metodología.
1.3. DESCRIPCIÓN DEL MÉTODO DE EVALUACIÓN
1.3.1. Preparativos
1.3.1.1. Condiciones de alojamiento y alimentación
Los animales deben ser alojados de manera individual. La temperatura de los animalarios debe ser de 22 oC (±3 oC). Aunque la humedad relativa debe ser del 30 % como mínimo y preferiblemente no superar el 70 %, excepto durante la limpieza del animalario, el objetivo debe ser el 50-60 %. La iluminación será artificial, con 12 horas de luz y 12 de oscuridad. Para la alimentación se podrán utilizar dietas de laboratorio convencionales, con suministro ilimitado de agua para beber.
1.3.1.2. Preparación de los animales
Se eligen los animales al azar, se les marca para su identificación individual (pero no en las orejas) y permanecen en sus jaulas al menos 5 días antes de la administración, para que se aclimaten a las condiciones del laboratorio. Antes de iniciar el tratamiento todos los animales serán examinados para comprobar que no presentan lesiones cutáneas observables.
1.3.2. Condiciones del ensayo
1.3.2.1. Animales de experimentación
El ratón es la especie elegida para este ensayo. Se utilizan hembras adultas jóvenes de las cepas CBA/Ca o CBA/J, nulíparas y que no estén preñadas. Al comienzo del estudio los animales deben tener una edad de 8 a 12 semanas; la variación del peso de los animales debe ser mínima, sin superar el 20 % del peso medio. Se podrán utilizar otras cepas y machos, cuando se aporten datos suficientes para demostrar que no existen diferencias significativas específicas de la cepa o del sexo en la respuesta LLNA.
1.3.2.2. Verificación de la fiabilidad
Se utilizan controles positivos para demostrar que el funcionamiento del ensayo es adecuado, y la competencia del laboratorio para realizarlo con éxito. El control positivo debe dar una respuesta LLNA positiva con un nivel de exposición calculado para producir un aumento del índice de estimulación (IE) > 3 con respecto al grupo de control negativo. La dosis del control positivo debería elegirse de tal manera que la inducción sea evidente, pero no excesiva. Las sustancias preferidas son el aldehído hexilcinámico (CAS No 101-86-0, EINECS no 202-983-3) y el mercaptobenzotiazol (CAS No 149-30-4, EINECS no 205-736-8). En algunas circunstancias se pueden utilizar otras sustancias de control, mediante una justificación adecuada y siempre que se cumplan los criterios antes mencionados. Aunque normalmente se necesita un grupo de control positivo en cada ensayo, puede haber situaciones en las que los laboratorios de ensayos dispongan de datos históricos de controles positivos, que demuestren la uniformidad de una respuesta satisfactoria durante un período de 6 meses o más. En tales situaciones puede estar indicado hacer menos ensayos con controles positivos, en intervalos que no superen los 6 meses. Aunque la sustancia de control positivo debe ser evaluada en un vehículo conocido por provocar una respuesta uniforme (por ejemplo, acetona: aceite de oliva), puede haber ciertas situaciones legales en las que también sea necesario evaluar en un vehículo no estándar (formulación relevante desde el punto de vista clínico o químico). En tal caso habrá que evaluar la posible interacción del control positivo con este vehículo no convencional.
1.3.2.3. Número de animales, niveles de dosis y selección del vehículo
Se utilizará un mínimo de 4 animales por grupo de dosis, con un mínimo de 3 concentraciones de la sustancia en estudio, más un grupo de control negativo tratado solo con el vehículo utilizado con la sustancia analizada y en su caso, un control positivo. Cuando se vayan a recoger datos de animales individuales será necesario un mínimo de 5 animales por grupo de dosis. Excepto por la ausencia de tratamiento con la sustancia de ensayo, los animales de los grupos control deben ser manejados y tratados de manera idéntica a la utilizada con los animales de los grupos tratados.
La elección de la dosis y el vehículo debe basarse en las recomendaciones que se recogen en la referencia (1). Las dosis se eligen a partir de series de concentraciones del 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 %, etc. Para elegir las 3 concentraciones consecutivas se tendrán en cuenta los datos existentes sobre toxicidad aguda e irritación cutánea, en su caso, de forma que la concentración más alta logre la máxima exposición evitando la toxicidad sistémica y la excesiva irritación cutánea local (2) (11).
El vehículo se elegirá de forma que se logre la máxima concentración y solubilidad de la sustancia analizada con una relación solución/suspensión adecuada para la aplicación de la sustancia. Los vehículos recomendados son, por orden de preferencia, acetona/aceite de oliva (4:1 v/v), dimetilformamida, metil etil cetona, propilenglicol y dimetilsulfóxido (2) (10), pero se pueden utilizar otros si se aportan la correspondiente justificación científica. En determinadas situaciones puede ser necesario utilizar un disolvente relevante desde el punto de vista clínico o la formulación comercial en que se vende la sustancia analizada, como control adicional. Se tendrá especial cuidado en garantizar que se incorporan materiales hidrófilos al sistema del vehículo, pues humedecen la piel y no se pierden inmediatamente. Por eso se evitarán los vehículos totalmente acuosos.
1.3.3. Procedimiento de ensayo
1.3.3.1. Calendario del experimento
El calendario del experimento es el siguiente:
|
|
Dia 1: Se identifica cada animal y se toma nota del peso. En el dorso de cada oreja se hace una aplicación abierta de 25 μl de la apropiada dilución de la sustancia en estudio, del vehículo solo o del control positivo (si procede). |
|
|
Días 2 y 3: Se repite el procedimiento de aplicación del día 1. |
|
|
Días 4 y 5: Sin tratamiento. |
|
|
Día 6: Se toma nota del peso de cada animal. Se inyectan 250 μl de suero salino tamponado con fosfato (PBS) que contenga 20 μCi (7,4e + 8 Bq) de 3H-metil timidina a todos los ratones (de evaluación y de control) a través de la vena de la cola. También se pueden inyectar 250 μL de PBS con 2 μCi (7,4e + 7 Bq) de 125I-yododesoxiuridina y fluorodesoxiuridina 10-5 M a todos los ratones a través de la vena de la cola. Cinco horas después se sacrifica a los animales. Se extirpan los ganglios linfáticos auriculares de cada oreja y se agrupan en PBS por cada grupo de experimentación (enfoque de agrupación por grupo de tratamiento); también se pueden extirpar pares de ganglios linfáticos de animales individuales y agruparlos en PBS por animal (enfoque de animales individuales). En el anexo I de la referencia 10 se recogen los detalles de la identificación y disección de los ganglios, con dibujos explicativos. |
1.3.3.2. Preparación de suspensiones celulares
Se prepara una única suspensión de células de ganglios linfáticos (CGL) a partir de los grupos de tratamiento agrupados o bilateralmente de animales individuales, mediante disgregación mecánica suave con una malla de acero inoxidable de 200 μm. Se lavan las CGL dos veces con PBS en exceso y se hacen precipitar con ácido tricloroacético (ATC) al 5 % a 4 oC durante 18 horas (1). A continuación el sedimento se vuelve a suspender en 1 ml de ATC y se traslada a viales de gammagrafía con 10 ml de líquido de gammagrafía para el recuento de 3H, o se transfieren directamente a tubos de recuento gamma para el recuento de 125I.
1.3.3.3. Determinación de la proliferación celular (radiactividad incorporada)
La incorporación de 3H-metil timidina se mide por gammagrafía-β, que cuenta las desintegraciones por minuto (DPM). La incorporación de 125I-yododesoxiuridina se mide por el recuento de 125I y también se expresa en DPM. Dependiendo del enfoque utilizado, la incorporación se expresará como DPM/grupo de tratamiento (enfoque acumulado) o como DPM/animal (enfoque individual).
1.3.3.4. Observaciones
1.3.3.4.1. Observaciones clínicas
Una vez al día se realizará una observación minuciosa de los animales en busca de signos clínicos, bien de irritación local en el punto de aplicación o de toxicidad sistémica. Todas las observaciones se anotarán sistemáticamente en los registros individuales abiertos para cada animal.
1.3.3.4.2. Peso corporal
Como se indica en el punto 1.3.3.1 hay que medir el peso corporal individual de los animales al empezar el ensayo y en el momento del sacrifico de los animales.
1.3.4. Cálculo de resultados
Los resultados se expresan por el índice de estimulación (IE). Si se emplea el enfoque por acumulación, el IE se obtiene dividiendo la incorporación de radiactividad acumulada en cada grupo de tratamiento por la incorporación acumulada en el grupo de control con vehículo; así se obtiene un IE medio. Si se emplea el enfoque individual, el IE se obtiene dividiendo la media de DPM/animal de cada grupo de tratamiento y la del control positivo por la media de DPM/animal del grupo de control con vehículo o disolvente. De esta forma, el IE promedio para controles tratados con vehículo es 1.
El uso del enfoque individual para calcular el IE permite realizar un análisis estadístico de los datos. Para elegir el método de análisis estadístico adecuado el investigador debe conocer las posibles irregularidades de las variancias y otros problemas asociados, que pueden exigir una transformación de los datos o el uso de un análisis estadístico no paramétrico. Un enfoque adecuado para interpretar los datos consiste en evaluar todos los datos individuales de los animales tratados y los de control con vehículo, y derivar de ellos la curva de respuesta a la dosis más adecuada, teniendo en cuenta los límites de confianza (8) (12) (13). No obstante el investigador debe estar alerta a las posibles respuestas «fuera de los límites» en animales individuales de un grupo, que pueden obligar a utilizar otra forma de medir la respuesta (por ejemplo, la mediana en lugar de la media) o a eliminar a los que quedan fuera de los límites.
El proceso de decisiones con respecto a la respuesta positiva incluye un índice de estimulación >3 junto con la consideración de la respuesta a la dosis y de la significación estadística, en su caso (3) (6) (8) (12) (14).
Si fuera necesario aclarar los resultados obtenidos, se tendrán en cuenta diversas propiedades de la sustancia analizada, como si tiene relación estructural con sensibilizantes cutáneos conocidos, si produce irritación cutánea excesiva y la naturaleza de la respuesta a la dosis observada. Estas y otras consideraciones se comentan con detalle en otro punto (7).
2. DATOS
Los datos se resumirán en tablas, con los valores medios e individuales de DPM y los índices de estimulación de cada grupo de dosis (incluido el de control con vehículo).
3. NOTIFICACIÓN
3.1. INFORME DEL ENSAYO
El informe del ensayo contendrá la siguiente información:
|
|
Sustancia analizada:
|
|
|
Vehículo:
|
|
|
Animales de experimentación:
|
|
|
Condiciones del análisis:
|
|
|
Verificación de la fiabilidad:
|
|
|
Resultados:
|
|
|
evolución cronológica del inicio y signos de toxicidad, incluida la irritación cutánea en el punto de administración en cada animal, en su caso.
|
4. REFERENCIAS
|
1. |
Kimber, I. and Basketter, D.A. (1992). The murine local lymph node assay; collaborative studies and new directions: A commentary. Food and Chemical Toxicology 30, 165-169. |
|
2. |
Kimber, I, Derman, R.J., Scholes E.W, and Basketter, D.A. (1994). The local lymph node assay: developments and applications. Toxicology, 93, 13-31. |
|
3. |
Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C.A., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E., Hastings, K.L. (1998). Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. Journal of Toxicology and Environmental Health, 53, 563-79. |
|
4. |
Testing Method B.6. |
|
5. |
Chamberlain, M. and Basketter, D.A. (1996). The local lymph node assay: status of validation. Food and Chemical Toxicology, 34, 999-1002. |
|
6. |
Basketter, D.A., Gerberick, G.F., Kimber, I. and Loveless, S.E (1996). The local lymph node assay- A viable altemative to currently skin sensitisation tests. Food and Chemical Toxicology, 34, 985-997. |
|
7. |
Basketter, D.A., Gerberick, G.F. and Kimber, I. (1998). Strategies for identifying false positive responses in predictive sensitisation tests. Food and Chemical Toxicology. 36, 327-33. |
|
8. |
Van Och, F.M.M, Slob, W., De Jong, W.H., Vandebriel, R.J., Van Loveren, H. (2000). A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employement of a regression method that includes determination of uncertainty margins. Toxicology, 146,49-59. |
|
9. |
Dearman, R.J., Hilton, J., Evans, P., Harvey, P., Basketter, D.A. and Kimber, I. (1998). Temporal stability of local lymph node assay responses to hexyl cinnamic aldehyde. Journal of Applied Toxicology, 18, 281-4. |
|
10. |
National Institute of Environmental Health Sciences (1999). The. Murine Local Lymph Node Assay: A Test Method for Assessing the Allergic Contact Dermatitis Potential of Chemicals/Compounds: The Results of an Independent Peer Review Evaluation Coordinated by the Interagency Coordinating Committee on the Validation of Altemative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Altemative Toxicological Methods (NICETAM). NIH Publication No: 99-4494, Research Triangle Park, N.C. (http://iccvam.niehs.nih.gov). |
|
11. |
Testing method B.4. |
|
12. |
Basketter, D.A., Selbie, E., Scholes, E.W. Lees, D. Kimber, I. and Botham, P.A. (1993) Results with OECD recommended positive control sensitisers in the maximisation, Buehler and local lymph node assays. Food and Chemical Toxicology, 31, 63-67. |
|
13. |
Basketter D.A., Lea L.J., Dickens A., Briggs D., Pate I., Dearman R.J., Kimber I. (1999). A comparison of statistical approaches to the derivation of EC3 values from local lymph node assay dose responses. J. Appl. Toxicology, 19, 261-266. |
|
14. |
Basketter DA, Blaikie L, Derman RJ, Kimber I, Ryan CA, Gerberick GF, Harvey P, Evans P, White IR and Rycroft RTG (2000). Use of local lymph node assay for the estimation of relative contact allergenic potency. Contact Dermatitis 42 ,344-48. |
B.43. ESTUDIO DE NEUROTOXICIDAD EN ROEDORES
1. MÉTODO
El presente método reproduce las directrices de ensayo de la OCDE TG 424 (1997).
Este método de ensayo tiene por objeto obtener la información necesaria para confirmar o caracterizar de manera más precisa la posible neurotoxicidad de sustancias químicas en animales adultos. Puede combinarse con los actuales métodos de ensayo de los estudios de toxicidad por administración continuada o bien realizarse como estudio independiente. Se recomienda consultar el documento orientativo de la OCDE sobre estrategias y métodos de ensayo de neurotoxicidad (1) (OECD Guidance Document on Neurotoxicity Testing Strategies and Methods), que puede servir de ayuda para la concepción de estudios basados en este método de ensayo. Esta consulta es especialmente importante cuando se recomienden modificaciones de las observaciones y procedimientos de ensayo para el uso habitual del presente método. El documento orientativo se ha preparado a fin de facilitar la selección de otros procedimientos de ensayo que puedan emplearse en determinadas circunstancias.
En el presente método no se trata la evaluación de la neurotoxicidad para el desarrollo.
1.1. INTRODUCCIÓN
En la evaluación de las características tóxicas de las sustancias químicas, es importante considerar el potencial de efectos neurotóxicos. El método de ensayo de toxicidad sistémica por administración continuada incluye ya observaciones para detectar la posible neurotoxicidad. El presente método de ensayo puede utilizarse para preparar un estudio destinado a obtener más información sobre los efectos neurotóxicos observados en los estudios de toxicidad sistémica por administración continuada o para confirmar tales efectos. Sin embargo, según la valoración que se haga de la posible neurotoxicidad de ciertas clases de sustancia químicas puede considerarse más conveniente evaluarlas mediante este método sin indicación previa de la posible neurotoxicidad a partir de estudios de toxicidad sistémica por administración continuada. Entre los aspectos que deben tenerse en cuenta para ello pueden citarse los siguientes:
|
— |
observación de signos neurológicos o lesiones neuropatológicas en estudios de toxicidad distintos de los estudios de toxicidad sistémica por administración continuada, o |
|
— |
relación estructural u otro tipo de información que los relacione con neurotóxicos conocidos. |
Además puede haber otros casos en los que sea adecuado utilizar este método de ensayo, para más información véase (1).
El presente método se ha concebido de manera que pueda ajustarse a distintas necesidades concretas a la hora de confirmar la neurotoxicidad histopatológica y comportamental de una sustancia, y de aportar una caracterización y cuantificación de las respuestas neurotóxicas.
Anteriormente, la neurotoxicidad se equiparaba a la neuropatía que implicaba lesiones neuropatológicas o disfunciones neurológicas, como convulsiones, parálisis o temblores. Aunque la neuropatía es una manifestación importante de la neurotoxicidad, actualmente se sabe que hay muchos otros signos que indican toxicidad en el sistema nervioso (por ejemplo, pérdida de coordinación motora, deficiencias sensoriales y disfunciones del aprendizaje y la memoria) y que pueden no aparecer en estudios de neuropatía o en otros tipos de estudios.
El presente método de ensayo de neurotoxicidad está pensado para detectar efectos neuropatológicos y neurocomportamentales en roedores adultos. Aunque los efectos en el comportamiento, incluso en ausencia de cambios morfológicos, pueden dar cuenta de un impacto nocivo en el organismo, no todas las alteraciones del comportamiento están relacionadas con el sistema nervioso. Por lo tanto, cualquier cambio observado tiene que evaluarse junto con datos correlativos de tipo bioquímico, hematológico e histopatológico, y con datos sobre otros tipos de toxicidad sistémica. Las pruebas prescritas en este método para obtener la caracterización y cuantificación de las respuestas neurotóxicas incluyen procedimientos comportamentales e histopatológicos que pueden ir apoyados de manera complementaria por investigaciones bioquímicas y/o electrofisiológicas (1) (2) (3) (4).
Los neurotóxicos pueden actuar sobre un número de dianas dentro del sistema nervioso y mediante una gran variedad de mecanismos. Dado que no hay ningún conjunto de ensayos que, por sí solo, sea capaz de evaluar perfectamente el potencial neurotóxico de todas las sustancias, puede ser necesario utilizar otros ensayos in vivo o in vitro aplicables concretamente al tipo de neurotoxicidad observada o prevista.
Este método de ensayo puede aplicarse también junto con las orientaciones establecidas en el documento de la OCDE denominado OECD Guidance Document on Neurotoxicity Testing Strategies and Methods (1) a fin de preparar estudios destinados a caracterizar de manera más precisa la cuantificación dosis-respuesta o a aumentar la sensibilidad de esta, de manera que pueda estimarse más exactamente un nivel sin efectos adversos observados o comprobar peligros sospechados o conocidos de la sustancia. Por ejemplo, pueden prepararse estudios para determinar y evaluar los mecanismos neurotóxicos o para complementar los datos ya disponibles a partir de los procedimientos básicos de observación neuropatológica y neurocomportamental. No es necesario que estos estudios generen de nuevo datos que se obtendrían también a partir de los procedimientos estándar recomendados en el presente método, si estos datos ya son conocidos y no se consideran necesarios para la interpretación de los resultados del estudio.
Este estudio de neurotoxicidad, cuando se realiza solo o en combinación con otros, aporta información que puede:
|
— |
determinar si el sistema nervioso está afectado de manera permanente o reversible por la sustancia ensayada; |
|
— |
contribuir a la caracterización de las alteraciones del sistema nervioso relacionadas con la exposición a la sustancia y a la comprensión de los mecanismos que intervienen; |
|
— |
determinar las relaciones entre dosis y tiempo de respuesta a fin de estimar un nivel sin efectos adversos observados (que pueda utilizarse con objeto de establecer criterios de seguridad para la sustancia). |
En este método de ensayo se administra la sustancia estudiada por vía oral. Otras vías de administración (por ejemplo, por vía dérmica o por inhalación) pueden resultar más adecuadas y requerir modificaciones de los procedimientos recomendados. La elección de la vía de administración depende del perfil de exposición humana y de la información cinética y toxicológica disponible.
1.2. DEFINICIONES
Efecto adverso: toda alteración respecto a una situación de referencia, relacionada con el tratamiento, que disminuya la capacidad del organismo para sobrevivir, reproducirse o adaptarse al entorno.
Dosis: cantidad de sustancia de ensayo administrada. Se expresa en peso (g, mg), en peso de sustancia de ensayo por unidad de peso del animal (por ejemplo, mg/kg) o en concentración constante en la dieta (ppm).
Posología: término general que abarca la dosis administrada, su frecuencia y duración.
Neurotoxicidad: alteración adversa de la estructura o función del sistema nervioso debida a la exposición a un agente físico, biológico o químico.
Neurotóxico: cualquier agente físico, biológico o químico que pueda provocar neurotoxicidad.
NOAEL: sigla inglesa referente a la dosis de exposición sin efectos adversos observados, es decir, la dosis más alta a la que no se observa ningún efecto adverso debido al tratamiento.
1.3. PRINCIPIO DEL MÉTODO DE ENSAYO
Se administra por vía oral una serie de dosis de la sustancia a varios grupos de roedores de laboratorio. Normalmente se requiere una administración continuada y el período de administración puede ser 28 días, 90 días (toxicidad subcrónica) o 1 año o más (toxicidad crónica). Los procedimientos establecidos en este método de ensayo pueden aplicarse también a un estudio de neurotoxicidad aguda. Los animales se someten a ensayo para detectar anomalías neurológicas o del comportamiento o para caracterizarlas. Durante cada período de observación se evalúa una gama de comportamientos que podría estar afectada por neurotóxicos. Al final del ensayo, se hace una perfusión in situ a un subgrupo de animales de cada sexo y de cada grupo, y se preparan y examinan secciones de los nervios periféricos y de la médula espinal y el cerebro.
Cuando se trate de un estudio único independiente destinado a detectar neurotoxicidad o a caracterizar los efectos neurotóxicos, los animales de cada grupo no empleados para perfusión y posterior histopatología (véase el cuadro 1) pueden utilizarse para otros procedimientos concretos de tipo electrofisiológico, neuroquímico, neuropatológico o neurocomportamental que puedan complementar los datos obtenidos a partir de los exámenes estándar prescritos en el presente método (1). Estos procedimientos complementarios pueden ser especialmente útiles cuando las observaciones empíricas o los efectos previstos indiquen un tipo determinado o una diana de neurotoxicidad química. Otra posibilidad es utilizar los animales restantes para evaluaciones como las prescritas en los métodos de ensayo para estudios de toxicidad por administración continuada en roedores.
Cuando los procedimientos del presente método de ensayo se combinen con los de otros métodos, se necesita un número suficiente de animales para que puedan cumplirse todos los requisitos de las observaciones de ambos estudios.
1.4. DESCRIPCIÓN DEL MÉTODO
1.4.1. Selección de la especie animal
La especie de preferencia entre los roedores es la rata, aunque pueden utilizarse otras especies de roedores. Deben usarse animales adultos jóvenes y sanos de cepas utilizadas habitualmente en laboratorio. Las hembras deben ser nulíparas y no grávidas. La administración ha de empezar lo antes posible tras el destete, preferiblemente, antes de que los animales tengan 6 semanas de edad y, en cualquier caso, antes de que tengan 9 semanas. No obstante, cuando este estudio se combine con otros, podrían tener que ajustarse estos requisitos de edad. Al principio del estudio, la diferencia de peso entre los animales empleados no debe superar el ± 20 % del peso medio de cada sexo. Cuando se realice un estudio de la toxicidad oral por administración continuada como fase previa de un estudio de toxicidad a largo plazo, deben utilizarse en ambos estudios animales procedentes de la misma cepa y del mismo origen.
1.4.2. Alojamiento y alimentación
El cuarto de experimentación ha de estar a una temperatura de 22 oC (± 3 oC). Aunque la humedad relativa ha de ser, como mínimo, del 30 % y preferiblemente no superior al 70 %, salvo durante la limpieza del local, lo ideal es que esté comprendida entre el 50 y el 60 %. Se aplicará una iluminación artificial en una secuencia de 12 horas de luz y 12 horas de oscuridad. Debe mantenerse al mínimo el ruido fuerte intermitente. Puede proporcionarse una dieta alimentaria corriente para animales de laboratorio y agua a voluntad. Si la sustancia de ensayo se administra con los alimentos, es preciso obtener una mezcla adecuada, lo cual puede influir en la elección de la dieta. Los animales pueden enjaularse por separado o en pequeños grupos del mismo sexo.
1.4.3. Preparación de los animales
Se eligen al azar animales jóvenes y sanos y se reparten en los lotes testigo y tratado. Las jaulas se colocan de manera que los posibles efectos debidos a la posición de las mismas sean mínimos. Los animales se identifican individualmente y se mantienen en sus jaulas durante al menos 5 días antes del inicio del ensayo, a fin de permitir su aclimatación a las condiciones del laboratorio.
1.4.4. Vía de administración y preparación de las dosis
En este método de ensayo se administra la sustancia estudiada por vía oral. La administración de la sustancia por vía oral puede hacerse mediante una sonda, la dieta, el agua o cápsulas. Pueden usarse otras vías de administración (por ejemplo: cutánea o inhalación) pero en este caso pueden tener que modificarse los procedimientos recomendados. La elección de la vía de administración depende del perfil de exposición humana y de la información cinética y toxicológica disponible. Deberá justificarse adecuadamente la elección de la vía de administración, así como las consiguientes modificaciones de los procedimientos del método de ensayo.
En caso necesario, la sustancia estudiada se disolverá o suspenderá en un vehículo adecuado. Se recomienda considerar en primer lugar el uso de una solución o suspensión acuosa, después el uso de una solución o emulsión oleosa (por ejemplo, en aceite de maíz) y, por último, la posible solución o suspensión en otros vehículos. Las características tóxicas del vehículo deben ser conocidas. Además, deben valorarse las siguientes características del vehículo: efectos en la absorción, la distribución, el metabolismo o la retención de la sustancia estudiada que puedan alterar sus características tóxicas; y efectos en el consumo de alimentos y agua o en el estado nutricional de los animales.
1.5. PROCEDIMIENTOS
1.5.1. Número y sexo de los animales
Cuando se trate de un estudio único independiente, se utilizarán al menos 20 animales (10 hembras y 10 machos) en cada grupo tratado y de control para la evaluación de las observaciones clínicas y funcionales, que deben ser detalladas. Como mínimo 5 machos y 5 hembras, seleccionados entre estos 10 machos y 10 hembras, deben ser perfundidos in situ y utilizados para hacer una histopatología detallada al final del estudio. En los casos en que solo se observe un número limitado de animales en un grupo determinado para detectar efectos neurotóxicos, se considerará la inclusión de estos animales entre los seleccionados para perfusión. Cuando el estudio se lleve a cabo en combinación con un estudio de toxicidad por administración continuada, deberá usarse un número adecuado de animales para que puedan alcanzarse los objetivos de ambos estudios. En el cuadro 1 se da el número mínimo de animales por grupo para varias combinaciones de estudios. Si van a sacrificarse animales durante el experimento o están previstos grupos para observar la reversibilidad, persistencia o aparición retardada de efectos tóxicos con posterioridad al tratamiento, o cuando se plantee la realización de observaciones complementarias, habrá de aumentarse el número de animales para asegurar que se dispone de los animales necesarios para observación e histopatología.
1.5.2. Lote tratado y lote testigo
Deben utilizarse, al menos, tres grupos para tratamiento y uno para control. No obstante, si, a partir de la evaluación de otros datos, no son previsibles efectos a una dosis continuada de 1 000 mg/kg de peso corporal, podrá realizarse un ensayo límite. Si no se dispone de datos apropiados, puede realizarse un estudio para ayudar a determinar la gama de dosis que debe emplearse. A excepción de la administración de la sustancia de ensayo, los animales del lote testigo deben ser tratados de la misma manera que los de los lotes de ensayo. Si se utiliza un vehículo para la administración de la sustancia de ensayo, el lote testigo recibirá el mayor volumen utilizado de dicho vehículo.
1.5.3. Control de fiabilidad
El laboratorio que lleve a cabo el estudio debe presentar datos que demuestren su capacidad de realizarlo y la sensibilidad de los procedimientos que emplee. Tales datos han de acreditar su capacidad de detectar y cuantificar, en su caso, los cambios en los diferentes parámetros cuya observación se recomienda, como signos neurovegetativos, reactividad sensorial, fuerza de prensión y actividad motriz. En la bibliografía (referencias 2 a 29) puede encontrarse información sobre sustancias que causan diferentes tipos de respuestas neurotóxicas y que pueden usarse para controles positivos. Pueden utilizarse datos de referencia anteriores si se mantienen los aspectos esenciales de los procedimientos experimentales. Se recomienda la actualización periódica de los datos de referencia anteriores. Deben obtenerse nuevos datos que demuestren que los procedimientos mantienen la sensibilidad requerida cuando el laboratorio haya modificado alguno de los aspectos esenciales del ensayo o los procedimientos.
1.5.4. Selección de la dosis
Las dosis deben seleccionarse teniendo en cuenta la toxicidad previamente observada y los datos cinéticos y de toxicidad disponibles referentes a la sustancia de ensayo o productos afines. La dosis más elevada debe seleccionarse con el propósito de inducir efectos neurotóxicos o efectos tóxicos sistémicos evidentes. Posteriormente, debe seleccionarse una secuencia descendente de dosis destinadas a demostrar cualquier posible relación dosis-respuesta y la ausencia de efectos adversos observados con la dosis inferior. En principio, las dosis deben fijarse de manera que los efectos tóxicos primarios en el sistema nervioso puedan distinguirse de los relacionados con la toxicidad sistémica. Los intervalos del doble al triple suelen ser óptimos para establecer las dosis decrecientes y a menudo es preferible añadir un cuarto lote de ensayo en lugar de utilizar intervalos muy amplios (por ejemplo, con un factor superior a 10) entre dosis. Si se dispone de una estimación razonable sobre la exposición del hombre, esta deberá también tenerse en cuenta.
1.5.5 Ensayo límite
Si en un estudio con una dosis de, al menos, 1 000 mg/kg de peso corporal/día, siguiendo el procedimiento descrito, no se produce ningún efecto neurotóxico observable y si, a la luz de los datos de sustancias estructuralmente afines, no cabe esperar efectos tóxicos, puede considerarse innecesario realizar un estudio completo con 3 dosis. Según el grado previsto de exposición humana, puede ser preciso administrar una dosis oral superior en el ensayo límite. Si se trata de otras vías de administración, como la inhalación o la aplicación cutánea, suelen ser las características fisicoquímicas de la sustancia de ensayo las que determinan el grado máximo de exposición que puede alcanzarse. Para la realización de un estudio de toxicidad oral aguda, la dosis de un ensayo límite debe ser, al menos, 2 000 mg/kg.
1.5.6. Administración de las dosis
Las dosis de sustancia se administran a los animales todos y cada uno de los días del período de 28; es necesario justificar la aplicación de una posología de solo 5 días por semana o un período de exposición más corto. Si la sustancia de ensayo se administra por sonda, debe hacerse en una sola dosis y con una sonda gástrica o una cánula de intubación adecuada. El volumen máximo de líquido que puede administrarse de una sola vez depende del tamaño del animal. El volumen no debe superar 1 ml/100 g de peso corporal; no obstante, en el caso de las soluciones acuosas puede considerarse la posibilidad de administrar 2 ml/100 g de peso corporal. Salvo en el caso de sustancias irritantes o corrosivas, que por lo general producen efectos exacerbados con concentraciones mayores, deberá reducirse al mínimo la variabilidad del volumen de ensayo ajustando la concentración de manera que el volumen sea el mismo en todas las dosis.
Si la sustancia se administra con los alimentos o el agua de bebida, es importante cerciorarse de que las cantidades de sustancia de ensayo administradas no interfieren la nutrición normal ni el equilibrio hídrico. Cuando la sustancia de ensayo se administre con los alimentos, puede utilizarse una concentración constante en la dieta (en ppm) o bien una dosis constante en relación con el peso corporal de los animales; debe precisarse el método escogido. Si la sustancia se administra por sonda, la dosis debe darse todos los días a la misma hora y ajustarse según sea necesario para mantener una dosis constante en relación con el peso corporal del animal. Cuando se utilice un estudio de administración continuada como fase previa de un estudio a largo plazo, deberá utilizarse en ambos estudios una dieta similar. Para estudios de toxicidad aguda, si no es posible utilizar una dosis única, podrá administrarse la sustancia en fracciones menores a lo largo de un período que no excederá de 24 horas.
1.6. OBSERVACIONES
1.6.1. Frecuencia de las observaciones y ensayos
En los estudios de administración continuada, el período de observación debe cubrir todo el período de administración. En los estudios de toxicidad aguda, debe haber un período de observación de 14 días posterior al tratamiento. En el caso de los animales de grupos satélite que se mantengan sin exposición durante el período posterior al tratamiento, las observaciones deben cubrir también este período.
Las observaciones deben hacerse con la frecuencia suficiente para que sea máxima la probabilidad de detección de cualquier anomalía neurológica o del comportamiento. Las observaciones deben hacerse preferiblemente a las mismas horas cada día prestando atención al período más agudo de los efectos previstos tras la administración. En el cuadro 2 se resume la frecuencia de las observaciones clínicas y las pruebas funcionales. Si, a partir de estudios anteriores, se dispone de datos cinéticos o de otro tipo que muestren la necesidad de utilizar tiempos diferentes para las observaciones, pruebas o períodos post-observación, se aplicará otro programa a fin de conseguir el máximo de información. Los cambios en el programa habrán de justificarse debidamente.
1.6.1.1. Observaciones del estado general de salud y de la mortalidad/morbilidad
Deberán observarse cuidadosamente todos los animales al menos una vez al día para comprobar su estado de salud y al menos dos veces al día para comprobar la mortalidad y la morbilidad.
1.6.1.2. Observaciones clínicas detalladas
Se someterán a observación clínica detallada todos los animales seleccionados con este fin (véase el cuadro 1) una vez antes de la primera exposición (para poder hacer comparaciones con un mismo sujeto) y después a diferentes intervalos según la duración del estudio (véase el cuadro 2). Deberán hacerse observaciones clínicas detalladas sobre lotes satélite de recuperación al final del período de recuperación. Las observaciones clínicas detalladas se harán fuera de la jaula de alojamiento en un ambiente normal y se registrarán cuidadosamente utilizando sistemas de puntuación que incluyan criterios o escalas de puntuación para cada medición de las observaciones. Los criterios o escalas deberán ser definidos explícitamente por el laboratorio que lleve a cabo el ensayo. Debe procurarse que las variaciones en las condiciones de ensayo sean mínimas (no relacionadas sistemáticamente con el tratamiento) y que las observaciones sean realizadas por observadores especializados ajenos al tratamiento.
Se recomienda que las observaciones se hagan de manera estructurada aplicando sistemáticamente criterios bien definidos (incluida la definición del «intervalo» normal) a cada animal en cada momento de observación. El «intervalo normal» deberá documentarse adecuadamente. Se registrarán todos los signos observados. Siempre que sea factible, se registrará también la magnitud de los signos observados. Las observaciones clínicas deben incluir, entre otras cosas, los cambios de la piel, pelo, ojos, membranas mucosas, presencia de secreciones y excreciones y actividad neurovegetativa (por ejemplo, lagrimeo, piloerección, tamaño de la pupila, así como respiración por la boca, cualquier signo de orina o defecación y orina descolorida).
También habrá de anotarse cualquier respuesta no habitual en lo que se refiere a la posición del cuerpo, el nivel de actividad (por ejemplo, aumento o disminución de la exploración del ambiente normal en el que se mueva) y la coordinación de los movimientos. Deben registrarse también los cambios observados en la marcha (por ejemplo, contoneo o ataxia), postura (por ejemplo, espalda encorvada) y respuesta a la manipulación, la colocación u otros estímulos ambientales, así como la presencia de movimientos clónicos o tónicos, convulsiones o temblores, estereotipos (por ejemplo, realización excesiva de movimientos de limpieza, movimientos anómalos de la cabeza, recorridos repetitivos en círculo) o comportamientos anómalos (por ejemplo, lamido excesivo o mordiscos, automutilación, marcha hacia atrás y vocalización) o agresión.
1.6.1.3. Pruebas funcionales
De manera semejante a las observaciones clínicas detalladas, deben efectuarse también pruebas funcionales una vez antes de la exposición y frecuentemente a continuación en todos los animales seleccionados con este fin (véase el cuadro 1). La frecuencia de estas pruebas depende también de la duración del estudio (véase el cuadro 2). Además de los períodos de observación establecidos en el cuadro 2, se llevarán a cabo observaciones funcionales de lotes satélite de recuperación lo más cercanas que sea posible al sacrificio final. Las pruebas funcionales deben incluir la reactividad sensorial frente a estímulos de diferentes tipos [por ejemplo, auditivos, visuales y propioceptivos (5) (6) (7)], y la evaluación de la fuerza de prensión (8) y de la actividad motriz (9). La actividad motriz se medirá con un dispositivo automático capaz de detectar tanto aumentos como disminuciones de actividad. Si se usa otro sistema definido, este deberá ser cuantitativo y su sensibilidad y fiabilidad deberán demostrarse. Se probará cada dispositivo para asegurar la fiabilidad a lo largo del tiempo y la concordancia entre diferentes dispositivos. En las referencias respectivas se da más información sobre los procedimientos que pueden seguirse. Si no hay datos (por ejemplo, relaciones estructura-actividad, datos epidemiológicos, otros estudios toxicológicos...) que indiquen los posibles efectos neurotóxicos, debe considerarse la inclusión de pruebas más especializadas de funciones sensoriales o motrices o del aprendizaje y la memoria, para estudiar estos posibles efectos más detalladamente. En (1) se da más información sobre pruebas más especializadas y su aplicación.
Excepcionalmente, puede omitirse la prueba a animales que muestren signos de toxicidad en un grado tal que interfiera de manera significativa la prueba funcional. La eliminación de animales de cualquier prueba funcional se justificará debidamente.
1.6.2. Peso corporal y consumo de alimento y agua
Para estudios de hasta 90 días de duración, tienen que pesarse todos los animales al menos una vez por semana y deben hacerse mediciones del consumo de alimentos (consumo de agua, cuando la sustancia estudiada se administre por este medio) al menos semanalmente. Para estudios a largo plazo, deben pesarse todos los animales al menos una vez por semana durante las primeras 13 semanas y al menos una vez cada 4 semanas a continuación. Se medirá el consumo de alimentos (consumo de agua, cuando la sustancia estudiada se administre por este medio) al menos semanalmente durante las primeras 13 semanas y luego a intervalos de unos 3 meses, a menos que el estado de salud o el peso corporal aconsejen otra cosa.
1.6.3. Oftalmología
Para estudios de más de 28 días de duración, debe realizarse una exploración oftalmológica con un oftalmoscopio o equipo equivalente adecuado antes de administrar la sustancia de ensayo y al término del estudio, preferiblemente, de todos los animales, y al menos del lote con la dosis más alta y del lote de control. Si se observan cambios oculares o si los signos clínicos lo exigen, deben examinarse todos los demás animales. Para estudios a largo plazo, debe efectuarse una exploración oftalmológica a las 13 semanas. Las exploraciones oftalmológicas no serán necesarias si se conocen ya estos datos a partir de otros estudios de duración parecida y a dosis semejantes.
1.6.4. Hematología y bioquímica clínica
Cuando el estudio de neurotoxicidad se haga en combinación con un estudio de toxicidad sistémica por administración continuada, se practicarán exámenes hematológicos y determinaciones bioquímicas clínicas según lo indicado en el respectivo método del estudio de toxicidad sistémica. La recogida de muestras debe hacerse de tal manera que se reduzcan al mínimo los posibles efectos en el neurocomportamiento.
1.6.5. Histopatología
Los análisis neuropatológicos tienen que estar pensados de manera que complementen y amplíen las observaciones efectuadas durante la fase in vivo del estudio. Se fijarán in situ tejidos de, al menos, 5 animales por sexo y por grupo (véase el cuadro 1 y el parágrafo siguiente), utilizando técnicas de fijación y perfusión ampliamente reconocidas (véase la referencia 3 del capítulo 5 y la referencia 4 del capítulo 50). Deben registrarse todas las modificaciones macroscópicas observables. Cuando se trate de un estudio único independiente destinado a detectar neurotoxicidad o a caracterizar efectos neurotóxicos, el resto de los animales puede utilizarse o bien para otros procedimientos concretos de tipo electrofisiológico (10) (11) (16) (17), neuroquímico (10) (11) (14) (15), neuropatológico (10) (11) (12) (13) o neurocomportamental (10) (11) que puedan complementar los procedimientos y análisis descritos aquí o bien para aumentar el número de sujetos examinados para detectar histopatologías. Estos procedimientos complementarios pueden ser especialmente útiles cuando las observaciones empíricas o los efectos previstos indiquen un tipo determinado o una diana de neurotoxicidad (2) (3). Otra posibilidad es utilizar el resto de los animales para las evaluaciones patólogicas habituales descritas en el método sobre estudios de toxicidad por administración continuada.
Se aplicará un procedimiento general de tinción, como hematoxilina-eosina, en todas las muestras de tejido incluidas en parafina y se hará un examen microscópico. Cuando se observen o se sospechen signos de neuropatía periférica, deberán examinarse muestras incluidas en plástico de tejido de nervios periféricos. Según los signos clínicos que aparezcan podrán examinarse otros tejidos o utilizarse procedimientos de tinción especiales. En (3) y (4) se dan orientaciones sobre otros tejidos que pueden examinarse. Pueden ser útiles también (18) otros colorantes especiales para demostrar determinados tipos de alteraciones patológicas.
Se someterán a examen histológico secciones representativas de los sistemas nerviosos central y periférico (véase la referencia 3 del capítulo 5 y la referencia 4 del capítulo 50). Las zonas examinadas tienen que incluir normalmente: el prosencéfalo, el centro del cerebro, incluida una sección del hipocampo, el mesencéfalo, el cerebelo, la protuberancia anular, el bulbo raquídeo, el ojo con el nervio óptico y la retina, la médula espinal en los engrosamientos cervical y lumbar, los ganglios de la raíz dorsal, las fibras de la raíz ventral y dorsal, el nervio ciático en su parte próxima (en la rodilla) y el nervio tibial en sus ramificaciones del músculo de la pantorrilla. Las secciones de la médula espinal y de los nervios periféricos tienen que incluir tanto una sección transversal como una longitudinal. Debe prestarse atención a la vasculatura del sistema nervioso. Se examinará también una muestra de músculo esquelético, especialmente del de la pantorrilla. Hay que prestar especial atención a las zonas de los sistemas nerviosos central y periférico con estructura celular y fibrosa que se sabe que resultan particularmente afectadas por neurotóxicos.
En las referencias (3) y (4) se dan orientaciones sobre las alteraciones neuropatológicas que derivan normalmente de la exposición a sustancias tóxicas. Se recomienda un examen por fases de las muestras de tejido empezando por una comparación de las secciones del lote de dosis alta con las del lote testigo. Si no se observan alteraciones neuropatológicas en las muestras de estos lotes, no se requieren más análisis. Si se observan alteraciones neuropatológicas en el lote de dosis alta, deben codificarse y examinarse secuencialmente muestras de cada uno de los tejidos posiblemente afectados pertenecientes a los lotes de dosis intermedia y baja.
Si en el examen cualitativo se encuentran pruebas de alteraciones neuropatológicas, se realizará otro examen en todas las regiones del sistema nervioso que muestren estas alteraciones. Deberán codificarse y examinarse al azar, sin conocimiento del código, secciones de todos los lotes de cada una de las regiones posiblemente afectadas. Debe registrarse la frecuencia y gravedad de cada lesión. Después de que todos los lotes hayan sido clasificados, puede revelarse el código y entonces pueden hacerse análisis estadísticos para evaluar relaciones dosis-respuesta. Deberán darse ejemplos que muestren diferentes niveles de gravedad de cada lesión.
Los resultados neuropatológicos tienen que evaluarse en el contexto de observaciones y mediciones del comportamiento, así como en relación con otros datos de estudios de toxicidad sistémica de la sustancia anteriores y realizados al mismo tiempo.
2. RESULTADOS
2.1. TRATAMIENTO DE LOS RESULTADOS
Deben proporcionarse datos de cada animal. Además, tienen que resumirse todos los datos en un cuadro que recoja, para cada lote de ensayo o testigo, el número de animales al inicio del ensayo, el número de animales hallados muertos durante el mismo o sacrificados por razones compasivas, el momento de la muerte o sacrificio, el número de animales que presenten signos de toxicidad, una descripción de los signos observados, con inclusión del momento de su aparición, la duración y la gravedad de los efectos tóxicos, el número de animales que presenten lesiones, y el tipo y la gravedad de las lesiones.
2.2. EVALUACIÓN E INTERPRETACIÓN DE LOS RESULTADOS
Los resultados del estudio deben evaluarse en lo que se refiere a la incidencia, gravedad y correlación de los efectos neurocomportamentales y neuropatológicos (así como a los efectos electrofisiológicos y neuroquímicos si se incluyen exámenes complementarios) y a cualquier otro efecto adverso observado. Siempre que sea posible, los resultados numéricos deberán evaluarse mediante un método estadístico adecuado y de amplia aceptación. Los métodos estadísticos deben seleccionarse durante el diseño del estudio.
3. INFORME
3.1. INFORME DEL ENSAYO
El informe del ensayo deberá incluir la información siguiente:
|
|
Sustancia estudiada:
|
|
|
Vehículo (si procede):
|
|
|
Animales sometidos a ensayo:
|
|
|
Condiciones de ensayo:
|
|
|
Procedimientos de observación y prueba:
|
|
|
Resultados:
|
|
|
Discusión de los resultados
|
|
|
Conclusiones
|
4. REFERENCIAS
|
1. |
OEDC Guidance Document on Neurotoxicity Testing Strategies and Test Methods. OCDE, Paris. In preparation. |
|
2. |
Test Guideline for a Developmental Neurotoxicity Study, OECD Guidelines for the Testing of Chemicals. In preparation. |
|
3. |
World Health Organization (WHO) (1986). Environmental Health Criteria document 60: Principies and Methods for the Assessment of Neurotoxicity associated with Exposure to Chemicals. |
|
4. |
Spencer, P.S. and Schaumburg, H.H. (1980). Experimental and Clinical Neurotoxicology. Eds. Spencer, P.S. and Schaumburg, H.H. eds. Williams and Wilkins, Baltimore/London. |
|
5. |
Tupper, D.E. and Wallace, R.B. (1980). Utility of the Neurological Examination in Rats. Acta Neurobiol. Exp., 40, 999-1003. |
|
6. |
Gad, S.C. (1982). A Neuromuscular Screen for Use in Industrial Toxicology. J. Toxicol. Environ. Health, 9,691-704. |
|
7. |
Moser, V.C., McDaniel, K.M. and Phillips, P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of amitraz. Toxic. Appl. Pharmacol., 108, 267-283. |
|
8. |
Meyer, O.A., Tilson, H.A., Byrd, W.C. and Riley, M.T. (1979). A Method for the Routine Assessment of Fore- and Hind- limb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233-236. |
|
9. |
Crofton, K.M., Haward, J.L., Moser, V.C., Gill, M.W., Reirer, L.W., Tilson, H.A. and MacPhail, R.C. (1991) Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol., 13, 599-609. |
|
10. |
Tilson, H.A., and Mitchell, C.L. eds. (1992). Neurotoxicology Target Organ Toxicology Series. Raven Press, New York. |
|
11. |
Chang, L.W., ed. (1995). Principies of Neurotoxicology. Marcel Dekker, New York. |
|
12. |
Broxup, B. (1991). Neuopathology as a screen for Neurotoxicity Assessment. J. Amer. Coll. Toxicol., 10, 689-695. |
|
13. |
Moser, V.C., Anthony, D.C., Sette, W.F. and MacPhail, R.C. (1992). Comparison of Subchronic Neurotoxicity of 2-Hydroxyethyl Acrylate and Acrylamide in Rats. Fund. Appl.ToxicoL, 18, 343-352. |
|
14. |
O'Callaghan, J.P. (1988). Neurotypic and Gliotypic Proteins as Biochemical Markers of Neurotoxicity. Eurotoxicol. Teratol., 10, 445-452. |
|
15. |
O'Callaghan J.P. and Miller, D.B. (1988). Acute Exposure of the Neonatal Rat to Triethyltin Results in Persistent Changes in Neurotypic and Gliotypic Proteins. J. Pharmacol. Exp. Ther., 244, 368-378. |
|
16. |
Fox. D.A., Lowndes, H.E. and Birkamper, G.G. (1982). Electrophysiological Techniques in Neurotoxicology. In: Nervous System Toxicology. Mitchell, C.L. ed. Raven Press, New York, pp 299-335. |
|
17. |
Johnson, B.L. (1980). Electrophysiological Methods in neurotoxicity Testing. In: Experimental and Clinical Neurotoxicology. Spencer, P.S. and Schaumburg, H.H. eds., Williams and Wilkins Co., Baltimore/London, pp. 726-742. |
|
18. |
Bancroft, J.D. and Steven A. (1990). Theory and Pratice of Histological Techniques. Chapter 17, europathological Techniques. Lowe, James and Cox, Gordon eds. Churchill Livingstone. |
Cuadro 1
Número mínimo de animales por grupo cuando el estudio de neurotoxicidad se realiza por separado o en combinación con otros estudios
|
|
ESTUDIO DE NEUROTOXICIDAD |
|||
|
Como estudio aparte |
Combinado con el estudio de 28 días |
Combinado con el estudio de 90 días |
Combinado con el estudio de toxicidad crónica |
|
|
Número total de animales por lote |
10 machos y 10 hembras |
10 machos y 10 hembras |
15 machos y 15 hembras |
25 machos y 25 hembras |
|
Número de animales seleccionados para pruebas funcionales incluidas observaciones clínicas detalladas |
10 machos y 10 hembras |
10 machos y 10 hembras |
10 machos y 10 hembras |
10 machos y 10 hembras |
|
Número de animales seleccionados para perfusión in situ y neurohistopatología |
5 machos y 5 hembras |
5 machos y 5 hembras |
5 machos y 5 hembras |
5 machos y 5 hembras |
|
Número de animales seleccionados para observaciones de toxicidad por administración continuada/subcrónica/crónica, hematología, bioquímica clínica, histopatología, etc., según lo indicado en las respectivas Orientaciones. |
|
5 machos y 5 hembras |
10 machos (10) y 10 hembras (10) |
20 machos (10) y 20 hembras (10) |
|
Observaciones complementarias, según corresponda |
5 machos y 5 hembras |
|
|
|
Cuadro 2
Frecuencia de las observaciones y pruebas funcionales
|
Tipo de observaciones |
Duración del estudio |
||||||||||||||||||||||||||
|
T. aguda |
28 días |
90 días |
T. crónica |
||||||||||||||||||||||||
|
En todos los animales |
Estado general de salud |
diariamente |
diariamente |
diariamente |
diariamente |
||||||||||||||||||||||
|
Mortalidad/morbilidad |
dos veces al día |
dos veces al día |
dos veces al día |
dos veces al día |
|||||||||||||||||||||||
|
En animales seleccionados para observaciones funcionales |
Observaciones clínicas detalladas |
|
|
|
|
||||||||||||||||||||||
|
Pruebas funcionales |
|
|
|
|
B.44. ABSORCIÓN CUTÁNEA: MÉTODO IN VIVO
1. MÉTODO
El presente método de ensayo es equivalente a las directrices de ensayo de la OCDE TG 427 (2004).
1.1. INTRODUCCIÓN
La exposición a muchas sustancias se produce principalmente a través de la piel, pero en la mayoría de los estudios toxicológicos efectuados con animales de laboratorio se utiliza la administración por vía oral. El estudio de absorción cutánea in vivo que se recoge en las presentes directrices permite la extrapolación a partir de los estudios orales cuando se efectúa la evaluación de la seguridad tras una exposición dérmica.
Antes de llegar a la circulación, las sustancias deben atravesar muchas capas de células de la piel. Para la mayoría de las sustancias, la capa que limita la tasa de esa incorporación es la capa córnea, formada por células muertas. La permeabilidad a través de la piel depende no solo de la lipofilia de la sustancia y del espesor de la capa exterior de la epidermis, sino también de otros factores, como el peso molecular y la concentración de la sustancia. En general, la piel de ratas y conejos es más permeable que la humana, mientras que la permeabilidad cutánea de cobayas y monos es más parecida a la nuestra.
Los métodos para medir la absorción cutánea pueden clasificarse en dos categorías: in vivo e in vitro. Los métodos in vivo pueden proporcionar buena información sobre la absorción cutánea de varias especies de laboratorio. Más recientemente se han elaborado métodos in vitro. Estos utilizan el transporte a través de piel humana o animal, con un espesor bien íntegro o bien parcial, hasta un depósito de líquidos. En otro método aparte (1) se describe el ensayo in vitro. Se recomienda consultar el documento orientativo de la OCDE sobre realización de estudios de absorción cutánea (2) para seleccionar el método más adecuado en cada situación, ya que en él se da más información sobre la idoneidad de los métodos tanto in vivo como in vitro.
El método in vivo, descrito en el presente texto, permite la determinación de la penetración de la sustancia problema a través de la piel hasta el compartimento sistémico. La técnica se utiliza ampliamente desde hace muchos años (3) (4) (5) (6) (7). Aunque en muchos casos pueden ser adecuados los estudios in vitro de absorción a través de la piel, es posible que haya situaciones en las que solo se puedan obtener los datos necesarios mediante estudios in vivo.
Las ventajas del método in vivo radican en que utiliza un sistema fisiológica y metabólicamente intacto, recurre a una especie común a muchos estudios de toxicidad y puede modificarse para utilizarlo con otras especies. Sus desventajas consisten en el uso de animales vivos, en la necesidad de utilizar sustancias radiomarcadas para conseguir unos resultados fiables, en las dificultades para determinar la fase de absorción precoz y en las diferencias de permeabilidad de la especie preferida (rata) respecto a la piel humana. La piel animal suele ser más permeable y, por tanto, puede llevar a una sobreestimación de la absorción a través de la piel humana (6) (8) (9). No deben ensayarse con animales vivos las sustancias cáusticas o corrosivas.
1.2. DEFINICIONES
Se entenderá por:
Dosis no absorbida: la dosis lavada de la superficie cutánea tras la exposición y la presente en las protecciones no oclusivas, incluida la que se haya visto que se volatiliza de la piel durante la exposición.
Dosis absorbida (in vivo): la dosis presente en la orina, líquidos de lavado de la jaula, heces, aire espirado (si se mide), sangre, tejidos (si se recogen) y el resto del cuerpo, tras la eliminación de la piel correspondiente al lugar de aplicación.
Dosis absorbible: la dosis presente en la superficie o en el interior de la piel tras el lavado.
1.3. PRINCIPIO DEL MÉTODO DE ENSAYO
La sustancia problema, preferentemente radiomarcada, se aplica a la piel esquilada de los animales, a una o más dosis apropiadas, en forma de preparado representativo en uso. Se deja el preparado problema en contacto con la piel durante un plazo fijo bajo una protección adecuada (no oclusiva, semioclusiva u oclusiva) para evitar la ingestión del preparado. Al final del plazo de exposición, se quita la protección y se limpia la piel con un agente adecuado (tanto la protección como el material de lavado se conservan para su análisis), y se aplica otra protección. Los animales están alojados antes, durante y después del plazo de exposición en jaulas de metabolismo individuales y las excretas y el aire espirado en estos periodos se recogen para su análisis. Puede omitirse la recogida del aire espirado si se dispone de información suficiente sobre la ausencia o poca importancia de la formación de metabolitos radiactivos volátiles. En cada estudio participan normalmente varios grupos de animales que se exponen al preparado problema. Un grupo se sacrifica al final del período de exposición. Otros grupos se sacrifican posteriormente a intervalos programados (2). Al final del tiempo de muestreo se sacrifican los animales restantes, se recoge su sangre para analizarla, se extrae la piel de los lugares de aplicación para su análisis y se analiza también el resto del cuerpo para buscar las posibles sustancias no excretadas. Se efectúa la determinación de las muestras con los medios adecuados y se calcula el grado de absorción a través de la piel (6) (8) (9).
1.4. DESCRIPCIÓN DEL MÉTODO
1.4.1. Selección de la especie animal
La rata es la especie más utilizada, pero también pueden emplearse cepas y especies sin pelo que presenten unas tasas de absorción cutánea más parecidas a las del hombre (3) (6) (7) (8) (9). Deben emplearse animales adultos jóvenes, con buena salud, de un solo sexo (en principio, machos) de cepas de laboratorio utilizadas normalmente. Al empezar el estudio, la variación del peso de los animales utilizados no debe superar el ± 20 % del peso medio. Por ejemplo, son adecuadas las ratas machos de 200 g–250 g, especialmente de la mitad superior de esta banda.
1.4.2. Número y sexo de los animales
Para cada preparado problema y cada tiempo programado de sacrificio debe utilizarse un grupo de al menos 4 animales del mismo sexo. Cada grupo de animales se sacrificará tras un plazo diferente, por ejemplo al final del plazo de exposición (normalmente 6 o 24 horas) y en momentos posteriores (por ejemplo, 48 y 72 horas). Si se dispone de datos que demuestren la existencia de diferencias importantes en cuanto a la toxicidad dérmica entre machos y hembras, se elegirá el sexo más sensible. En ausencia de tales datos, puede utilizarse cualquiera de los sexos.
1.4.3. Alojamiento y alimentación
El cuarto de animales de experimentación ha de estar a una temperatura de 22 oC (± 3 oC). Aunque la humedad relativa ha de ser, como mínimo, del 30 % y preferentemente no superar el 70 %, salvo durante la limpieza del local, lo ideal es que esté comprendida entre el 50 y el 60 %. Se aplicará iluminación artificial en una secuencia de 12 horas de luz y 12 horas de oscuridad. Puede proporcionarse una dieta alimentaria corriente para animales de laboratorio; la cantidad no debe estar limitada, y tampoco el suministro de agua potable. Durante el estudio y, de preferencia, también durante la fase de aclimatación, los animales se alojarán en jaulas de metabolismo individuales. Como el vertido de agua y piensos puede afectar a los resultados, debe reducirse al mínimo la probabilidad de que ocurra.
1.4.4. Preparación de los animales
Los animales se marcan para identificarlos individualmente y se mantienen en sus jaulas durante al menos 5 días antes del inicio del estudio, a fin de permitir su aclimatación a las condiciones del laboratorio.
Tras la fase de aclimatación, y unas 24 horas antes de la administración de la sustancia, se esquila una zona de piel de los hombros y del dorso de cada animal. Las propiedades de permeabilidad de la piel lesionada son diferentes de las de la piel intacta, por lo que debe procurarse no excoriar la piel. Una vez efectuado el esquileo y unas 24 horas antes de la aplicación de la sustancia problema a la piel (véase el punto 1.4.7), la superficie cutánea debe limpiarse con acetona para eliminar el sebo cutáneo. No se recomienda limpiar con agua y jabón porque un eventual resto de jabón podría aumentar la absorción de la sustancia problema. La superficie debe ser lo bastante grande para permitir un cálculo fiable de la cantidad absorbida de sustancia problema por centímetro cuadrado de piel, por lo que, de preferencia, ha de tener un mínimo de 10 cm2. Esta superficie puede conseguirse con ratas de 200-250 g de peso corporal. Tras esta preparación, los animales se devuelven a las jaulas de metabolismo.
1.4.5. Sustancia problema
La sustancia problema es aquella cuyas características de penetración se van a estudiar. Lo mejor es que la sustancia problema esté radiomarcada.
1.4.6. Preparación de las pruebas
El preparado de la sustancia problema (por ejemplo, material puro, diluido o formulado que contiene la sustancia problema y que se aplica a la piel) debe ser el mismo (o un análogo realista) que aquel al que pueda verse expuesto el hombre u otra posible especie diana. Deben justificarse las eventuales desviaciones respecto al preparado «en uso». En caso necesario, la sustancia problema se disuelve o suspende en un vehículo adecuado. En caso de que el vehículo sea distinto al agua, deben conocerse sus características de absorción y su posible interacción con la sustancia problema.
1.4.7. Aplicación a la piel
Se delimita en la piel el lugar de aplicación, con una superficie específica. Después se aplica de forma homogénea en ese lugar una cantidad conocida del preparado problema. Esta cantidad debe reflejar en principio la posible exposición humana, normalmente de 1 a 5 mg/cm2 en caso de preparado sólido o hasta 10 μl/cm2 en caso de líquidos. Si se emplean otras cantidades, hay que justificarlo en función de las condiciones previstas de uso, de los objetivos del estudio o de las características físicas del preparado problema. Tras la aplicación, hay que proteger el sitio tratado para que el animal no se lo frote. En la figura 1 se muestra un ejemplo de dispositivo clásico. Normalmente, el lugar de aplicación ha de llevar una protección no oclusiva (por ejemplo, una gasa de nailon permeable). Sin embargo, si la dosis es muy grande hay que ocluir el lugar de aplicación. En caso de que la evaporación de sustancias problema semivolátiles reduzca la tasa de recuperación de la sustancia problema hasta extremos inaceptables (véase también el párrafo primero del punto 1.4.10), será necesario recoger la sustancia evaporada en un filtro de carbón activo que recubra el dispositivo de aplicación (véase la figura 1). Es importante que los eventuales dispositivos no lesionen la piel, ni absorban el preparado problema ni reaccionen con él. Los animales se devuelven a sus jaulas de metabolismo individuales para recoger las excretas.
1.4.8. Duración de la exposición y toma de muestras
La duración de la exposición es el plazo entre la aplicación y la eliminación del preparado problema mediante lavado de la piel. El plazo de exposición debe tener una duración apropiada (normalmente 6 o 24 horas), según la duración prevista de la exposición humana. Tras la exposición, los animales se mantienen en las jaulas de metabolismo hasta el momento previsto para su sacrificio. De forma periódica a lo largo de toda la duración del estudio, hay que observar si los animales presentan signos de toxicidad o reacciones anormales. Al final de la exposición, hay que ver si la piel tratada presenta signos visibles de irritación.
Las jaulas de metabolismo deben permitir la recogida aparte de la orina y las heces a lo largo de todo el estudio. También deben permitir la recogida de dióxido de carbono 14C y de compuestos volátiles de carbono 14C, que deben analizarse si se producen en gran cantidad (> 5 %). La orina, las heces y los fluidos recogidos (por ejemplo, dióxido de carbono 14C y compuestos volátiles de carbono 14C) deben tomarse por separado de cada grupo y en cada momento de muestreo. Si se sabe con suficiente seguridad que la formación de metabolitos radiactivos volátiles es nula o muy limitada, pueden utilizarse jaulas abiertas.
Las excretas se recogen durante la exposición, hasta 24 horas tras el contacto inicial con la piel, y después cada día hasta el final del experimento. Aunque en principio es suficiente con tres intervalos de recogida de excretas, la finalidad prevista del preparado problema o los eventuales datos cinéticos pueden indicar la necesidad de utilizar intervalos adicionales o más adecuados para el estudio.
Al final de la exposición se retira el dispositivo protector de cada animal, y se conserva aparte para su análisis. La piel tratada de todos los animales debe lavarse al menos tres veces con un agente de limpieza, utilizando hisopos adecuados. Debe procurarse no contaminar otras partes del cuerpo. El agente de limpieza debe ser representativo de la práctica higiénica normal; por ejemplo, puede consistir en una solución acuosa de jabón. Por último, hay que secar la piel. Todos los hisopos y líquidos de lavado se guardan para su análisis. Antes de devolver los animales a sus jaulas individuales, se les aplica una protección nueva en el lugar del tratamiento, en caso de que formen parte de grupos que se vayan a estudiar posteriormente.
1.4.9. Procedimiento final
Los distintos animales de cada grupo se sacrificarán en el momento previsto y se recogerá su sangre para analizarla. El apósito o dispositivo protector se recogerá para su análisis. De cada animal se tomará la piel del lugar de aplicación y una superficie similar de piel esquilada que no haya recibido la sustancia, y se analizarán por separado. El lugar de aplicación podrá fraccionarse para separar la capa córnea de la epidermis subyacente, a fin de obtener más información sobre la distribución de la sustancia. La determinación de esta distribución a lo largo del tiempo tras la exposición debe indicar el destino de cualquier sustancia problema en la capa córnea. Para facilitar el fraccionamiento de la piel (tras el lavado final de esta y el sacrificio del animal), se retirarán todos los dispositivos protectores. Se extirpará de la rata la piel del lugar de aplicación, con una banda anular de piel adyacente, y se sujetará en un tablero con alfileres. Con una presión suave se aplicará a la superficie de la piel un trozo de cinta adhesiva, que se retirará junto con parte de la capa córnea. Se aplicarán trozos sucesivos de cinta hasta que esta deje de adherirse a la superficie de la piel, momento en que se habrá retirado la totalidad de la capa córnea. Todos los trozos de cinta procedentes de un mismo animal podrán reunirse en un solo envase al que se añadirá un agente para digerir tejidos, a fin de solubilizar la capa córnea. Los posibles tejidos diana se podrán recoger para someterlos a mediciones aparte antes de proceder a la determinación de la dosis absorbida en el cuerpo restante. Se conservarán los cuerpos de los distintos animales para su análisis. Normalmente será suficiente realizar el análisis del contenido total. Los órganos diana se podrán recoger para someterlos a análisis aparte (si así lo indican otros estudios). La orina presente en la vejiga en el momento del sacrificio programado se añadirá a la orina recogida previamente. Tras la recogida de las excretas de las jaulas de metabolismo en el momento del sacrificio programado, las jaulas y los colectores se lavarán con un disolvente adecuado. El resto del material que pueda estar contaminado se analizará análogamente.
1.4.10. Análisis
En todos los estudios debe conseguirse una recuperación adecuada (es decir, una media del 100 ± 10 % de la radiactividad). Deberán justificarse los casos en que la recuperación no esté en dicha banda. La cantidad de la dosis administrada presente en cada muestra se analizará mediante métodos validados adecuadamente.
El estudio estadístico debe incluir una medida de la varianza entre las réplicas de cada aplicación.
2. DATOS
Deben hacerse las siguientes mediciones con cada animal, en cada tiempo de muestreo, respecto a la sustancia problema y sus metabolitos (además de los datos individuales, deben comunicarse en forma de media los datos agrupados por tiempos de muestreo):
|
— |
cantidad relativa a los dispositivos protectores, |
|
— |
cantidad que puede extraerse de la piel, |
|
— |
cantidad en el interior o en la superficie de la piel que no puede retirarse mediante lavado, |
|
— |
cantidad en la sangre de la muestra, |
|
— |
cantidad en las excretas y en el aire espirado (en su caso), |
|
— |
cantidad restante en el cuerpo y en los eventuales órganos tomados para su análisis aparte. |
La cantidad de sustancia problema y sus metabolitos presente en las excretas, aire espirado, sangre y cuerpo permitirá la determinación de la cantidad total absorbida en cada intervalo. También puede calcularse la cantidad de sustancia problema absorbida por centímetro cuadrado de piel expuesta a la sustancia problema a lo largo del tiempo de exposición.
3. INFORMES
3.1. INFORME DEL ENSAYO
El informe del ensayo debe incluir los requisitos establecidos en el protocolo, con una justificación del sistema de ensayo utilizado y con los datos siguientes:
|
|
Sustancia de ensayo:
|
|
|
Preparado problema:
|
|
|
Animales de ensayo:
|
|
|
Condiciones de ensayo:
|
|
|
Resultados:
|
|
|
Discusión de los resultados. |
|
|
Conclusiones. |
4. REFERENCIAS
|
1. |
Método de ensayo B.45. Absorción cutánea: método in vitro. |
|
2. |
OECD (2002). Guidance Document for the Conduct of Skin Absorption Studies. OECD, Paris. |
|
3. |
ECETOC (1993) Percutaneous Absorption. European Centre for Ecotoxicology and Toxicology of Chemicals, Monograph No. 20. |
|
4. |
Zendzian RP (1989) Skin Penetration Method suggested for Environmental Protection Agency Requirements. J. Am. Coll. Toxicol. 8(5), 829-835. |
|
5. |
Kemppainen BW, Reifenrath WG (1990) Methods for skin absorption. CRC Press Boca Raton, FL, USA. |
|
6. |
EPA (1992) Dermal Exposure Assessment: Principles and Applications. Exposure Assessment Group, Office of Health and Environmental Assessment. |
|
7. |
EPA (1998) Health Effects Test Guidelines, OPPTS 870-7600, Dermal Penetration. Office of Prevention, Pedsticides and Toxic Substances. |
|
8. |
Bronaugh RL, Wester RC, Bucks D, Maibach HI and Sarason R (1990) In vivo percutaneous absorption of fragrance ingredients in reshus monkeys and humans. Fd. Chem. Toxic. 28, 369-373. |
|
9. |
Feldman RJ and Maibach HI (1970) Absorption of some organic compounds through the skin in man. J. Invest Dermatol. 54, 399-404. |
Figura 1
Ejemplo de diseño de dispositivo clásico utilizado para definir y proteger el lugar de aplicación dérmica en los estudios in vivo de absorción a través de la piel

B.45. ABSORCIÓN CUTÁNEA: MÉTODO IN VITRO
1. MÉTODO
El presente método de ensayo es equivalente a las directrices de ensayo de la OCDE TG 428 (2004).
1.1. INTRODUCCIÓN
El objetivo del presente método es proporcionar información sobre la absorción de una sustancia problema aplicada a la piel extirpada. Puede combinarse con el método in vivo del ensayo de absorción cutánea (1) o realizarse independientemente. Se recomienda consultar el documento orientativo de la OCDE sobre realización de estudios de absorción cutánea (2), que puede servir de ayuda para la concepción de estudios basados en este método de ensayo. El documento orientativo se ha preparado para facilitar la selección de los procedimientos in vitro adecuados según las distintas circunstancias, a fin de garantizar la fiabilidad de los resultados obtenidos con este método.
Los métodos para medir la absorción cutánea y la transferencia dérmica pueden clasificarse en dos categorías: in vivo e in vitro. Los métodos in vivo están bien comprobados y proporcionan información farmacocinética con una serie de especies animales. En otro método aparte (1) se describe un ensayo in vivo. Se lleva muchos años utilizando también métodos in vitro para medir la absorción cutánea. Aunque no se han efectuado estudios oficiales de validación de los ensayos in vitro correspondientes al presente método de ensayo, los expertos de la OCDE consideraron en 1999 que se disponía de suficientes datos evaluados para apoyar el método in vitro (3). En el documento orientativo (2) se incluye más información que avala este apoyo, con inclusión de un número significativo de comparaciones directas de métodos in vitro e in vivo. Hay una serie de monografías que examinan este aspecto y proporcionan datos pormenorizados sobre el uso de un método in vitro (4) (5) (6) (7) (8) (9) (10) (11) (12). Los métodos in vitro miden la difusión de sustancias a la piel y, a través de esta, hasta un depósito de líquidos, y pueden utilizar piel muerta para medir solo la difusión, o piel reciente, activa metabólicamente, para medir simultáneamente la difusión y el metabolismo cutáneo. Tales métodos se utilizan particularmente en el cribado para comparar la transferencia de sustancias a la piel y a través de esta desde distintas formulaciones, y también pueden proporcionar modelos útiles para la evaluación de la absorción a través de la piel humana.
Es posible que el método in vitro no se pueda aplicar a todas las situaciones ni a todas las clases de sustancias. Quizá se pueda utilizar el método de ensayo in vitro para una evaluación cualitativa inicial de la penetración cutánea. En algunos casos puede ser necesario completarla con datos in vivo. Debe consultarse el documento orientativo (2) para considerar las situaciones en que pueda ser adecuado el método in vitro. En (3) se encuentra más información detallada para avalar la decisión.
El presente método presenta los principios generales para medir la absorción cutánea y la transferencia de una sustancia problema utilizando piel extirpada. Puede utilizarse piel procedente de muchas especies de mamíferos, incluido el hombre. Las propiedades de permeabilidad de la piel se mantienen tras su extirpación del cuerpo porque la principal barrera contra la difusión es la capa córnea, formada por células muertas, ya que no se ha encontrado transporte activo de sustancias a través de la piel. Se ha visto que la piel es capaz de metabolizar ciertas sustancias durante la absorción a través de ella (6), pero este proceso no es limitante en cuanto a la tasa de la dosis absorbida realmente, aunque puede afectar a la naturaleza de la sustancia que llega a la corriente sanguínea.
1.2. DEFINICIONES
Se entenderá por:
dosis no absorbida la dosis lavada de la superficie cutánea tras la exposición y la presente en las protecciones no oclusivas, además de la que se haya visto que se volatiliza de la piel durante la exposición;
dosis absorbida (in vitro) la masa de sustancia problema que llega al líquido receptor o a la circulación sistémica dentro de un plazo especificado;
dosis absorbible (in vitro) la dosis presente en la superficie o en el interior de la piel tras el lavado.
1.3. PRINCIPIO DEL MÉTODO DE ENSAYO
La sustancia problema, que puede estar radiomarcada, se aplica a la superficie de una muestra de piel que separa las dos cámaras de una célula de difusión. La sustancia permanece sobre la piel durante un tiempo dado, en condiciones determinadas, antes de eliminarse mediante un procedimiento adecuado de limpieza. Se toman muestras del líquido receptor a ciertos intervalos a lo largo del experimento y se determina su contenido en sustancia problema y metabolitos de esta.
Cuando se utilizan sistemas metabólicamente activos, pueden analizarse con métodos adecuados los metabolitos de la sustancia problema. Al final del experimento se cuantifica la distribución de la sustancia problema y sus metabolitos, en su caso.
Aplicando las condiciones adecuadas, descritas en este método y en el documento orientativo (2), se mide la absorción de la sustancia problema durante un plazo determinado mediante el análisis del líquido receptor y de la piel tratada. La sustancia problema que permanece en la piel debe considerarse absorbida, salvo que se demuestre que la absorción se puede determinar a partir de solo los valores del líquido receptor. El análisis de los otros componentes (material extraído de la piel por lavado y material restante dentro de las capas de la piel) permite una evaluación de los datos más avanzada, con inclusión de la distribución de toda la sustancia problema y el porcentaje de recuperación.
Para demostrar la eficacia y fiabilidad del sistema de ensayo en el laboratorio que lo aplique, debe disponerse de los resultados correspondientes a sustancias de referencia apropiadas, los cuales deben concordar con los publicados en la bibliografía en relación con el método utilizado. Este requisito puede cumplirse sometiendo a ensayo una sustancia de referencia apropiada (de preferencia con una lipofilia similar a la de la sustancia problema) en paralelo con esta, o bien presentando datos previos adecuados sobre una serie de sustancias de referencia de lipofilia diferente (por ejemplo, cafeína, ácido benzoico y testosterona).
1.4. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
1.4.1. Célula de difusión
Una célula de difusión consiste en una cámara donadora y una cámara receptora, entre las que se coloca la piel (en la figura 1 se muestra un ejemplo de disposición normal). La célula debe quedar bien cerrada alrededor de la piel, permitir con facilidad la toma de muestras y la mezcla adecuada de la solución receptora en contacto con la parte inferior de la piel, así como un buen control de la temperatura de la célula y de su contenido. Son válidas tanto las células de difusión estática como las de difusión dinámica. En principio, las cámaras donadoras se dejan sin ocluir durante la exposición a una dosis pequeña de un preparado problema. Sin embargo, con las aplicaciones de grandes dosis, y en ciertos casos de pequeñas dosis, es posible que las cámaras donadoras queden ocluidas.
1.4.2. Líquido receptor
Se recomienda el uso de un líquido receptor fisiológicamente adecuado, aunque también es posible utilizar otros en casos justificados. Debe indicarse la composición exacta del líquido receptor. Debe demostrarse que la sustancia problema es suficientemente soluble en el líquido receptor, de forma que este no actúe de barrera contra la absorción. Además, el líquido receptor no debe afectar a la integridad del preparado de piel. En un sistema dinámico, el caudal no debe dificultar la difusión de la sustancia problema al líquido receptor. En un sistema estático, hay que agitar continuamente el líquido y tomar muestras de él periódicamente. Si se estudia el metabolismo, el líquido receptor debe permitir que la piel siga estando viva a lo largo de todo el experimento.
1.4.3. Preparados de piel
Puede utilizarse piel de origen humano o animal. Se sabe que el uso de piel humana es objeto de condiciones y consideraciones éticas tanto nacionales como internacionales. Aunque se prefiere la piel viva, también puede utilizarse piel muerta, siempre que pueda demostrarse su integridad. Son válidas las membranas epidérmicas (separadas enzimática, térmica o químicamente) o la piel con la epidermis y parte de la dermis (normalmente de 200 a 400 μm de espesor), preparadas con un dermatomo. Puede utilizarse piel completa (con la epidermis y toda la dermis), pero ha de evitarse el espesor excesivo (aprox. > 1 mm), salvo que sea necesario específicamente para la determinación de la sustancia problema en las capas de la piel. Deben justificarse la elección de la especie, el lugar anatómico y la técnica de preparación. Es necesario conseguir datos aceptables de un mínimo de cuatro ensayos en paralelo de cada preparado problema.
1.4.4. Integridad del preparado de piel
Es fundamental que la piel se prepare adecuadamente. Un tratamiento inapropiado puede provocar lesiones en la capa córnea, por lo que debe comprobarse la integridad de la piel preparada. Si se investiga el metabolismo de la piel, debe utilizarse lo antes posible piel recién extirpada, y en condiciones que permitan la actividad metabólica. Como norma general, la piel recién extirpada debe utilizarse en el plazo de 24 horas, pero el plazo de conservación aceptable puede variar en función del sistema enzimático que intervenga en la metabolización y de la temperatura de conservación (13). En caso de que los preparados de piel no sean recientes cuando se vayan a utilizar, deben presentarse pruebas de que mantienen la función de barrera.
1.4.5. Sustancia problema
La sustancia problema es aquella cuyas características de penetración se van a estudiar. Lo mejor es que la sustancia problema esté radiomarcada.
1.4.6. Preparado problema
El preparado de la sustancia problema (por ejemplo, material puro, diluido o formulado que contiene la sustancia problema y que se aplica a la piel) debe ser el mismo (o un análogo realista) que aquel al que pueda verse expuesto el hombre u otra posible especie diana. Deben justificarse las eventuales desviaciones respecto al preparado «en uso».
1.4.7. Concentraciones y formulaciones de las sustancias problema
Normalmente se utiliza más de una concentración de la sustancia problema, en la banda superior de la posible exposición humana. Análogamente, debe considerarse someter a ensayo una gama de formulaciones representativas.
1.4.8. Aplicación a la piel
En condiciones normales de exposición humana a las sustancias químicas, se suelen encontrar dosis pequeñas. Por tanto, debe aplicarse una cantidad que refleje la exposición humana, normalmente 1-5 mg/cm2 de piel en caso de sólidos y hasta 10 μl/cm2 en caso de líquidos. La cantidad debe justificarse en función de las condiciones previstas de uso, de los objetivos del estudio o de las características físicas del preparado problema. Por ejemplo, las aplicaciones a la superficie cutánea pueden ser de grandes dosis cuando se aplican volúmenes importantes por unidad de superficie.
1.4.9. Temperatura
La difusión pasiva de sustancias (y, en consecuencia, su absorción por la piel) depende de la temperatura. La cámara de difusión y la piel deben mantenerse a temperatura constante, cerca de la temperatura normal de la piel (32 ± 1 oC). Según el diseño de la célula, será necesario tener temperaturas diferentes en el baño de agua o en el bloque de calentamiento para garantizar que el receptor y la piel están en condiciones fisiológicas. La humedad debe situarse de preferencia entre el 30 y el 70 %.
1.4.10. Duración de la exposición y toma de muestras
La exposición de la piel al preparado problema puede mantenerse durante todo el experimento o bien durante plazos más breves (para imitar un tipo específico de exposición humana). El exceso del preparado problema debe lavarse de la piel con un agente de limpieza adecuado y los líquidos de lavado se deben recoger para su análisis. El procedimiento de eliminación del preparado problema dependerá de las condiciones previstas de uso, y debe justificarse. Normalmente hace falta tomar muestras durante un período de 24 horas, para poder caracterizar de forma adecuada la variación de la absorción. Como la integridad de la piel puede empezar a fallar a partir de las 24 horas, el muestreo no debe durar en principio más de ese tiempo. En caso de sustancias problema que penetran rápidamente en la piel, esto puede no ser necesario, pero con sustancias problema que penetran despacio es posible que haga falta un plazo más prolongado. La frecuencia de la toma de muestras de líquido receptor debe permitir la representación gráfica de la variación de la absorción de la sustancia problema.
1.4.11. Procedimiento final
Hay que analizar todos los componentes del sistema de ensayo y determinar la recuperación. Se incluyen la cámara donadora, los líquidos de lavado de la superficie de la piel, el preparado de piel y la cámara con el líquido receptor. En ciertos casos, la piel puede fraccionarse en superficie expuesta y en superficie bajo las pestañas de la célula, y en capa córnea, epidermis y dermis, para efectuar análisis aparte.
1.4.12. Análisis
En todos los estudios debe conseguirse una recuperación adecuada (el objetivo es conseguir una media del 100 ± 10 % de la radiactividad, y habrá que justificar las eventuales desviaciones). Debe analizarse la cantidad de sustancia problema que se encuentra en el líquido receptor, en el preparado de piel, en los líquidos de lavado de la superficie de la piel y en el líquido de lavado del material, utilizando una técnica adecuada.
2. DATOS
Deben indicarse el análisis del líquido receptor, la distribución de la sustancia problema en el sistema de ensayo y la variación de la absorción a lo largo del tiempo. Cuando se utilizan condiciones de exposición con dosis pequeñas, deben calcularse la cantidad lavada de la piel, la cantidad ligada a la piel (y a las diferentes capas de piel, si se analizan por separado) y la cantidad presente en el líquido receptor (tasa, y cantidad o porcentaje de la dosis aplicada). La absorción de la piel puede expresarse a veces utilizando solo datos del líquido receptor. Sin embargo, si al final del estudio queda sustancia en la piel, puede ser necesario tenerlo en cuenta para calcular la cantidad total absorbida [véase el punto 66 de la referencia (3)]. Cuando se utilizan condiciones de exposición con dosis grandes, los datos pueden permitir el cálculo de una constante de permeabilidad (Kp). En estas últimas condiciones, no es importante el porcentaje absorbido.
3. INFORMES
3.1. INFORME DEL ENSAYO
El informe del ensayo debe incluir los requisitos establecidos en el protocolo, con una justificación del sistema de ensayo utilizado y con los datos siguientes:
|
|
Sustancia problema:
|
|
|
Preparado problema:
|
|
|
Condiciones de ensayo:
|
|
|
Resultados:
|
|
|
Discusión de los resultados. |
|
|
Conclusiones. |
4. REFERENCIAS
|
1. |
Método de ensayo B.44. Absorción cutánea: método in vivo. |
|
2. |
OECD (2002). Guidance Document for the Conduct of Skin Absorption Studies. OECD, Paris. |
|
3. |
OECD (2000). Report of the Meeting of the OECD Extended Steering Committee for Percutaneous Absorption Testing, Annex 1 to ENV/JM/TG(2000)5. OECD, Paris. |
|
4. |
Kemppainen BW and Reifenrath WG. (1990). Methods for skin absorption. CRC Press, Boca Raton. |
|
5. |
Bronaugh RL and Collier, SW. (1991). Protocol for In vitro Percutaneous Absorption Studies, in In Vitro Percutaneous Absorption: Principles, Fundamentals and Applications, RL Bronaugh and HI Maibach, Eds., CRC Press, Boca Raton, pp. 237-241. |
|
6. |
Bronaugh RL and Maibach HI. (1991). In vitro Percutaneous Absorption: Principles, Fundamentals and Applications. CRC Press, Boca Raton. |
|
7. |
European Centre for Ecotoxicology and Toxicology of Chemicals (1993). Monograph No. 20, Percutaneous Absorption, ECETOC, Brussels. |
|
8. |
Diembeck W, Beck H, Benech-Kieffer F, Courtellemont P, Dupuis J, Lovell W, Paye M, Spengler J, Steiling W (1999). Test Guidelines for In Vitro Assessment of Dermal Absorption and Percutaneous Penetration of Cosmetic Ingredients, FdChem Tox, 37, 191-205. |
|
9. |
Recommended Protocol for In vitro Percutaneous Absorption Rate Studies (1996). US Federal Register, Vol. 61, No. 65. |
|
10. |
Howes D, Guy R, Hadgraft J, Heylings JR et al. (1996). Methods for assessing Percutaneous absorption. ECVAM Workshop Report ATLA 24, 81 R10. |
|
11. |
Schaefer H and Redelmeier TE. (1996). Skin barrier: principles of percutaneous absorption. Karger, Basel. |
|
12. |
Roberts MS and Walters KA. (1998). Dermal absorption and toxicity assessment. Marcel Dekker, New York. |
|
13. |
Jewell, C., Heylings, JR., Clowes, HM. And Williams, FM. (2000). Percutaneous absorption and metabolism of dinitrochlorobenzene in vitro. Arch Toxicol 74: 356-365. |
Figura 1
Ejemplo de diseño clásico de célula de difusión estática para estudios in vitro de absorción a través de la piel
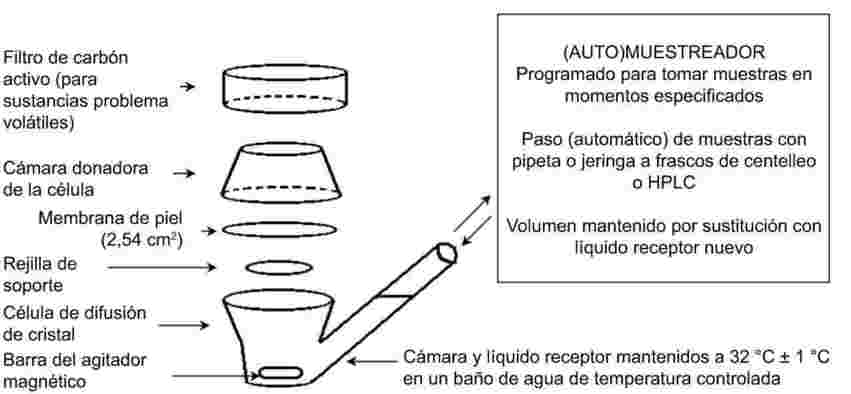
(1) Hay que tomar nota de la superficie de la opacidad corneal.
(2) El ayuno desde la víspera es preferible para ciertas medidas del suero y del plasma, sobre todo para la determinación de glucosa. La razón principal de esta preferencia es que el aumento de la variabilidad que provocaría necesariamente la toma de alimentos podría enmascarar efectos más sutiles y hacer más difícil la interpretación. Por otra parte, no obstante, el ayuno desde la víspera puede interferir con el metabolismo general de los animales y, especialmente en estudios de alimentación, puede alterar la exposición diaria a la sustancia estudiada. Si se adopta el ayuno desde la víspera, deben realizarse determinaciones bioquímicas clínicas después de la realización de las observaciones funcionales en la cuarta semana del estudio.
(3) El ayuno desde la víspera es preferible para ciertas medidas en el suero y el plasma, sobre todo para la determinación de glucosa. La razón principal es que el aumento de la variabilidad que provocaría necesariamente la toma de alimentos podría enmascarar efectos más sutiles y dificultar la interpretación. Por otra parte, no obstante, el ayuno desde la víspera puede inferir con el metabolismo general de los animales y, especialmente en los estudios en que la sustancia de ensayo se administra en los alimentos, puede alterar la exposición diaria a dicha sustancia. Si se opta por el ayuno desde la víspera, las determinaciones de bioquímica clínica deben realizarse después de las observaciones funcionales del estudio.
(4) Conocida actualmente como alaninaminotransferasa sérica.
(5) Conocida actualmente como aspartatoaminotransferasa sérica.
(6) Estos órganos, de 10 animales por sexo y grupo en los roedores y de todos los no roedores, se pesarán, lo mismo que la tiroides (con las paratiroides) de todos los roedores.
(7) Se pesarán estos órganos, de 10 animales por sexo y grupo de roedores.
(8) En este método, se entiende por grupo de referencia aquel al que se administra la sustancia analizada por otra vía que garantiza una disponibilidad completa de la dosis.
(9) Disolvente utilizado para medir la absorción.
(10) Incluye 5 animales seleccionados para pruebas funcionales y observaciones clínicas detalladas dentro del estudio de neurotoxicidad.
PARTE C: MÉTODOS PARA LA DETERMINACIÓN DE LA ECOTOXICIDAD
ÍNDICE
|
C.1. |
TOXICIDAD AGUDA EN PECES |
|
C.2. |
ENSAYO DE INMOVILIZACIÓN AGUDA DE DAPHNIA SP |
|
C.3. |
ENSAYO DE INHIBICIÓN DE ALGAS |
|
C.4. |
DETERMINACIÓN DE LA BIODEGRADABILIDAD «FÁCIL» |
|
PARTE I. |
CONSIDERACIONES GENERALES |
|
PARTE II. |
ENSAYO BASADO EN LA PÉRDIDA DE COD (Método C.4-A) |
|
PARTE III. |
ENSAYO DE DETECCIÓN DE LA OCDE MODIFICADO (Método C.4-B) |
|
PARTE IV. |
ENSAYO DE DESPRENDIMIENTO DE CO2 (Método C.4-C) |
|
PARTE V. |
RESPIROMETRÍA MANOMÉTRICA (Método C.4-D) |
|
PARTE VI. |
ENSAYO DEL FRASCO CERRADO (Método C.4-E) |
|
PARTE VII. |
ENSAYO DEL M.I.T.I. (Método C.4-F) |
|
C.5 |
DEGRADACIÓN: DEMANDA BIOQUÍMICA DE OXÍGENO |
|
C.6. |
DEGRADACIÓN: DEMANDA QUÍMICA DE OXÍGENO |
|
C.7. |
DEGRADACIÓN — DEGRADACIÓN ABIÓTICA: HIDRÓLISIS EN FUNCIÓN DEL PH |
|
C.8. |
TOXICIDAD PARA GUSANOS DE TIERRA |
|
C.9. |
BIODEGRADACIÓN — PRUEBA ZAHN-WELLENS |
|
C.10. |
BIODEGRADACIÓN — ENSAYO DE SIMULACIÓN CON LODO ACTIVADO |
|
C.11. |
BIODEGRADACIÓN — LODO ACTIVADO: PRUEBA DE INHIBICIÓN DE LA RESPIRACIÓN |
|
C.12. |
BIODEGRADACIÓN — PRUEBA LASC MODIFICADA |
|
C.13 |
BIOCONCENTRACIÓN: ENSAYO DINÁMICO CON PECES |
|
C.14. |
ENSAYO DE CRECIMIENTO EN PECES JUVENILES |
|
C.15. |
ENSAYO DE TOXICIDAD A CORTO PLAZO EN EMBRIONES DE PEZ Y ALEVINES |
|
C.16. |
ENSAYO DE TOXICIDAD ORAL AGUDA EN ABEJAS |
|
C.17. |
ENSAYO DE TOXICIDAD AGUDA POR CONTACTO EN ABEJAS |
|
C.18. |
ADSORCIÓN/DESORCIÓN SEGÚN UN MÉTODO EQUILIBRADO POR LOTES |
|
C.19. |
CÁLCULO DEL COEFICIENTE DE ADSORCIÓN (KOC) EN SUELOS Y EN LODOS DE AGUAS RESIDUALES MEDIANTE CROMATOGRAFÍA LÍQUIDA DE ALTA RESOLUCIÓN (HPLC) |
|
C.20 |
ENSAYO DE REPRODUCCIÓN EN DAPHNIA MAGNA |
|
C.21. |
MICROORGANISMOS DEL SUELO: ENSAYO DE TRANSFORMACIÓN DEL NITRÓGENO |
|
C.22. |
MICROORGANISMOS DEL SUELO: ENSAYO DE TRANSFORMACIÓN DEL CARBONO |
|
C.23. |
TRANSFORMACIÓN AEROBIA Y ANAEROBIA EN EL SUELO |
|
C.24. |
TRANSFORMACIÓN AEROBIA Y ANAEROBIA EN SISTEMAS DE SEDIMENTOS ACUÁTICOS |
C.1. TOXICIDAD AGUDA EN PECES
1. MÉTODO
1.1. INTRODUCCIÓN
El objeto de este ensayo es determinar la toxicidad letal aguda de una sustancia en peces de agua dulce. Es conveniente disponer, en la medida de lo posible, de una amplia información sobre la hidrosolubilidad, la presión de vapor, la estabilidad química, las constantes de disociación y la biodegradabilidad de la sustancia de ensayo, con el fin de seleccionar el método de ensayo más apropiado (estático, semiestático, dinámico), que permita garantizar concentraciones constantes de la sustancia de ensayo durante todo el período de ensayo.
Tanto en la planificación del ensayo como en la interpretación de los resultados deberá tenerse en cuenta otro tipo de información suplementaria (por ejemplo, la fórmula estructural, el grado de pureza, la naturaleza y porcentaje de impurezas significativas, la presencia y cantidad de aditivos, así como el coeficiente de reparto n-octanol/agua)
1.2. DEFINICIÓN Y UNIDADES
La toxicidad aguda es el efecto adverso, discernible, inducido en un organismo como consecuencia de la exposición a una sustancia dada durante un corto período de tiempo (días). En el presente ensayo, la toxicidad aguda viene expresada como la concentración letal media (CL50), es decir, la concentración que, en el agua, causa la muerte del 50 % de los peces de un lote sometido a ensayo durante un período de exposición continuo que debe indicarse.
Todas las concentraciones de la sustancia de ensayo se expresan en peso por volumen (mg/l). Pueden expresarse también en peso por peso (mg/kg-1).
1.3. SUSTANCIAS DE REFERENCIA
Se podrá efectuar un ensayo con una sustancia de referencia a fin de demostrar que, en las condiciones experimentales de laboratorio, la respuesta de la especie sometida al ensayo no ha variado significativamente.
Para este ensayo no se especifica ninguna sustancia de referencia.
1.4. PRINCIPIO DEL MÉTODO DE ENSAYO
Puede realizarse una prueba límite a 100 mg/l, para demostrar que la CL50 es mayor que esta concentración.
Se exponen los peces a la sustancia o sustancias de ensayo añadidas al agua, durante un período de 96 horas y en un intervalo de concentraciones determinado. Se registra la mortalidad, como mínimo, cada 24 horas y se calculan, si es posible, las concentraciones causantes de la muerte del 50 % de los peces (CL50) en cada período de observación.
1.5. CRITERIOS DE CALIDAD
Los criterios de calidad se aplicarán tanto al ensayo límite como al método de ensayo definitivo.
La mortalidad de los testigos, no debe sobrepasar el 10 % al final del ensayo (o un pez si se utilizan menos de 10).
La concentración de oxígeno disuelto debe ser superior al 60 % del valor de saturación, a la largo de todo el ensayo.
La concentración de la sustancia de ensayo debe ser superior al 80 % de la concentración inicial durante todo el ensayo.
Para las sustancias que se disuelven fácilmente en el medio de ensayo, produciendo soluciones estables, es decir, que no se volatilizan, degradan, hidrolizan ni adsorben en cantidad significativa, la concentración inicial puede considerarse equivalente a la concentración nominal. Se demostrará que se han mantenido las concentraciones a lo largo del ensayo y de que se han satisfecho los criterios de calidad.
En cuanto a las sustancias
|
i) |
poco solubles en el medio de ensayo, o |
|
ii) |
capaces de formar emulsiones o dispersiones estables, o |
|
iii) |
no estables en soluciones acuosas, |
se tomará como concentración inicial la concentración medida en la solución (o, si no es técnicamente posible, medida en la columna de agua) al comienzo del ensayo. La concentración se determinará tras un período de equilibrio pero antes de introducir los peces de ensayo.
En cualquiera de estos casos se harán mediciones complementarias, durante el ensayo, con el fin de confirmar la concentración de exposición real o que se han mantenido los criterios de calidad.
El pH no variará más de 1 unidad.
1.6. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
Pueden utilizarse tres tipos de procedimientos.
Ensayo estático
Ensayo de toxicidad durante el cual no hay renovación de la solución de ensayo {las soluciones no se renuevan durante todo el ensayo).
Ensayo semiestático
Ensayo sin renovación continua de la solución de ensayo, pero con renovación periódica, de las soluciones de ensayo después de períodos prolongados (por ejemplo, cada 24 horas).
Ensayo dinámico
Ensayo de toxicidad durante el cual se va renovando constantemente el agua de los recipientes de ensayo. El producto químico sometido a ensayo se transporta con el agua utilizada para renovar el medio de ensayo.
1.6.1. Reactivos
1.6.1.1. Soluciones de las sustancias de ensayo
Las soluciones madre a las concentraciones requeridas se preparan disolviendo la sustancia en agua desionizada o en agua según el punto 1.6.1.2.
Las concentraciones elegidas para el ensayo se preparan mediante dilución de la solución madre. Si se procede a un ensayo a concentraciones elevadas, la sustancia puede disolverse directamente en el agua de dilución.
En general, las sustancias se ensayaran solo hasta su límite de solubilidad. En el caso de algunas sustancias (por ejemplo, aquellas que son poco hidrosolubles, que tienen un elevado Pow o que forman en el agua dispersiones estables más que verdaderas soluciones), se puede aceptar una concentración de ensayo por encima del límite de solubilidad de la sustancia a fin de garantizar que se obtiene la máxima concentración soluble/estable. Es importante, sin embargo, que esta concentración no altere por otra parte el sistema de ensayo (por ejemplo, formación en la superficie del agua de una película de la sustancia que impida la oxigenación del agua, etc.)
Puede utilizarse dispersión ultrasónica, solventes orgánicos, emulgentes o dispersantes para favorecer la preparación de las soluciones madre de las sustancias poco hidrosolubles o para que dichas sustancias se dispersen mejor en el medio de ensayo. Cuando se utilicen estas sustancias auxiliares, todas las concentraciones de ensayo deberán contener la misma cantidad de sustancia auxiliar y se expondrá un lote de peces como testigo suplementario a la misma concentración de la sustancia auxiliar que la utilizada en las series del ensayo. La concentración de esas sustancias auxiliares debe reducirse al mínimo, y en ningún caso será superior a 100 mg por litro en el medio de ensayo.
El ensayo debe realizarse sin ajustar el pH. Si se produjera un cambio significativo del pH, debe repetirse el ensayo ajustando el pH y registrar los resultados. En ese caso, el valor del pH de la solución madre debe ajustarse al valor del pH del agua de dilución, excepto que existan razones concretas que lo impidan. Para ajustar el pH se utilizará, preferentemente, HC1 o NaOH. Este ajuste del pH debe efectuarse de forma que no se modifique significativamente la concentración de la sustancia de ensayo en la solución madre. Si el ajuste ocasiona una reacción química o una precipitación física de la sustancia de ensayo, deberá mencionarse en el informe.
1.6.1.2. Agua de mantenimiento y de dilución
Se podrá utilizar agua potable (no contaminada por concentraciones potencialmente peligrosas de cloro, metales pesados u otras sustancias), agua natural de buena calidad o agua reconstituida (véase el Apéndice 1). Se preferirán las aguas cuya dureza total esté comprendida entre 10 y 250 mg por litro (como CaCO3), y un pH comprendido entre 6,0 y 8,5.
1.6.2. Equipo
El equipo debe ser de material químicamente inerte:
|
— |
sistema de dilución automático (para el ensayo dinámico), |
|
— |
aparato para medir el oxígeno, |
|
— |
aparato para determinar la dureza del agua, |
|
— |
equipo apropiado para el control de la temperatura, |
|
— |
pHmetro. |
1.6.3. Peces del ensayo
Los peces deben gozar de buena salud y estar exentos de malformaciones visibles.
Se recomienda que la especie utilizada se elija en función de criterios prácticos como disponibilidad inmediata durante todo el año, fácil mantenimiento, comodidad para el ensayo, sensibilidad relativa a las sustancias químicas, y todos los demás factores significativos económicos, biológicos o ecológicos. Debe tenerse también en cuenta, al seleccionar la especie de peces, la necesidad de que los datos sean comparables y la existencia de una armonización internacional en vigor (referencia 1).
Las especies que se recomiendan para la realización de este ensayo figuran en el apéndice 2; las especies preferidas son el pez cebra y la trucha.
1.6.3.1. Mantenimiento
Los peces que se someten al ensayo deberán provenir preferentemente de un solo lote, siendo todos los individuos de longitud y edad similares. Deben mantenerse, al menos durante 12 días, en las siguientes condiciones.
Carga:
apropiada al sistema (recirculación o ensayo dinámico) y a la especie de peces.
Agua:
véase el punto 1.6.1.2.
Luz:
fotoperíodo de 12 a 16 horas por día.
Concentración de oxigeno disuelto:
al menos 80 % del valor de saturación.
Alimentación:
diariamente o 3 veces por semana; la alimentación debe ser interrumpida 24 horas antes de comenzar el ensayo.
1.6.3.2. Mortalidad
Se registrará la mortalidad después de un período inicial de 48 horas. Se juzgará la calidad del lote según los siguientes criterios:
|
— |
mortalidad superior al 10 % de la población en 7 días: se rechaza todo el lote, |
|
— |
mortalidad al cabo de 7 días comprendida entre el 5 y el 10 % de la población: se prolongará el período de observación 7 días más. Si no aparecen nuevas mortalidades, se admite el lote, en caso contrario debe rechazarse, |
|
— |
mortalidad inferior al 5 % de la población: se admite el lote. |
1.6.4. Adaptación
Los peces deben mantenerse en las condiciones de ensayo (calidad y temperatura del agua), durante al menos 7 días antes de su utilización.
1.6.5. Procedimiento del ensayo
Antes del ensayo definitivo se puede proceder a un ensayo para determinar el intervalo de concentraciones eficaces, el cual proporcionará información sobre el intervalo de concentraciones que deberán utilizarse en el ensayo definitivo.
Se efectuará un control (o testigo), sin sustancia de ensayo y, si se considera importante, un control (o testigo) con la sustancia auxiliar, además de las concentraciones de ensayo propiamente dichas.
Se elegirá un método estático, semiestático o dinámico, en función de las propiedades físicas y químicas de la sustancia de ensayo, para cumplir los criterios de calidad.
Los peces estarán expuestos a la sustancia de ensayo en las siguientes condiciones:
|
— |
duración: 96 horas, |
|
— |
número de animales: al menos 7 por concentración, |
|
— |
recipientes: de una capacidad apropiada, en función de la carga recomendada, |
|
— |
carga: se recomienda una carga máxima de 1,0 g por litro para los ensayos estáticos y semiestáticos; para los ensayos dinámicos se puede aceptar una carga más elevada, |
|
— |
concentraciones de ensayo: al menos cinco concentraciones de ensayo que difieran en un factor constante no mayor de 2,2 y abarcando si es posible el intervalo de 0 a 100 % de mortalidad, |
|
— |
agua: véase el punto 1.6.1.2, |
|
— |
luz: fotoperíodo de 12 a 16 horas por día, |
|
— |
temperatura: apropiada para la especie elegida (apéndice 2), pero en los límites de ± 1 oC, en cualquier ensayo concreto, |
|
— |
concentración en oxígeno disuelto: al menos el 60 % del valor de la saturación a la temperatura elegida, |
|
— |
alimento: ninguno. |
Los peces se observaran después de las 2 a 4 primeras horas y, al menos, a intervalos de 24 horas. Los peces son considerados muertos si el toque del pedúnculo caudal no produce reacción alguna y no se percibe ningún movimiento respiratorio. Los peces muertos se retirarán en el momento de cada observación y se registrará la mortalidad. Se registrarán las anomalías visibles (por ejemplo, pérdida de equilibrio, cambios en el comportamiento natatorio, función respiratoria, pigmentación, etc.).
Diariamente se medirá el pH, el oxígeno disuelto y la temperatura.
Ensayo límite
Puede realizarse un ensayo límite a 100 mg por litro, utilizando los procedimientos descritos en este método de ensayo, con el fin de demostrar que la CL50 es mayor que dicha concentración.
Si la naturaleza de la sustancia no permite alcanzar una concentración de 100 mg por litro en el agua de ensayo, el ensayo límite debe ser realizado a una concentración igual a la solubilidad de la sustancia (o a la máxima concentración que forme una dispersión estable) en el medio utilizado (véase también el punto 1.6.1.1).
El ensayo límite se realizará utilizando de 7 a 10 peces, y el mismo número en los controles (o testigos). (La teoría binomial indica que cuando se utilizan 10 peces y la mortalidad es cero, hay un limite de confianza del 99,9 % de que la CL50 es mayor que la concentración usada en el ensayo límite. Si se utilizan 7, 8 o 9 peces, la ausencia de mortalidad proporciona un límite de confianza del 99 % de que la CL50 es mayor que la concentración usada.)
Si aparece mortalidad, debe ser realizado un estudio completo. Si se observan efectos subletales, deben registrarse.
2. RESULTADOS Y EVALUACIÓN
Se trazará sobre papel log-probit el porcentaje de mortalidad en función de la concentración, para cada período en que se registren observaciones (24, 48, 72 y 96 horas).
Cuando sea posible y para cada tiempo de observación, se calculará la CL50 con sus límites de confianza (p = 0,05) utilizando procedimientos estandardizados; estos valores se redondearán a una o, como mucho, dos cifras significativas (ejemplos de redondeo a dos cifras: 170 en lugar de 173,5; 0,13 en lugar de 0,127; 1,2 en lugar de 1,21).
Si la pendiente de la curva de concentración/porcentaje de respuesta es demasiado acentuada como para permitir calcular la CL50, bastará con una estimación gráfica de este valor.
Si dos concentraciones consecutivas, con una razón de 2,2, dan solo 0 y 100 % de mortalidad, estos dos valores serán suficientes para indicar el rango en el que se encuentra la CL50.
Si se observa que la estabilidad o la homogeneidad de la sustancia de ensayo no puede mantenerse, se mencionará en el informe y se tendrá en cuenta al interpretar los resultados.
3. INFORME
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
información acerca del pez de ensayo (nombre científico, cepa, proveedor, tratamiento previo eventual, tamaño y número utilizado en cada concentración de ensayo), |
|
— |
origen del agua de dilución y sus principales características químicas (pH, dureza, temperatura), |
|
— |
en el caso de una sustancia poco hidrosoluble, método de preparación de la solución madre y de las soluciones de ensayo, |
|
— |
concentraciones de las eventuales sustancias auxiliares, |
|
— |
relación de las concentraciones utilizadas y cualquier información disponible sobre la estabilidad, a dichas concentraciones, de la sustancia en la solución de ensayo, |
|
— |
si se realizan análisis químicos, los métodos utilizados y los resultados obtenidos, |
|
— |
resultados del ensayo límite, si se ha realizado, |
|
— |
motivos de la elección y los detalles del procedimiento de ensayo utilizado (por ejemplo, sistema estático, semiestático, dinámico, velocidad de administración, tasa de renovación, aireación o no, carga de peces, etc.), |
|
— |
descripción del equipo de ensayo, |
|
— |
régimen de iluminación, |
|
— |
concentraciones del oxígeno disuelto, valores de pH y temperaturas de las soluciones de ensayo cada 24 horas, |
|
— |
pruebas de que se han respetado los criterios de calidad, |
|
— |
tabla que muestre la mortalidad acumulada para cada concentración y para el testigo (y, en su caso, el testigo con producto auxiliar) para cada uno de los tiempos de observación recomendados, |
|
— |
representación gráfica de los porcentajes de mortalidad en función de las concentraciones al final del ensayo, |
|
— |
si es posible, valores de la CL50 en cada uno de los tiempos de observación recomendados (límite de confianza del 95 %), |
|
— |
métodos estadísticos utilizados para determinar los valores de la CL50, |
|
— |
si se utiliza una sustancia de referencia, resultados obtenidos, |
|
— |
concentración máxima utilizada que no haya causado mortalidad durante el período de ensayo, |
|
— |
concentración mínima utilizada que haya causado un 100 % de mortalidad durante el período de ensayo. |
4. BIBLIOGRAFÍA
|
(1) |
OCDE, París 1981, Test Guideline 203, Decisión del Consejo C(81) 30 final, y actualizaciones. |
|
(2) |
AFNOR — Determination of the acute toxicity of a substance to Bracbydanio rerio — Static and Flow Through methods — NFT 90-303 June 1985. |
|
(3) |
AFNOR — Determination of the acute toxicity of a substance to Salmo gairdneri — Static and Flow Through methods — NFT 90-305 June 1985. |
|
(4) |
ISO 7346/1,/2 and/3 — Water Quality — Determination of the acute lethal toxicity of substances to a fresh water fish (Bracbydanio rerio Hamilton-Buchanan — Teleostei, Cyprinidae). Part 1: Static method. Part 2: Semi-static method. Part 3: Flow-through method. |
|
(5) |
Eidgenössisches Department des Innern, Suiza, Richtlinien für Probenahme und Normung von Wasserun- tersuchungsmethoden — Part II 1974. |
|
(6) |
DIN Testverfahren mit Wasserorganismen, 38 412 (L1) und L (15). |
|
(7) |
JIS K 0102, Acute toxicity test for fish. |
|
(8) |
NEN 6506 — Water — Bepaling van de akute toxiciteit met behulp van Poecilia reticulata, 1980. |
|
(9) |
Environmental Protection Agency, Methods for the acute toxicity tests with fish, macroinvertebrates and amphibians. The Committee on Methods for Toxicity Tests with Aquatic Organisms, Ecological Research Series EPA-660-75-009, 1975. |
|
(10) |
Environmental Protection Agency, Environmental monitoring and support laboratory, Office of Research and Development, EPA-600/4-78-012, January 1978. |
|
(11) |
Environmental Protection Agency, Toxic Substance Control, Part IV, 16 March 1979. |
|
(12) |
Standard methods for the examination of water and wastewater, 14th edition, APHA-AWWA-WPCF, 1975. |
|
(13) |
Comisión de las Comunidades Europeas, Inter-Laboratory test programme concerning the study of the ecotoxicity of a chemical substance with respect to the fish. EEC Study D.8368, 22 March 1979. |
|
(14) |
Verfahrensvorschlag des Umweltbundesamtes zum akuten Fisch-Test. Rudolph, P. und Boje, R. Ökotoxikologie, Grundlagen für die Ökotoxikologische Bewertung von Umweltchemikalien nach dem Chemikaliengesetz, ecomed 1986. |
|
(15) |
Litchfield, J.T. y Wilcoxon, F., A simplified method for evaluating dose effects experiments, J. Pharm. Exp. Therap., 1949, vol. 96, 99. |
|
(16) |
Finney, D.J. Statistical Methods in Biological Assay. Griffin, Weycombe, U.K, 1978. |
|
(17) |
Sprague, J.B. Measurement of pollutant toxicity to fish. I Bioassay methods for acute toxicity. Water Res., 1969, vol.3, 793–821. |
|
(18) |
Sprague, J.B. Measurement of pollutant toxicity to fish. Il Utilising and applying bioassay results. Water Res., 1970, vol. 4, 3-32. |
|
(19) |
Stephan, C.E. Methods for calculating an LC50. In Aquatic Toxicology and Hazard Evaluation (edited by F.I. Mayer and J.L. Hamelink), American Society for Testing and Materials, ASTM STP 634, 1977, 65-84. |
|
(20) |
Stephan, C.E., Busch, K.A., Smith, R., Burke, J. y Andrews, R.W. A computer program for calculating an LC50. US EPA. |
Apéndice 1
Agua reconstituida
Ejemplo de agua de dilución adecuada
Los productos químicos deben ser de calidad analítica.
El agua debe ser agua destilada de buena calidad, o agua desionizada de una conductividad inferior a 5 μScm-1.
El aparato para la destilación del agua no contendrá ningún componente de cobre.
Soluciones madre
|
CaCl2. 2H2O (cloruro de calcio dihidratado): disolver y enrasar a un litro con agua. |
11,76 g |
|
MgSO4. 7H2O (sulfato de magnesio heptahidratado): disolver y enrasar a un litro con agua. |
4,93 g |
|
NaHCO3 (carbonato ácido de sodio): disolver y enrasar a un litro con agua. |
2,59 g |
|
KCl (cloruro de potasio): disolver y enrasar a un litro con agua. |
0,23 g |
Agua de dilución reconstituida
Mezclar 25 ml de cada una de las cuatro soluciones madre y completar hasta un litro con agua.
Airear hasta que la concentración de oxígeno disuelto sea igual al valor de saturación del aire.
El pH debe ser 7,8 ± 0,2.
Si es necesario, ajustado con NaOH (hidróxido de sodio) o HCl (ácido clorhídrico).
El agua de dilución así preparada se deja reposar durante unas 12 horas y no debe ser aireada posteriormente.
La suma de iones Ca/Mg en esta solución es igual a 2,5 mmol/l. La relación de los iones Ca:Mg es de 4:1, la de los iones Na:K de 10:1. La alcalinidad total de esta solución es igual a 0,8 mmol/1.
Cualquier desviación en la preparación del agua de dilución no debe modificar su composición ni sus propiedades.
Apéndice 2
Especies de peces recomendadas para el ensayo
|
Especies recomendadas |
Intervalo recomendado de temperaturas de ensayo ( oC) |
Longitud total recomandada del pez de ensayo (cm) |
|
Bracbydanio rerio (Teleostei, Cyprinidae) (Hamilton-Buchanan) Pez cebra |
20 a 24 |
3,0 ± 0,5 |
|
Pimephales prometas (Teleostei, Cyprinidae) (Rafinesque) Pez cabeza gorda |
20 a 24 |
5,0 ± 2,0 |
|
Cyprinus carpio (Teleostei, Cyprinidae) (Linnaeus 1758) Carpa común |
20 a 24 |
6,0 ± 2,0 |
|
Oryzias latipes (Teleostei, Poeciliiae) Cyprinodontidae (Tomminck y Schlegel 1850) Medaka |
20 a 24 |
3,0 ± 1,0 |
|
Poecilia reticulata (Teleostei, Poeciliidae) (Peters 1859) Guppy |
20 a 24 |
3,0 ± 1,0 |
|
Lepomis macrochirus (Teleostei, Centrarchidae) (Rafínesque Linnaeus 1758) Agallas azules |
20 a 24 |
5,0 ± 2,0 |
|
Onchorhynchus mykiss (Teleostei, Salmonidae) (Walbaum 1988) Trucha arco iris |
12 a 17 |
6,0 ± 2,0 |
|
Leuciscus idus (Teleostei, Cyprinidae) (Linnaeus 1758) Caho |
20 a 24 |
6,0 ± 2,0 |
Aprovisionamiento
Los peces que se mencionan arriba son fáciles de criar y se puede disponer fácilmente de ellos durante todo el año. Pueden reproducirse y desarrollarse en explotaciones piscícolas o en el laboratorio, en condiciones sanitarias controladas, de forma que el animal que se someta al ensayo esté sano y su origen sea conocido. Estos peces están disponibles en casi todo el mundo.
Apéndice 3
Ejemplo de curva de procentaje de mortalidad/concentración
Ejemplo de determinación de la CL50 en un papel log-probit

C.2. ENSAYO DE INMOVILIZACIÓN AGUDA DE DAPHNIA SP.
1. MÉTODO
El presente método de ensayo de inmovilización aguda es equivalente a las directrices de ensayo de la OCDE TG 202 (2004).
1.1. INTRODUCCIÓN
El presente método describe un ensayo de toxicidad aguda para evaluar los efectos de las sustancias químicas en los dáfnidos. En la medida de lo posible se han utilizado los métodos de ensayo existentes (1) (2) (3).
1.2. DEFINICIONES
A efectos del presente método, se entenderá por:
|
|
«CE 50»: la concentración estimada que inmoviliza al 50 % de los dáfnidos en un plazo de exposición definido; si se utiliza otra definición, deberá indicarse junto con su referencia, |
|
|
«inmovilización»: la situación de los animales incapaces de nadar en el plazo de 15 segundos tras una agitación suave del recipiente de ensayo (aunque puedan seguir moviendo las antenas). |
1.3. PRINCIPIO DEL MÉTODO DE ENSAYO
Se exponen dáfnidos jóvenes, de menos de 24 horas de edad al inicio del ensayo, a una gama de concentraciones de la sustancia problema durante 48 horas. Se registra la inmovilización a las 24 y a las 48 horas, y se compara con los valores de los controles. Se analizan los resultados para calcular la CE50 a las 48 horas (véanse las definiciones del punto 1.2). La determinación de la CE50 a las 24 horas es optativa.
1.4. INFORMACIÓN SOBRE LA SUSTANCIA PROBLEMA
Hay que conocer la solubilidad en agua y la presión de vapor de la sustancia problema; también se debe disponer de un método analítico fiable para cuantificar la sustancia en las soluciones problema con la eficiencia de recuperación y el límite de determinación establecidos. Entre la información útil se incluyen la fórmula estructural de la sustancia problema, su pureza, estabilidad en el agua y a la luz, Pow, y los resultados de un ensayo de biodegradabilidad fácil (véase el método C.4).
Nota: En la referencia (4) se ofrece orientación sobre los ensayos de sustancias con propiedades fisicoquímicas que dificultan estos ensayos.
1.5. SUSTANCIAS DE REFERENCIA
Puede estudiarse la CE50 de una sustancia de referencia como forma de garantizar la fiabilidad de las condiciones del ensayo. Con este fin se recomienda utilizar sustancias tóxicas empleadas en ensayos interlaboratorios internacionales (1)(5) (1). La frecuencia de los ensayos con una sustancia de referencia debe ser preferentemente mensual, y al menos semestral.
1.6. CRITERIOS DE CALIDAD
Para que un ensayo sea válido, los resultados deben cumplir los siguientes criterios:
|
— |
en los controles, incluido el que contiene el agente solubilizante, no debe quedar inmovilizado más del 10 % de los dáfnidos, |
|
— |
la concentración de oxígeno disuelto al final del ensayo debe ser ≥ 3 mg/l en los recipientes de control y de ensayo. |
Nota: Respecto al primer criterio, no más del 10 % de los dáfnidos de control pueden presentar inmovilización u otros signos de enfermedad o presión, por ejemplo, decoloración o comportamiento anómalo, como quedar retenidos en la superficie del agua.
1.7. DESCRIPCIÓN DEL MÉTODO
1.7.1. Equipo
Los recipientes de ensayo y demás equipo que hayan de entrar en contacto con las soluciones problema serán íntegramente de vidrio o de otro material químicamente inerte. Los recipientes de ensayo consistirán normalmente en tubos de ensayo o vasos de precipitados; deben lavarse antes de cada utilización siguiendo procedimientos normalizados de laboratorio. Los recipientes de ensayo deben cubrirse de forma no hermética para reducir la pérdida de agua debida a la evaporación y para evitar la entrada de polvo en las soluciones. Los ensayos con sustancias volátiles deben realizarse en recipientes cerrados y completamente llenos, de volumen suficiente para evitar que el oxígeno llegue a un nivel limitante o demasiado bajo (véanse el punto 1.6 y el párrafo primero del punto 1.8.3).
Además se utilizarán algunos de los instrumentos siguientes o todos ellos: oxigenómetro (con microelectrodo u otro equipo que sea conveniente para medir el oxígeno disuelto en muestras de pequeño volumen), pH-metro, instrumento adecuado para controlar la temperatura, equipo para determinar la concentración de carbono orgánico total (COT), equipo para determinar la demanda química de oxígeno (DQO), aparato para determinar la dureza, etc.
1.7.2. Organismo de ensayo
La especie recomendada es Daphnia magna Straus, aunque pueden utilizarse en este ensayo otras especies adecuadas de Daphnia (por ejemplo, Daphnia pulex). Los animales tendrán una edad inferior a 24 horas al principio del ensayo y, para reducir la variabilidad, se recomienda fervientemente que no sean progenie de primera nidada. Han de proceder de una población sana (es decir, sin signos de presión, tales como mortalidad elevada, presencia de machos y efipios, retraso de la producción de la primera nidada, animales decolorados, etc.). Todos los organismos utilizados para un ensayo concreto deben proceder de cultivos establecidos a partir de la misma población de dáfnidos. Los animales del cultivo madre deben mantenerse en condiciones (luz, temperatura, medio) similares a las que se vayan a usar en el ensayo. Si el medio de cultivo de dáfnidos que se va utilizar en el ensayo es diferente del utilizado en cultivos normales de dáfnidos, es adecuado incluir una fase de aclimatación previa al ensayo. Con este objetivo, los dáfnidos de la nidada deben mantenerse en agua de dilución a la temperatura del ensayo durante al menos 48 horas antes del inicio del ensayo.
1.7.3. Agua de mantenimiento y de dilución
Puede utilizarse como agua de mantenimiento y de dilución el agua natural (superficial o subterránea), el agua reconstituida o el agua del grifo desclorada, siempre que los dáfnidos sobrevivan en ella durante las fases de cultivo, aclimatación y ensayo sin mostrar signos de presión. Es adecuada como agua de ensayo cualquier agua que se ajuste a las características químicas de un agua de dilución aceptable según se indica en el anexo 1. La calidad del agua ha de ser constante a lo largo de todo el ensayo. El agua reconstituida puede obtenerse añadiendo al agua desionizada o destilada cantidades específicas de reactivos de pureza analítica reconocida. En el anexo 2 y en las referencias (1) y (6) se dan ejemplos de agua reconstituida. Obsérvese que, para el ensayo de sustancias con metales, ha de evitarse utilizar medios que contengan agentes quelantes conocidos, como los medios M4 y M7 del anexo 2. El pH debe encontrarse en la banda de 6 a 9. La dureza recomendada es de entre 140 y 250 mg/l (en CaCO3) para Daphnia magna, pero con otras especies de Daphnia también puede ser adecuada una dureza menor. El agua de dilución tiene que airearse antes de su utilización en el ensayo, de forma que la concentración de oxígeno disuelto llegue a la saturación.
Si se utiliza agua natural, deben medirse los parámetros de calidad al menos dos veces al año o siempre que se sospeche la posibilidad de algún cambio significativo en estas características (véanse el párrafo anterior y el anexo 1). Debe medirse también la concentración de metales pesados (por ejemplo, Cu, Pb, Zn, Hg, Cd, Ni). Si se utiliza agua del grifo desclorada, es recomendable hacer cada día un análisis del cloro. Si el agua de dilución es de una fuente de agua superficial o subterránea, deben medirse la conductividad y el carbono orgánico total (COT) o la demanda química de oxígeno (DQO).
1.7.4. Soluciones problema
Las soluciones problema con las concentraciones elegidas se preparan normalmente por dilución de una solución madre. Preferentemente, estas soluciones madre deben prepararse disolviendo la sustancia problema en agua de dilución. Debe evitarse, en la medida de lo posible, el uso de disolventes, emulgentes o dispersantes. Sin embargo, estos compuestos pueden ser necesarios en algunos casos para obtener una solución madre a la concentración deseada. En la referencia (4) se dan orientaciones sobre disolventes, emulgentes y dispersantes adecuados. En cualquier caso, la cantidad de sustancia problema contenida en las soluciones problema no debe sobrepasar el límite de solubilidad en el agua de dilución.
El ensayo debe realizarse sin ajustar el pH. Si el pH no queda en la banda de 6 a 9, puede realizarse un segundo ensayo en el que se ajuste el pH de la solución madre al del agua de dilución antes de la adición de la sustancia problema. El ajuste del pH debe hacerse de forma que la concentración de la solución madre no cambie de forma significativa y no se produzca ninguna reacción química ni precipitación de la sustancia problema. Las sustancias recomendadas son HCl y NaOH.
1.8. PROCEDIMIENTO
1.8.1. Condiciones de exposición
1.8.1.1. Grupos de ensayo y controles
Los recipientes de ensayo se llenan con volúmenes adecuados de agua de dilución y de soluciones de la sustancia problema. La proporción de volumen de aire y de agua en el recipiente debe ser la misma en los grupos de ensayo y de control. A continuación se ponen los dáfnidos en los recipientes de ensayo. Con cada concentración de ensayo y cada control deben utilizarse al menos 20 animales, de preferencia divididos en cuatro grupos de cinco animales cada uno. Para cada animal deben preverse al menos 2 ml de solución problema (es decir, un volumen de 10 ml para cinco dáfnidos en cada recipiente de ensayo). La prueba puede realizarse con un sistema de renovación semiestática o dinámica si no es estable la concentración de la sustancia problema.
Además de la serie de tratamiento, debe utilizarse una serie de control con el agua de dilución y, si procede, otra serie de control con el agente solubilizante.
1.8.1.2. Concentraciones de ensayo
Puede efectuarse un experimento previo para determinar la gama de concentraciones que se han de utilizar en el ensayo definitivo, salvo que se disponga de información sobre la toxicidad de la sustancia problema. A tal fin, los dáfnidos se exponen a una serie de concentraciones de la sustancia problema muy diferentes entre sí. Deben exponerse cinco dáfnidos a cada concentración de ensayo durante un máximo de 48 horas, y no es necesario efectuar ninguna prueba en paralelo. Es posible abreviar el plazo de exposición (por ejemplo, a 24 horas o menos) si pueden obtenerse en menos tiempo datos adecuados para los fines del experimento de determinación de la gama de concentraciones.
Se emplearán al menos cinco concentraciones de ensayo, que formarán una progresión geométrica con una razón preferentemente no superior a 2,2. Si se usan menos de cinco concentraciones, hay que justificarlo. La mayor concentración estudiada debe provocar de preferencia una inmovilización del 100 %, y la menor concentración estudiada no debe provocar de preferencia ningún efecto observable.
1.8.1.3. Condiciones de incubación
La temperatura debe estar entre 18 oC y 22 oC, y en cada uno de los ensayos debe mantenerse constante, con una variación máxima de ± 1 oC. Se recomienda un ciclo de 16 horas de luz y 8 horas de oscuridad. También puede aceptarse la oscuridad completa, especialmente si la sustancia problema es inestable a la luz.
Los recipientes de ensayo no deben airearse durante el ensayo. El ensayo se realiza sin ajustar el pH. No se debe proporcionar alimento a los dáfnidos durante el ensayo.
1.8.1.4. Duración
La duración del ensayo es de 48 horas.
1.8.2. Observaciones
Debe comprobarse si hay dáfnidos inmovilizados en cada recipiente de ensayo a las 24 y 48 horas desde el inicio del ensayo (véanse las definiciones del punto 1.2). Además de la inmovilidad, debe indicarse todo eventual aspecto o comportamiento anormal.
1.8.3. Mediciones analíticas
El oxígeno disuelto y el pH se miden al inicio y al final del ensayo en los recipientes de los controles y en los de la mayor concentración de sustancia problema. La concentración de oxígeno disuelto de los controles debe cumplir el criterio de validez (véase el punto 1.6). El pH no debe variar en principio más de 1,5 unidades en ninguno de los ensayos. La temperatura se suele medir en los recipientes de control o en el aire ambiente y debe registrarse preferentemente de forma continua durante el ensayo o, como mínimo, al inicio y al final del mismo.
La concentración de la sustancia problema debe medirse, como mínimo, en los recipientes con la concentración más elevada y más baja, al principio y al final del ensayo (4). Se recomienda basar los resultados en las concentraciones medidas. No obstante, si los datos disponibles muestran que la concentración de la sustancia problema se ha mantenido debidamente a lo largo de todo el ensayo en un intervalo de ± 20 % de la concentración inicial nominal o medida, los resultados pueden fundarse en estos valores iniciales nominales o medidos.
1.9. ENSAYO LÍMITE
Siguiendo los procedimientos descritos en el presente método, puede efectuarse un ensayo límite con 100 mg/l de sustancia problema o hasta su límite de solubilidad en el medio de ensayo, si este es inferior, a fin de demostrar que la CE50 es mayor que esta concentración. El ensayo límite debe hacerse utilizando 20 dáfnidos (preferentemente divididos en cuatro grupos de cinco), con igual número en el control o controles. Si se observa alguna inmovilización, debe realizarse un estudio completo. Deben registrarse los eventuales comportamientos anormales observados.
2. DATOS
Los datos deben resumirse en forma de cuadro, indicando respecto a cada grupo de tratamiento y de control el número de dáfnidos utilizados y la inmovilización en cada observación. Deben representarse gráficamente los porcentajes de inmovilización a las 24 y 48 horas frente a las concentraciones de sustancia problema. Los datos se analizan mediante métodos estadísticos apropiados (por ejemplo, análisis de probit, etc.) para calcular las pendientes de las curvas y la CE50 con unos límites de confianza del 95 % (p = 0,05) (7) (8).
Cuando los métodos normales de cálculo de la CE50 no sean aplicables a los datos obtenidos, se utilizarán la concentración más elevada que no provoque inmovilidad y la concentración más baja que provoque un 100 % de inmovilidad para estimar la CE50, considerada como la media geométrica de estas dos concentraciones.
3. INFORMES
3.1. INFORME DEL ENSAYO
El informe del ensayo debe incluir la información siguiente:
Sustancia problema:
|
— |
naturaleza física y propiedades fisicoquímicas pertinentes, |
|
— |
identificación química, incluida la pureza. |
Especie de ensayo:
|
— |
procedencia y especie de Daphnia, proveedor original (si se conoce) y condiciones de cultivo utilizadas (con inclusión de la procedencia, el tipo y la cantidad del pienso, así como la frecuencia de la alimentación). |
Condiciones de ensayo:
|
— |
descripción de los recipientes de ensayo: tipo de recipientes, volumen de solución, número de dáfnidos por recipiente de ensayo, número de recipientes de ensayo (en paralelo) por concentración, |
|
— |
métodos de preparación de las soluciones madre y problema, incluido el eventual uso de dispersantes; concentraciones utilizadas, |
|
— |
origen del agua de dilución y características cualitativas del agua (pH, dureza, proporción Ca/Mg, proporción Na/K, alcalinidad, conductividad, etc.); composición del agua reconstituida si se utiliza, |
|
— |
condiciones de incubación: temperatura, intensidad y periodicidad de la luz, oxígeno disuelto, pH, etc. |
Resultados:
|
— |
número y porcentaje de dáfnidos inmovilizados o que presentan cualquier efecto nocivo (incluido un comportamiento anormal) en los controles y en cada grupo de tratamiento, en cada momento de observación, y descripción de la naturaleza de los efectos observados, |
|
— |
resultados y fecha del ensayo efectuado con la sustancia de referencia, si se conocen, |
|
— |
concentraciones nominales de ensayo y resultados de todos los análisis para determinar la concentración de la sustancia problema en los recipientes de ensayo; también deben indicarse la eficiencia de recuperación del método y el límite de determinación, |
|
— |
todas las mediciones fisicoquímicas de temperatura, pH y oxígeno disuelto efectuadas durante el ensayo, |
|
— |
la CE50 de inmovilización a las 48 horas, con intervalos de confianza y gráficos del modelo ajustado utilizado para su cálculo, las pendientes de las curvas dosis-respuesta y su error típico, métodos estadísticos utilizados para la determinación de la CE50 (también deben indicarse estos datos en relación con la inmovilización a las 24 horas si se han obtenido), |
|
— |
explicación de las eventuales desviaciones respecto al método de ensayo y de la medida en que afecten a los resultados de ensayo. |
4. REFERENCIAS
|
1. |
ISO 6341. (1996). Water quality — Determination of the inhibition of the mobility of Daphnia magna Straus (Cladocera, Crustacea) — Acute toxicity test. Third edition, 1996. |
|
2. |
EPA OPPTS 850.1010. (1996). Ecological Effects Test Guidelines — Aquatic Invertebrate Acute Toxicity Test, Freshwater Daphnids. |
|
3. |
Environment Canada. (1996) Biological test method. Acute Lethality Test Using Daphnia spp. EPS 1/RM/11. Environment Canada, Ottawa, Ontario, Canada. |
|
4. |
Guidance Document on Aquatic Toxicity Testing of Difficult Substances and Mixtures. OECD Environmental Health and Safety Publication. Series on Testing and Assessment. No. 23. Paris 2000. |
|
5. |
Comisión de las Comunidades Europeas. Study D8369. (1979). Inter-laboratory Test Programme concerning the study of the ecotoxicity of a chemical substance with respect to Daphnia. |
|
6. |
OECD Guidelines for the Testing of Chemicals. Guideline 211: Daphnia magna Reproduction Test, adopted September 1998. |
|
7. |
Stephan C.E. (1977). Methods for calculating an LC50. In Aquatic Toxicology and Hazard Evaluation (edited by F.I. Mayer and J.L. Hamelink). ASTM STP 634 — American Society for Testing and Materials. p. 65-84 |
|
8. |
Finney D.J. (1978). Statistical Methods in Biological Assay. 3rd ed. London. Griffin, Weycombe, UK. |
Anexo 1
ALGUNAS CARACTERÍSTICAS QUÍMICAS DE UN AGUA DE DILUCIÓN ACEPTABLE
|
Sustancia |
Concentración |
|
Partículas |
< 20 mg/l |
|
Carbono orgánico total |
< 2 mg/l |
|
Amoniaco no ionizado |
< 1 μg/l |
|
Cloro residual |
< 10 μg/l |
|
Plaguicidas organofosforados totales |
< 50 ng/l |
|
Plaguicidas organoclorados totales y bifenilos policlorados |
< 50 ng/l |
|
Cloro orgánico total |
< 25 ng/l |
Anexo 2
EJEMPLOS DE AGUA RECONSTITUIDA ACEPTABLE PARA EL ENSAYO
Agua de ensayo iso (1)
|
Soluciones madre (una sola sustancia) |
Para preparar el agua reconstituida, añádanse los volúmenes siguientes de soluciones madre a 1 litro de agua (2) |
|
|
Sustancia |
Cantidad añadida a 1 litro de agua (2) |
|
|
Cloruro de calcio CaCl2. 2H2O |
11,76 g |
25 ml |
|
Sulfato de magnesio MgSO4. 7H2O |
4,93 g |
25 ml |
|
Bicarbonato de sodio NaHCO3 |
2,59 g |
25 ml |
|
Cloruro de potasio KCl |
0,23 g |
25 ml |
Medios M7 y M4 de Elendt
Aclimatación a los medios M4 y M7 de Elendt
Algunos laboratorios han tenido dificultades para transferir directamente las dafnias a los medios M4 y M7. Sin embargo, se han obtenido resultados bastante buenos con una aclimatación gradual, esto es, cambiando del medio propio a Elendt al 30 %, luego al 60 % y por último al 100 %. El período de aclimatación necesario puede ser bastante largo, incluso de un mes.
Preparación
Oligoelementos
Se empieza por preparar soluciones madre distintas (I) de cada oligoelemento en agua de pureza apropiada (desionizada, destilada o tratada mediante ósmosis inversa). A partir de estas soluciones madre (I) se prepara una segunda solución madre única (II) que contiene todos los oligoelementos (solución combinada), de la forma siguiente:
|
Soluciones madre I (una sola sustancia) |
Cantidad añadida al agua (mg/l) |
Concentración (en relación con el medio M4) |
Para preparar la solución madre II combinada, se añade al agua la siguiente cantidad de solución madre I (ml/l) |
|
|
M4 |
M7 |
|||
|
H3BO3 |
57 190 |
20 000 veces |
1,0 |
0,25 |
|
MnCl2.4H2O |
7 210 |
20 000 veces |
1,0 |
0,25 |
|
LiCl |
6 120 |
20 000 veces |
1,0 |
0,25 |
|
RbCl |
1 420 |
20 000 veces |
1,0 |
0,25 |
|
SrCl2.6H2O |
3 040 |
20 000 veces |
1,0 |
0,25 |
|
NaBr |
320 |
20 000 veces |
1,0 |
0,25 |
|
Na2 MoO4.2H2O |
1 230 |
20 000 veces |
1,0 |
0,25 |
|
CuCl2.2H2O |
335 |
20 000 veces |
1,0 |
0,25 |
|
ZnCl2 |
260 |
20 000 veces |
1,0 |
1,0 |
|
CoCl2.6H2O |
200 |
20 000 veces |
1,0 |
1,0 |
|
KI |
65 |
20 000 veces |
1,0 |
1,0 |
|
Na2 SeO3 |
43,8 |
20 000 veces |
1,0 |
1,0 |
|
NH4VO3 |
11,5 |
20 000 veces |
1,0 |
1,0 |
|
Na2 EDTA.2H2O |
5 000 |
2 000 veces |
— |
— |
|
FeSO4.7H2O |
1 991 |
2 000 veces |
— |
— |
|
Las soluciones de Na2EDTA y FeSO4 se preparan por separado, después se reúnen y se esterilizan en autoclave inmediatamente. |
||||
|
Se obtiene así: |
||||
|
2 l de solución de Fe-EDTA |
|
1 000 veces |
20,0 |
5,0 |
Medios M4 y M7
Los medios M4 y M7 se preparan a partir de la solución madre II con los macronutrientes y las vitaminas que siguen:
|
|
Cantidad añadida al agua (mg/l) |
Concentración (en relación con el medio M4) |
Cantidad de solución madre II añadida para preparar el medio (ml/l) |
|
|
M4 |
M7 |
|||
|
Solución madre II (combinación de oligoelementos) |
|
20 veces |
50 |
50 |
|
Soluciones madre de macronutrientes (una sola sustancia) |
|
|
|
|
|
CaCl2 • 2H20 |
293 800 |
1 000 veces |
1,0 |
1,0 |
|
MgSO4 • 7H2O |
246 600 |
2 000 veces |
0,5 |
0,5 |
|
KCl |
58 000 |
10 000 veces |
0,1 |
0,1 |
|
NaHCO3 |
64 800 |
1 000 veces |
1,0 |
1,0 |
|
Na2 SiO3 • 9H2O |
50 000 |
5 000 veces |
0,2 |
0,2 |
|
NaNO3 |
2 740 |
10 000 veces |
0,1 |
0,1 |
|
KH2PO4 |
1 430 |
10 000 veces |
0,1 |
0,1 |
|
K2HPO4 |
1 840 |
10 000 veces |
0,1 |
0,1 |
|
Solución madre de vitaminas combinadas |
— |
10 000 veces |
0,1 |
0,1 |
|
La solución madre de vitaminas combinadas se prepara añadiendo las 3 vitaminas a 1 litro de agua como se indica a continuación: |
||||
|
Clorhidrato de tiamina |
750 |
10 000 veces |
|
|
|
Cianocobalamina (B12) |
10 |
10 000 veces |
|
|
|
Biotina |
7,5 |
10 000 veces |
|
|
La solución madre de vitaminas combinadas se conserva congelada en pequeñas partes alícuotas. Las vitaminas se incorporan a los medios poco antes de usarlos.
|
Nota |
: |
Para evitar la precipitación de sales al preparar los medios completos, se incorporan las partes alícuotas de las soluciones madre a aproximadamente 500-800 ml de agua desionizada y a continuación se completa hasta obtener 1 litro. |
|
Nota |
: |
La primera publicación del medio M4 puede encontrarse en Elendt, B.P. (1990). Selenium deficiency in crustacea; an ultrastructural approach to antennal damage in Daphnia magna Straus. Protoplasma, 154, 25-33. |
C.3. ENSAYO DE INHIBICIÓN DE ALGAS
1. MÉTODO
1.1. INTRODUCCIÓN
El objeto de este ensayo es determinar los efectos de una sustancia sobre el crecimiento de una especie de alga verde unicelular. Ensayos relativamente breves (72 horas) pueden valorar los efectos sobre varias generaciones. Este método puede adaptarse para ser utilizado con diferentes especies de algas unicelulares, en cuyo caso se proporcionará, junto con el informe del ensayo, una descripción del método utilizado.
Este método se aplica más fácilmente a sustancias hidrosolubles las cuales, en las condiciones del ensayo, van a permanecer probablemente en el agua.
El método puede ser utilizado para sustancias que no interfieren directamente con la medición del crecimiento de las algas.
Es conveniente disponer, si es posible, de información sobre la hidrosolubilidad, presión de vapor, estabilidad química, constantes de disociación y biodegradabilidad de las sustancias antes de comenzar el ensayo.
Debe tenerse en cuenta, tanto en la planificación de la prueba como en la interpretación de sus resultados, otro tipo de información adicional {por ejemplo, fórmula estructural, el grado de pureza, la naturaleza y el porcentaje de impurezas significativas, la presencia y cantidad de aditivos y el coeficiente de reparto n-octanol/agua).
1.2. DEFINICIONES Y UNIDADES
Densidad celular: número de células por mililitro.
Crecimiento: incremento de la densidad celular durante el período de ensayo.
Tasa de crecimiento: incremento de densidad celular por unidad de tiempo.
CE50: en este ensayo, es la concentración de la sustancia de ensayo que da lugar a una reducción del 50 %, bien en el crecimiento (C50Eb) bien en la tasa de crecimiento (C50Er) con respecto al testigo.
NOEC (concentración sin efecto observado): en este método, es la mayor concentración ensayada en la que no se observa una inhibición significativa del crecimiento respecto al testigo.
Todas las concentraciones de las sustancia de ensayo se expresan en peso por volumen (miligramos por litro). Pueden expresarse también en peso por peso (mg.kg-1).
1.3. SUSTANCIAS DE REFERENCIA
Puede someterse a ensayo una sustancia de referencia para demostrar que, en las condiciones de ensayo en laboratorio, la sensiblidad de la especie utilizada no se ha modificado de forma significativa.
Si se utiliza una sustancia de referencia, deben darse los resultados en el informe del ensayo. Puede utilizarse dicromato potásico como sustancia de referencia, pero su color puede afectar a la calidad e intensidad de la luz disponibles para las células, así como a las determinaciones espectrofotométricas, si se utilizan. Se ha usado el dicromato potásico en un ensayo internacional inter-laboratorios (véanse la referencia 3 y el apéndice 2).
1.4. PRINCIPIO DEL MÉTODO DE ENSAYO
Puede realizarse un ensayo límite a 100 mg/1 a fin de demostrar que la CE50 es mayor que esa concentración.
Se exponen cultivos de algas verdes seleccionadas en fase exponencial de crecimiento a diversas concentraciones de la sustancia de ensayo durante varias generaciones bajo condiciones definidas.
Las soluciones de ensayo se incuban durante un período de 72 horas, durante el cual se mide la densidad celular en cada una de ellas al menos cada 24 horas. Se determina la inhibición del crecimiento en relación a un cultivo testigo.
1.5. CRITERIOS DE CALIDAD
Los criterios de calidad se aplicarán tanto en el ensayo límite como en todo el método de ensayo.
La densidad celular en los cultivos testigo debe aumentar en un factor de al menos 16 en 3 días.
La concentración de la sustancia de ensayo debe mantenerse dentro del 80 % de la concentración inicia] durante toda la duración del ensayo.
En el caso de sustancias que se disuelven fácilmente en el medio de ensayo, produciendo soluciones estables, es decir, que no se volatilizan, degradan, hidrolizan ni adsorben en cantidad significativa, la concentración inicial puede considerarse como equivalente a la concentración nominal. Debe evidenciarse que se han mantenido las concentraciones a lo largo del ensayo y de que se han satisfecho los criterios de calidad.
En cuanto a las sustancias
|
i) |
poco solubles en el medio de ensayo, o |
|
ii) |
capaces de formar emulsiones o dispersiones estables, o |
|
iii) |
no estables en solución acuosa, |
se tomará como concentración inicial la concentración medida en la solución al comienzo del ensayo. La concentración se determinará tras un período de equilibrio.
En cualquiera de estos casos se harán mediciones complementarias, durante el ensayo, con el fin de confirmar la concentración de exposición real o que se han mantenido los criterios de calidad.
Se admite que durante el período del ensayo se incorporan a la biomasa algal, cantidades significativas de la sustancia de ensayo. Por ello, a fin de demostrar conformidad con los criterios de calidad anteriormente mencionados, se tendrá en cuenta tanto la cantidad de sustancia incorporada a la biomasa algal como la sustancia en solución (o, si no es posible técnicamente, medida en la columna de agua). No obstante, como la determinación de la concentración de la sustancia en la biomasa algal puede plantear problemas técnicos importantes, puede demostrarse que se han cumplido los criterios de calidad con un recipiente de ensayo que contenga la sustancia a la concentración más elevada pero sin algas y midiendo las concentraciones en la solución (o, si no es posible técnicamente, en la columna de agua) al comienzo y al final del período de ensayo.
1.6. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
1.6.1. Reactivos
1.6.1.1. Soluciones de las sustancias de ensayo
Las soluciones madre a las concentraciones requeridas se preparan disolviendo la sustancia en agua desionizada o en agua que responda a las condiciones establecidas en el punto 1.6.1.2.
Las concentraciones de ensayo elegidas se preparan añadiendo partes alícuotas adecuadas a precultivos de algas (véase el apéndice 1).
En general, las sustancias se ensayarán solo hasta el límite de su solubilidad. En el caso de algunas sustancias (por ejemplo, aquellas que son poco hidrosolubles, que tienen un elevado Pow o que forman en el agua dispersiones estables más que verdaderas soluciones), se puede aceptar una concentración de ensayo por encima del límite de solubilidad de la sustancia a fin de garantizar que se obtiene la máxima concentración soluble/estable. Es importante, sin embargo, que esta concentración no altere por otra parte el sistema de — ensayo (por ejemplo, formación en la superficie del agua de una película de la sustancia que impida la oxigenación del agua, etc.).
Puede utilizarse dispersión ultrasónica, solventes orgánicos, emulgentes o dispersantes para favorecer la preparación de las soluciones madre de las sustancias poco hidrosolubles o para que dichas sustancias se dispersen mejor en el medio de ensayo. Cuando se usen estas sustancias auxiliares, todas las concentraciones de ensayo deberán contener la misma cantidad de producto auxiliar y se expondrán testigos adicionales a la misma concentración del producto auxiliar que la utilizada en las series del ensayo. La concentración de estas sustancias auxiliares debe reducirse al mínimo, y en ningún caso será superior a 100 mg por litro en el medio de ensayo.
El ensayo debe realizarse sin ajustar el pH. Si se produjera un cambio significativo del pH, debe repetirse el ensayo ajusfando el pH y registrar los resultados. En ese caso, el valor del pH de la solución madre debe ajustarse al valor del pH del agua de dilución, excepto que existan razones concretas que lo impidan. Para ajustar el pH se utilizará, preferentemente, HC1 o NaOH. Este ajuste del pH debe efectuarse de tal forma que no se modifique significativamente la concentración de la sustancia de ensayo en la solución madre. Si el ajuste ocasiona una reacción química o una precipitación física de la sustancia de ensayo, deberá mencionarse en el informe.
1.6.1.2. Medio de ensayo
El agua debe ser agua destilada de buena calidad, o agua desionizada de una conductividad inferior a 5 μS.cm-1. El aparato para la destilación del agua no contendrá ningún componente de cobre.
Se recomienda el medio siguiente.
Se preparan cuatro soluciones madre, de acuerdo con la tabla siguiente. Las soluciones madre se esterilizan por filtración en membrana o por autoclave, y se conservan en la oscuridad a 4 oC. La solución madre no 4 se esterilizará solo por filtración en membrana. Estas soluciones madre se diluirán para conseguir las concentraciones finales de nutrientes en las soluciones de ensayo.
|
Nutriente |
Concentración en la solución madre |
Concentración final en la solución de ensayo |
|
Solución madre 1: macronutrientes |
||
|
NH4C1 |
1,5 g/l |
15 mg/l |
|
MgCl2.6H2O |
1,2 g/l |
12 mg/l |
|
CaCl2.2H2O |
1,8 g/l |
18 mg/l |
|
MgSO4.7H2O |
1,5 g/l |
15 mg/l |
|
KH2 PO4 |
0,16 g/l |
1,6 mg/l |
|
Solución madre: Fe-EDTA |
||
|
FeCl3.6H2O |
80 mg/l |
0,08 mg/l |
|
Na2EDTA.2H2O |
100 mg/l |
1 mg/l |
|
Solución madre 3: elementos traza |
||
|
H3BO3 |
185 mg/l |
0,185 mg/l |
|
MnCl2.4H2O |
415 mg/1 |
0,415 mg/l |
|
ZnCl2 |
3 mg/l |
3 × 10-3 mg/l |
|
CoCl2.6H20 |
1,5 mg/l |
1,5 × 10- 3 mg/l |
|
CuCl2.2H2O |
0,01 mg/l |
10-5 mg/l |
|
Na2MoO4.2H2O |
7 mg/l |
7 × 10-3 mg/l |
|
Solución madre 4: NaHCO 3 |
||
|
NaHCO3 |
50 g/l |
50 mg/l |
El pH del medio, después del equilibrio con el aire, es aproximadamente 8.
1.6.2. Equipo
|
— |
Equipo normal de laboratorio. |
|
— |
Matraces de ensayo de volumen adecuado (por ejemplo, serán adecuados matraces cónicos de 250 ml cuando el volumen de la solución de ensayo sea de 100 ml). Todos los matraces de ensayo serán idénticos en cuanto a material y dimensiones. |
|
— |
Equipo de cultivo: cámara en la que pueda mantenerse una temperatura de 21 a 25 oC con una oscilación de ± 2 oC, y con una iluminación uniforme continua, que proporcione un rango espectral de 400 a 700 nm. Si las algas en los cultivos testigo alcanzan las tasas de crecimiento recomendadas, puede suponerse que las condiciones para el crecimiento, incluida la intensidad luminosa, han sido adecuadas. Se recomienda utilizar, en el nivel medio de las soluciones de ensayo, una intensidad luminosa en el rango de 60 a 120 μE.m-2.s-1 (35 a 70 × 1018 fotones.m-2.s-1) medida en el rango de 400 a 700 nm utilizando un receptor adecuado. Cuando se utilizan instrumentos de medición de luminosidad calibrados en lux, se puede aceptar un rango equivalente de 6 000 a 10 000 lux. Puede obtenerse la intensidad luminosa utilizando de 4 a 7 lámparas fluorescentes de 30 W del tipo blanco universal (temperatura de color de aproximadamente 4 000 K), a una distancia de 0,35 m del cultivo de algas. |
|
— |
Las mediciones de densidad celular se harán utilizando un método directo de recuento de células vivas, por ejemplo, un microscopio con cámaras de recuento. No obstante, pueden utilizarse otros procedimientos (fotometría, turbidimetría, etc.) si son suficientemente sensibles y si muestran una correlación suficientemente buena con la densidad celular. |
1.6.3. Organismos de ensayo
Se sugiere que las especies de algas verdes utilizadas sean especies de crecimiento rápido adecuadas para cultivo y ensayo. Se prefieren las especies siguientes:
|
— |
Selenastrum caprkornutum, por ejemplo, ATCC 22662 or CCAP 278/4, |
|
— |
Scenedesmus subspicatus, por ejemplo, 86.81 SAG, |
Nota:.
|
ATCC |
= |
American Type Culture Collection (EEUU) |
|
CCAP |
= |
Culture Centre of Algae and Protozoa (GB) |
|
SAG |
= |
Colección de cultivos de algas (Gotinga, RFA) |
Si se utilizan otras especies, deben mencionarse las cepas en el informe.
1.6.4. Procedimiento del ensayo
Basándose en los resultados de las pruebas de tanteo se determinará el intervalo de concentraciones en el que se espera que ocurran los efectos.
Las dos medidas de crecimiento (biomasa y tasa de crecimiento) pueden dar lugar a medidas muy divergentes de inhibición de crecimiento; deben utilizarse ambas en la prueba de tanteo de rango para garantizar que la progresión geométrica de las concentraciones permitirá el cálculo de la C50Eb, y C50Er.
Densidad celular inicial
Se recomienda que la densidad celular inicial en los cultivos de ensayo sea de aproximadamente 104 células/ml para Selenastrum capricornutum y Scenedesmus subspicatus. Cuando se utilicen otras especies, la biomasa será comparable.
Concentraciones de la sustancia de ensayo
Para este ensayo se utilizarán al menos cinco concentraciones en progresión geométrica con un factor que no exceda de 2,2. La concentración mínima ensayada no tendrá efectos observables sobre el crecimiento de las algas. La concentración máxima ensayada inhibirá el crecimiento al menos un 50 % en relación con el testigo y, preferentemente, detendrá completamente el crecimiento.
Réplicas y controles
Cada concentración de ensayo incluirá tres réplicas. Se harán tres controles sin sustancia de ensayo y, si procede, tres controles que contengan la sustancia auxiliar. La prueba podrá modificarse, si se justifica, para aumentar el número de concentraciones y reducir el número de réplicas por concentración.
Realización del ensayo
Los cultivos de ensayo que contienen las concentraciones deseadas de la sustancia de ensayo y la cantidad deseada de inóculo de algas se preparan añadiendo partes alícuotas de soluciones madre de la sustancia de ensayo a cantidades adecuadas de precultivosde algas (véase el apéndice 1).
Se agitan los matraces de ensayo y se colocan en el aparato de cultivo. Se mantienen las algas en suspensión mediante agitación, removiendo o con burbujeo dé aire, para facilitar el intercambio de gases y reducir la variación del pH en las soluciones de ensayo. Los cultivos deberán mantenerse a una temperatura entre los 21 y los 25 oC, con variaciones máximas de ± 2 oC.
La densidad celular en cada matraz se determinará al menos a las 24, 48 y 72 horas después del comienzo del ensayo. Se utilizará el medio de cultivo de algas filtrado conteniendo la concentración adecuada del producto de ensayo para determinar el fondo, cuando se utilicen mediciones de densidad celular distintas de los métodos de recuento directo.
El pH se medirá al comienzo del ensayo y a las 72 horas.
El pH de los controles no debe desviarse, en general, más de 1,5 unidades durante el ensayo.
Ensayo de sustancias volátiles
Actualmente, no existe una forma generalmente aceptada para ensayar sustancias volátiles. Cuando se conozca que una sustancia tiende a volatilizarse, se podrán utilizar matraces cerrados con aumento del espacio superior (de cabeza). Hay que tener en cuenta la posibilidad de que falte CO2 cuando se calcule el espacio superior (de cabeza) de los matraces cerrados. Se han propuesto variaciones a este método [véase la referencia (4)].
Se debe intentar determinar la cantidad de sustancia que permanence en solución, y se aconseja gran precaución cuando se interpreten las ensayos de las pruebas con sustancias químicas si se usan sistemas cerrados.
Ensayo limite
Utilizando los procedimientos descritos en este método, puede realizarse un ensayo límite a 100 mg por litro a fin de demostrar que la CE50 es mayor que esa concentración.
Si la naturaleza de la sustancia no permite alcanzar una concentración de 100 mg por litro en el agua de ensayo, el ensayo límite debe ser realizado a una concentración igual a la solubilidad de la sustancia (o a la concentración máxima que forme una dispersión estable) en el medio usado (véase también el punto 1.6.1.1).
El ensayo límite debe ser realizado al menos por triplicado, con el mismo número de testigos. Deben usarse para el ensayo límite las dos medidas de crecimiento (biomasa y tasa de crecimiento).
Si se encuentra en un ensayo límite una disminución media igual o superior al 25 % en la biomasa o en la tasa de crecimiento, en relación con el testigo, debe llevarse a cabo un ensayo completo.
2. RESULTADOS Y EVALUACIÓN
Se tabularán las medidas de densidad celular en los cultivos de ensayo y en los controles, junto con las concentraciones de la sustancia de ensayo y los tiempos a los que se realizan las determinaciones. Se hará una gráfica del valor medio de la densidad celular para cada concentración de sustancia de ensayo, así como para los testigos, en función del tiempo (0-72 horas), a fin de conseguir curvas de crecimiento.
Para determinar la relación concentración/efecto, pueden utilizarse los dos enfoques siguientes. Algunas sustancias pueden estimular el crecimiento a bajas concentraciones. Solo se tendrán en cuenta los datos que indiquen una inhibición entre 0 y 100 %.
2.1. COMPARACIÓN DE LAS ÁREAS BAJO LAS CURVAS DE CRECIMIENTO
El área entre las curvas de crecimiento y la línea horizontal N = No puede calcularse de acuerdo con la fórmula:

donde:
|
A |
= |
área |
|
No |
= |
número de células/ml en el momento t0 (comienzo del ensayo, |
|
N1 |
= |
número medido de células/ml a t1, |
|
Nn |
= |
número medido de células/ml a tn, |
|
t1 |
= |
tiempo en que se realiza la primera medida tras el comienzo del ensayo, |
|
tn |
= |
tiempo en que se realiza la medida n tras el comienzo del ensayo, |
|
n |
= |
número de medidas hechas tras el comienzo del ensayo. |
El porcentaje de inhibición del crecimiento celular para cada concentración de sustancia de ensayo (IA) se calculará de acuerdo con la fórmula:
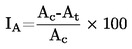
donde:
|
Ac |
= |
área entre la curva de crecimiento del testigo y la línea horizontal N = No. |
|
At |
= |
área entre la curva de crecimiento a la concentración t y la línea horizontal N = No. |
|
Los valores IA |
= |
se representarán en una gráfica en papelsemilogarítmico o en papel probit semilogarítmico en función de las concentraciones correspondientes. Si la gráfica se hace en papel probit, se unirán los puntos con una línea recta, bien de forma aproximada o mediante una regresión por ordenador. |
La CE50 se calcula a partir de la línea de regresión leyendo la concentración equivalente a la inhibición del 50 % (IA = 50 %). Para expresar este valor de forma inequívoca en relación a este método de cálculo, se propone utilizar el símbolo C 50Eb . ES esencial que C 50Eb vaya acompañada del período adecuado de exposición, por ejemplo, C50Eb (0-72 horas).
2.2. COMPARACIÓN DE LAS TASAS DE CRECIMIENTO
La tasa media de crecimiento específico (μ) de los cultivos con crecimiento exponencial puede calcularse como

donde t0 es el momento del comienzo del ensayo.
Por otra parte, la tasa media de crecimiento específico puede derivarse de la pendiente de la línea de regresión en una gráfica de In N en función del tiempo.
El porcentaje de inhibición de la tasa de crecimiento específico para cada concentración de la sustancia de ensayo (Iμr) se calcula de acuerdo con la fórmula:

donde
|
μc |
= |
tasa media de crecimiento específico de los testigos |
|
μt |
= |
tasa media de crecimiento específico a la concentración de ensayo t. |
El porcentaje de inhibición de la tasa media de crecimiento específico para cada concentración de sustancia de ensayo en relación con el valor de los testigos se representará en una gráfica en función de logaritmo de la concentración. La CE50 puede leerse en la gráfica resultante. Para expresar de forma inequívoca la CE50 obtenida por este método se propone utilizar el símbolo C50Er. Deben indicarse los momentos de medición, por ejemplo, si el valor de refiere a los momentos 0 y 72 horas, el símbolo será C50Er (0-72 horas).
Nota: La tasa de crecimiento específico es un término logarítmico, y pequeños cambios en la tasa de crecimiento pueden dar lugar a grandes cambios en la biomasa. Por ello, CEb y CEr son valores que no pueden compararse numéricamente.
2.3. CÁLCULO DE LA NOEC
La concentración sin efecto observado se determina mediante un procedimiento estadístico adecuado por comparación de muestras múltiples (por ejemplo, análisis de la varianza y prueba de Dunnett), utilizando los valores de las áreas bajo las curvas de crecimiento A (véase el punto 2.1) o las tasas de crecimiento específico μ (véase el punto 2.2) correspondientes a cada réplica individual.
3. INFORME
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
sustancia de ensayo: datos de identificación química, |
|
— |
organismos de ensayo: origen, cultivo de laboratorio, número de cepa, método de cultivo, |
|
— |
condiciones del ensayo:
|
|
— |
resultados:
|
4. REFERENCIAS
|
(1) |
OCDE, París, 1981, Test Guideline 201, Decision of the Council C(81)30 Final. |
|
(2) |
Umweltbundesamt, Berlin, 1984, Verfahrensvorschlag «Hemmung der Zellvermehrung bei der Grünalge Scenedesmus subspicatus», in: Rudolph/Boje: Ökotoxikologie, ecomed, Landsberg, 1986. |
|
(3) |
ISO 8692 — Water quality — Fresh water algal growth inhibition test with Scenedesmus subspicatus and Selenastrum capricornutum. |
|
(4) |
S. Galassi y M. Vighi — Chemosphere, 1981, vol. 10, 1123-1126. |
Apéndice 1
Ejemplo de procedimiento para el cultivo de algas
Observaciones generales
El objetivo del cultivo mediante el siguiente procedimiento es el de obtener cultivos de algas para ensayos de toxicidad.
Deberán utilizarse métodos apropiados que garanticen que los cultivos de algas no están contaminados por bacterias (ISO 4833). Puede ser conveniente utilizar cultivos axénicos, pero serán esenciales los cultivos de una sola especie de alga.
Todas las operaciones deberán llevarse a cabo en condiciones estériles, con el fin de evitar la contaminación por bacterias u otras especies de algas. Se eliminarán los cultivos contaminados.
Procedimiento para la obtención de algas
Preparación de soluciones nutritivas (medios)
El medio se puede preparar por dilución de las soluciones madre concentradas de nutrientes. Para un medio sólido, se añadirá un 0,8 % de agar. El medio utilizado será estéril. La esterilización por autoclave puede dar pérdida de NH3.
Cultivos de inóculo
Los cultivos de inóculo son pequeños cultivos de algas que se trasfieren periódicamente a un medio de cultivo fresco para que actúen como elemento inicial del ensayo. Si los cultivos no se utilizan regularmente, se mantienen en tubos de agar inclinado y se transfieren a un medio fresco al menos cada dos meses.
Los cultivos de inóculo se cultivan en matraces cónicos que contengan el medio apropiado (un volumen alrededor de 100 ml). Cuando las algas se incuban a 20 oC con iluminación continua, será necesario hacer una transferencia semanal.
Durante la transferencia se llevará, con pipetas estériles, una cantidad de cultivo «viejo» a matraces con medio fresco, de manera que, con las especies de crecimiento rápido, la concentración inicial sea unas 100 veces menor que en el cultivo viejo.
La tasa de crecimiento de una especie puede determinarse a partir de la curva de crecimiento. Si se conoce, será posible calcular la densidad a la cual deberá transferirse el cultivo al nuevo medio. Esto deberá realizarse antes de que el cultivo alcance su fase letal.
Precultivo
El precultivo está concebido para obtener una cantidad apropiada de algas para la inoculación de los cultivos de ensayo. Dicho precultivo deberá incubarse en las condiciones del ensayo y se utilizará cuando esté aún en fase de crecimiento exponencial, normalmente después de un período de incubación de 3 días. Si los cultivos de algas contienen células deformadas o anormales, deberán eliminarse.
Apéndice 2
La norma ISO 8692, «Calidad del agua; prueba de inhibición del crecimiento de algas en agua dulce utilizando Scenedesmus subspicatus y Selenastrum capricornutum», da los siguientes resultados en una prueba multicéntrica llevada a cabo por 16 laboratorios, utilizando como sustancia de prueba el dicromato potásico.
|
|
Medias (mg/l) |
Rango (mg/l) |
|
C50Er (0-72 h) |
0,84 |
de 0,60 a 1,03 |
|
C50Eb (0-72 h) |
0,53 |
de 0,20 a 0,75 |
C.4. DETERMINACIÓN DE LA BIODEGRADABILIDAD «FACIL»
PARTE I. CONSIDERACIONES GENERALES
I.1. INTRODUCCIÓN
Se describen seis métodos de ensayo que permiten detectar la biodegradabilidad fácil de los productos químicos en medio acuoso aerobio;
|
a) |
pérdida de carbono orgánico disuelto (COD) (Método C.4-A); |
|
b) |
prueba de detección de la OCDE modificada — Pérdida de COD (Método C.4-B); |
|
c) |
desprendimiento de dióxido de carbono (CO2) (prueba de Sturm modificada) (Método C.4-C); |
|
d) |
respirometría manométrica (Método C.4-D); |
|
e) |
frasco cerrado (Método C.4-E); |
|
f) |
MITI (Ministerio de Industria y Comercio Internacional de Japón) (Método C.4-F). |
En la Parte I del presente método se recogen consideraciones generales y consideraciones comunes a los seis ensayos. Los aspectos específicos de los distintos métodos se tratan en las partes II a VII. Los anexos contienen definiciones, fórmulas y material complementario.
Un estudio comparativo interlaboratorios, realizado por la OCDE en 1988, demostró la homogeneidad de los resultados obtenidos con estos métodos. Sin embargo, las características físicas de la sustancia problema pueden hacer que se prefiera un método a los otros.
I.2. SELECCIÓN DEL MÉTODO ADECUADO
Para seleccionar el método más adecuado es fundamental poseer información sobre la solubilidad, presión de vapor y características de absorción del producto químico. Debería conocerse la estructura química o la fórmula para calcular los valores teóricos o comprobar los valores obtenidos en las mediciones de parámetros, como DTO, CO2T, COD, COT o DQO (véanse los anexos I y II).
Las sustancias problema con una hidrosolubilidad no inferior a 100 mg/l pueden estudiarse con cualquier método, siempre que no sean volátiles ni sufran absorción. En el cuadro 1 se indican los métodos adecuados para los productos químicos poco hidrosolubles, volátiles o que sufran absorción. En el anexo III se describe cómo pueden tratarse las sustancias poco hidrosolubles y las volátiles. Las sustancias moderadamente volátiles pueden estudiarse con el método de pérdida de COD si hay bastante espacio para el gas en los recipientes utilizados (que debieran estar convenientemente cerrados). En este caso, es necesario utilizar un control abiótico para tener en cuenta las posibles pérdidas físicas.
Cuadro 1
Aplicabilidad de los métodos de ensayo
|
Prueba |
Método analítico |
Adecuación para sustancias: |
||
|
poco solubles |
volátiles |
absorbibles |
||
|
Pérdida de COD |
Carbono orgánico disuelto |
— |
— |
+/- |
|
Pérdida OECD modificada |
Carbono orgánico disuelto |
— |
— |
+/- |
|
Desprendimiento de CO2 |
Respirometría: desprendimiento de CO2 |
+ |
— |
+ |
|
Respirometría manométrica |
Respirometría manométrica: consumo de oxígeno |
+ |
+/- |
+ |
|
Frasco cerrado |
Respirometría: oxígeno disuelto |
+/- |
+ |
+ |
|
MITI |
Respirometría: consumo de oxígeno |
+ |
+/- |
+ |
Es necesario disponer de información sobre la pureza o las proporciones relativas de los principales componentes del producto problema para interpretar los resultados obtenidos, especialmente cuando estos son bajos o dudosos.
El disponer de datos sobre la toxicidad de la sustancia problema para bacterias (anexo IV) puede ser muy útil para seleccionar las concentraciones adecuadas para el ensayo y puede ser fundamental para interpretar correctamente los valores bajos de la biodegradación.
I.3. SUSTANCIAS DE REFERENCIA
Para comprobar el procedimiento se someten a ensayo sustancias de referencia que cumplan los criterios de biodegradabilidad fácil; para ello se introduce, en paralelo con el ensayo a realizar, una muestra con la sustancia de referencia adecuada.
La anilina (recién destilada), el acetato sódico y el benzoato sódico son sustancias adecuadas. Todas estas sustancias de referencia se degradan con estos métodos aunque no se añada inóculo deliberadamente.
Se pensó en buscar una sustancia de referencia que fuera fácilmente biodegradable pero que necesitara para su biodegradación la adición de inóculo. Se ha propuesto el uso del ftalato ácido de potasio pero es necesario disponer de más información sobre esta sustancia antes de que pueda ser aceptada como sustancia de referencia.
En el ensayo respirométrico, los compuestos que contengan nitrógeno pueden afectar a la captación de oxígeno debido a la nitrificación (véanse los anexos II y V).
I.4. PRINCIPIO DE LOS MÉTODOS DE ENSAYO
Se inocula una solución o suspensión de la sustancia problema en un medio mineral y se incuba en condiciones aerobias en la oscuridad o bajo luz difusa. La cantidad de COD en la solución problema debida al inóculo debería mantenerse lo más baja posible respecto a la cantidad de COD debida a la sustancia problema. Para tener en cuenta la actividad endógena del inóculo, se realizan ensayos paralelos en blanco, que contienen inóculo pero sin sustancia problema, si bien la actividad endógena de las células en presencia de la sustancia no corresponderá exactamente con la del control endógeno. Se trabajará en paralelo con una sustancia de referencia para controlar el funcionamiento de los procedimientos.
En general, la degradación se sigue mediante la determinación de parámetros, como el COD, la producción de CO2 y el consumo de oxígeno, tomando medidas con la frecuencia suficiente para permitir la identificación del comienzo y de la finalización de la biodegradación. Con los respirómetros automáticos, la medición es continua. El COD se mide a veces junto con otro parámetro, pero esto suele hacerse solo al principio y al final del ensayo. También puede utilizarse el análisis químico específico para evaluar la degradación primaria de la sustancia problema y determinar la concentración de las sustancias intermedias formadas eventualmente (esto es obligatorio en la prueba del MITI).
El ensayo dura normalmente 28 días. Sin embargo, es posible terminar los ensayos antes del día 28, por ejemplo en el caso de que la curva de biodegradación haya alcanzado un nivel constante en, al menos, 3 determinaciones. También es posible prolongar los ensayos mas de 28 días cuando la curva indique que la biodegradación se ha iniciado pero sin que se haya alcanzado el nivel constante el día 28.
I.5. CRITERIOS DE CALIDAD
I.5.1. Reproducibilidad
Debido a la naturaleza de la biodegradación y de las poblaciones bacterianas mixtas utilizadas como inóculos, las determinaciones deben realizarse al menos por duplicado.
La experiencia indica que la variación entre duplicados será menor cuanto mayor sea la concentración de microorganismos añadida inicialmente al medio de ensayo. También se ha visto en estudios interlaboratorios que pueden darse grandes variaciones entre los resultados obtenidos por diferentes laboratorios; no obstante, con los compuestos químicos que son fácilmente biodegradables se obtienen normalmente resultados concordantes.
I.5.2. Validez del ensayo
Un ensayo se considera válido si la máxima diferencia entre los duplicados respecto a los valores de la eliminación de la sustancia problema en la parte de la gráfica en forma de meseta, al final del test o al final del período de diez días, es inferior al 20 % y si la degradación porcentual de la sustancia de referencia alcanza el nivel de biodegradabilidad fácil antes de los 14 días. Si no se cumple alguna de estas condiciones, es necesario repetir el ensayo. Debido al rigor de los métodos, la obtención de bajos porcentajes de biodegradación no significa necesariamente que la sustancia problema no sea biodegradable en condiciones ambientales, sino que indica que será necesario realizar más estudios para establecer la biodegradabilidad.
Si en un ensayo de toxicidad, realizado con la sustancia problema junto con una sustancia de referencia, se obtiene en 14 días menos del 35 % de degradación (según el COD) o menos del 25 % (según la DTO o el CO2T), se supone que la sustancia de ensayo es inhibidora (véase también el anexo IV). La serie de ensayo debe repetirse, a ser posible, utilizando una concentración inferior de sustancia problema o una concentración superior de inóculo, que no exceda de 30 mg de sólidos/litro.
I.6. PREPARATIVOS Y PROCEDIMIENTOS GENERALES
En el cuadro 2 se resumen las condiciones generales aplicables a los ensayos. El material y las demás condiciones experimentales relativas específicamente a un ensayo concreto se describen después bajo el título de cada ensayo.
Cuadro 2
Condiciones de los ensayos
|
Ensayo |
Perdida de DOC |
Desprendimiento de CO2 |
Respiración manométrica |
Detección de la OCDE modificada |
Frasco cerrado |
MITI-(I) |
|||||
|
Concentración de sustancia problema |
|
|
|
|
|
|
|||||
|
mg/l |
|
|
100 |
|
2-10 |
100 |
|||||
|
mg COD/l |
10-40 |
10-20 |
|
10-40 |
|
|
|||||
|
mg DTO/l |
|
|
50-100 |
|
5-10 |
|
|||||
|
Concentración de inóculo (en células/l, aproximadamente) |
≤ 30 mg/l SS o ≤ 100 ml efluente/l (107-108) |
0,5 ml efluente secundario/l (105) |
≤ 5 ml efluente/l (104-106) |
30mg/l SS (107-108) |
|||||||
|
Concentración de elementos en el medio mineral (en mg/1): |
|
|
|
|
|
|
|||||
|
P |
116 |
11,6 |
29 |
||||||||
|
N |
1,3 |
0,13 |
1,3 |
||||||||
|
Na |
86 |
8,6 |
17,2 |
||||||||
|
K |
122 |
12,2 |
36,5 |
||||||||
|
Mg |
2,2 |
2,2 |
6,6 |
||||||||
|
Ca |
9,9 |
9,9 |
29,7 |
||||||||
|
Fe |
0,05-0,1 |
0,05-0,1 |
0,15 |
||||||||
|
pH |
7,4±0,2 |
preferentemente 7,0 |
|||||||||
|
Temperatura |
22 ± 2 oC |
25 ± 1 oC |
|||||||||
|
|
|
I.6.1. Agua de dilución
Se usará agua desionizada o destilada, exenta de sustancias tóxicas (por ejemplo, iones Cu+ +) a concentraciones inhibidoras. No debe contener más del 10 % del contenido en carbono orgánico introducido por la sustancia problema. Esta elevada pureza del agua es necesaria para no obtener valores elevados en la prueba en blanco. La contaminación puede proceder de impurezas inherentes, y también, de las resinas cambiadoras de iones y de residuos materiales procedentes de bacterias y algas. Para cada serie de ensayos debe utilizarse un solo lote de agua, comprobado previamente mediante análisis de COD. Esta comprobación no es necesaria para el ensayo del frasco cerrado, pero el consumo de oxígeno del agua debe ser bajo.
I.6.2. Soluciones madre de elementos minerales
Las soluciones de ensayo se prepararán a partir de soluciones madre de elementos minerales en concentración adecuada. Las siguientes soluciones madre pueden utilizarse (con diferentes factores de dilución) en los métodos de pérdida de COD, detección de la OCDE modificada, desprendimiento de CO2, respirometria manométrica y frasco cerrado.
Los factores de dilución y, en el caso de la prueba MITI, la preparación específica del medio mineral se recogen bajo el encabezamiento de cada prueba específica.
Soluciones madre:
Hay que preparar las siguientes soluciones madre, utilizando reactivos de grado analítico.
|
a) |
Ortofosfato diácido de potasio, KH2PO4 |
8,50 g |
|
Ortofosfato ácido de potasio, K2HPO4 |
21,75 g |
|
|
Ortofosfato ácido de, sodio dihidratado Na2HPO4 . 2 H2O |
33,40 g |
|
|
Cloruro amónico, NH4CL |
0,50 g |
|
|
Disolver en agua y enrasar a 1 litro. El pH de la solución debe ser 7,4 |
|
|
|
b) |
Cloruro calcico, anhidro, CaCl2 o |
27,50 g |
|
Cloruro cálcico dihidratado, CaCl2 - 2 H2O |
36,40 g |
|
|
Disolver en agua y enrasar a 1 litro |
|
|
|
c) |
Sulfato magnésico heptahidratado, MgSO4 . 7 H2O |
22,50 g |
|
Disolver en agua y enrasar a 1 litro |
|
|
|
d) |
Cloruro de hierro (III) hexahidratado, FeCl3. 6 H2O |
0,25 g |
|
Disolver en agua y enrasar a 1 litro |
|
Nota: Con el fin de no tener que preparar esta solución inmediatamente antes de su uso, añádase una gota de HC1 concentrado o 0,4 g de EDTA (sal disódica del ácido etilendiaminotetraacético) por litro.
I.6.3. Soluciones madre de productos químicos
Por ejemplo, disolver de 1 a 10 g, según se considere adecuado, de sustancia problema o de referencia en agua desionizada y enrasar a 1 litro cuando la solubilidad sea superior a 1 g/l. En caso contrario, preparar soluciones madre en el medio mineral o añadir los productos químicos directamente en el medio mineral. Para el tratamiento de productos químicos menos solubles, véase el anexo III, pero teniendo en cuenta que en la prueba del MITI (Método C.4-F) no pueden utilizarse ni disolventes ni emulgentes.
I.6.4. Inóculos
El inóculo puede proceder de diversas fuentes: lodo activado, aguas residuales efluentes (no cloradas), aguas superficiales, suelos o una mezcla de todo ello. En el caso de los ensayos de pérdida de COD, desprendimiento del CO2 y respirometría manométrica, si se utiliza lodo activado, este debe proceder de una planta depuradora o de una instalación de laboratorio que reciba predominantemente aguas residuales domésticas. Se ha visto que los inóculos procedentes de otras fuentes dan resultados más dispersos. En el caso de la prueba de detección de la OCDE modificada y de la prueba de frasco cerrado, es necesario un inóculo más diluido sin flóculos de lodo y la fuente preferida es un efluente secundario procedente de una depuradora de aguas residuales domésticas o de una instalación de laboratorio. En el caso del método MITI, el inóculo se obtiene mezclando material procedente de distintas fuentes y se describe bajo el encabezamiento específico de esta prueba.
I.6.4.1. Inóculo procedente de lodos activados
Se recoge una muestra de lodo activado recién obtenido del depósito de aireación de una planta depuradora de aguas residuales o de una instalación de laboratorio que trate predominantemente aguas residuales domésticas. En caso necesario, se retiran las partículas gruesas, por filtración a través de un tamiz fino y a continuación se mantiene el lodo en condiciones aerobias.
Otra posibilidad es dejar sedimentar o centrifugar (por ejemplo, a 1 100 g durante 10 minutos) después de eliminar las posibles partículas gruesas. Se desecha el sobrenadante. El lodo puede ser lavado en el medio mineral. Se suspende el lodo concentrado en medio mineral para obtener una concentración de 3 a 5 g de sólidos suspendidos/1 y se somete a aireación hasta que se utilice.
El lodo debería obtenerse de una planta convencional que trabaje adecuadamente. Si tiene que obtenerse el lodo de una planta depuradora de elevado rendimiento o cuando se píense que contiene inhibidores, debería lavarse el lodo. El lodo resuspendido se mezcla bien y se deja sedimentar o se centrifuga, se desecha el sobrenadante del lodo lavado y se vuelve a resuspender en otro volumen de medio mineral. Este procedimiento se repite hasta que se considere que el lodo queda exento de un exceso de sustrato o de inhibidores.
Después de conseguir la resuspensión completa, o con el lodo sin tratar, se toma una muestra justo antes de su utilización para determinar el peso seco de los sólidos en suspensión.
Otra posibilidad diferente es homogeneizar el lodo activado (de 3 a 5 g de sólidos suspendidos/1). Se trata el lodo en un agitador mecánico durante 2 minutos a velocidad media. El lodo agitado se deja sedimentar durante 30 minutos, o más en caso necesario, y se decanta el líquido para utilizarlo como inóculo en la proporción de 10 ml/l de medio mineral.
I.6.4.2. Otras fuentes de inóculos
Este inóculo puede obtenerse a partir del efluente secundario de una planta depuradora o de una instalación de laboratorio que reciba predominantemente aguas residuales domésticas. Se toma una muestra reciente y se mantiene en condiciones aerobias durante el transporte. Se deja sedimentar durante 1 hora o se filtra a través de papel de filtro grueso y el efluente decantado o el filtrado se mantiene en condiciones aerobias hasta su utilización. Pueden utilizarse hasta 100 ml de este tipo de inóculo por cada litro de medio.
Otra fuente adicional de inóculo es el agua superficial. En este caso, se recoge una muestra de un agua superficial adecuada como, por ejemplo, ríos o lagos, y se mantiene en condiciones aerobias hasta su utilización. En caso necesario, se concentra el inóculo por filtración o centrifugación.
I.6.5. Acondicionamiento previo de los inóculos
Los inóculos pueden estar preacondicionados a las condiciones experimentales, pero no preadaptados a la sustancia problema. El acondicionamiento previo consiste en la aireación del lodo activado en medio mineral o bien del efluente secundario durante 5 o 7 días a la temperarura del ensayo. El acondicionamiento previo mejora a veces la precisión de los métodos de ensayo reduciendo los valores del blanco. El acondicionamiento previo se considera innecesario para el inóculo del MITI.
I.6.6. Controles abióticos
Cuando sea necesario, se estudiará la posible degradación abiótica de la sustancia problema determinando la eliminación de COD, el consumo de oxígeno o el desprendimiento de dióxido de carbono en controles estériles que no contengan inóculo. La esterilización se consigue mediante filtración a través de membrana (0,2-0,45 micrómetros) o mediante la adición de una sustancia tóxica adecuada a la concentración conveniente. Si se utiliza una membrana de filtración, coger las muestras de forma aséptica para mantener la esterilidad. A no ser que la absorción de la sustancia problema haya sido excluida de antemano, las pruebas en las que se mide la biodegradación como la eliminación de COD, especialmente con lodo activado como inóculo, debieran incluir un control abiótico inoculado v envenenado.
I.6.7. Número de frascos
El número de frascos en un ensayo normal se describe bajo el encabezamiento de cada ensayo.
Pueden utilizarse los siguientes tipos de frasco:
|
— |
Suspensión de ensayo: conteniendo la sustancia problema y el inóculo. |
|
— |
Inóculo aislado: conteniendo únicamente inóculo. |
|
— |
Control del procedimiento: conteniendo sustancia problema e inóculo. |
|
— |
Control estéril abiótico: estéril, conteniendo la sustancia problema (véase el punto I.6.6). |
|
— |
Control de absorción: conteniendo la sustancia problema, inóculo y agente esterilizante. |
|
— |
Control de toxicidad: conteniendo la sustancia problema, la sustancia de referencia e inóculo. |
La determinación en la suspensión de ensayo y la determinación en el inóculo aislado deberían realizarse obligatoriamente en paralelo. Es aconsejable realizar las determinaciones en los otros frascos también en paralelo.
Sin embargo, esto puede no ser siempre posible. Hay que asegurarse de que se toman bastantes muestras o lecturas para tener la certeza de que se alcanza el porcentaje de eliminación adecuado durante el período de tiempo de 10 días.
I.7 DATOS Y EVALUACIÓN
Para el cálculo de Dt, degradación porcentual, se utilizan los valores medios de las medidas de los parámetros, realizadas por duplicado en los dos recipientes del ensayo y en el blanco. Las fórmulas vienen dadas en las secciones específicas correspondientes a cada ensayo. El curso de la degradación se representa gráficamente y se indica el período de observación de 10 días. Se calcula y se registra la pérdida porcentual al final del período de observación de 10 días y el valor en la fase estacionaria o al final del ensayo, según se considere adecuado.
En los ensayos respirométricos, los compuestos nitrogenados pueden afectar al consumo de oxígeno debido a la nitrificación (véanse los anexos II y V).
I.7.1. Medida de la degradación basada en la determinación del COD
El porcentaje de degradación (Dt) en cada momento en que se tome una muestra debiera calcularse separadamente para los frascos conteniendo la sustancia problema, utilizando los valores medios de las medidas por duplicado del COD con el fin de valorar la validez de la prueba (véase el punto 1.5.2). Se calcula utilizando la siguiente ecuación:

donde:
|
Dt |
= |
degradación porcentual en el tiempo t. |
|
Co |
= |
concentración media inicial de COD en el medio de cultivo inoculado con la sustancia problema (mg COD/l), |
|
Ct |
= |
concentración media de COD en el medio de cultivo inoculado con sustancia problema en el tiempo t (mg COD/l), |
|
Cbo |
= |
concentración media inicial de COD en el medio mineral en blanco inoculado (mg COD/l), |
|
Cbt |
= |
concentración media de COD en el medio mineral en blanco inoculado, en el tiempo t (mg COD/l). |
Todas las concentraciones son las obtenidas experimentalmente.
I.7.2. Degradación medida por análisis específicos
Cuando se tienen datos analíticos específicos, la biodegradación primaria se calcula con la fórmula siguiente:
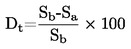
donde:
|
Dt |
= |
degradación porcentual en el tiempo t, normalmente 28 días |
|
Sa |
= |
cantidad residual de sustancia problema en el medio inoculado al final de la prueba (mg) |
|
Sb |
= |
cantidad residual de sustancia problema en la prueba en blanco con agua o medio a los que solo se ha añadido la sustancia problema (mg). |
I.7.3. Degradación abiótica
Cuando se utilice un control abiótico estéril, calcular el porcentaje de degradación abiótica utilizando:
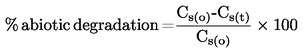
Donde:
|
Cs(o) |
= |
concentración COD en el control estéril el día 0, |
|
Cs(t) |
= |
concentración COD en el control estéril el día t. |
I.8. INFORME
El informe del ensayo incluirá, a ser posible, la siguiente información:
|
— |
sustancias problema y de referencia así como su pureza, |
|
— |
condiciones del ensayo, |
|
— |
inóculo: naturaleza y lugar o lugares de recogida, concentración y posible tratamiento de acondicionamiento previo, |
|
— |
proporción y naturaleza de los residuos industriales presentes en las aguas residuales, si se conocen, |
|
— |
tiempo de duración del ensayo y temperatura a la que se ha realizado, |
|
— |
en caso de sustancias problema poco solubles, tratamiento realizado, |
|
— |
método de ensayo utilizado; motivos científicos y explicación en caso de que se haya realizado algún cambio en el procedimiento, |
|
— |
ficha de recogida de datos, |
|
— |
cualquier fenómeno de inhibición que se haya observado, |
|
— |
cualquier degradación abiótica que se haya observado, |
|
— |
datos químicos analíticos específicos, si se tienen, |
|
— |
datos analíticos sobre los intermediarios, si se tienen, |
|
— |
gráfica de la degradación porcentual frente al tiempo para las sustancias problema y de referencia; hay que indicar claramente la fase de latencia, la fase de degradación, el período de observación de 10 días y la pendiente (anexo I). Si la prueba ha cumplido con los criterios de validez, podrá utilizarse para la gráfica la media de los porcentajes de degradación de los frascos que contengan la sustancia problema, |
|
— |
porcentaje de eliminación obtenido después del período de observación de 10 días, en la fase estacionaria y al final de la prueba. |
PARTE II. ENSAYO BASADO EN LA PÉRDIDA DE COD (Método C.4-A)
II.1. PRINCIPIO DEL MÉTODO
Un volumen medido de medio mineral inoculado con una concentración conocida de la sustancia problema (10-40 mg COD/l) como única fuente nominal de carbono orgánico se somete a aireación en la oscuridad o bajo luz difusa a 22 ± 2 oC.
La degradación se va siguiendo mediante el análisis del COD a intervalos frecuentes durante un período de 28 días. El grado de biodegradación se calcula expresando la concentración de COD eliminada (corregida con los valores obtenidos en el blanco con inóculo) como porcentaje de la concentración presente inicialmente. El grado de biodegradación primaria también puede calcularse mediante análisis químico complementario realizado al principio y al final de la incubación.
II.2. DESCRIPCIÓN DEL MÉTODO
II.2.1. Equipo
|
a) |
Matraces cónicos, por ejemplo de 250 ml hasta 2 l, según el volumen necesario para los análisis de COD. |
|
b) |
Agitador para matraces cónicos, bien con control automático de la temperatura o bien situado en una sala a temperatura constante. La agitación debe ser suficiente como para mantener condiciones aerobias en todos los matraces. |
|
c) |
Equipo de filtración con membranas adecuadas. |
|
d) |
Analizador de COD. |
|
e) |
Equipo para determinar el oxígeno disuelto. |
|
f) |
Centrífuga. |
II.2.2. Preparación del medio mineral
Para la preparación de la solución madre, véase el punto I.6.2.
Se mezclan 10 ml de solución (a) con 800 ml de agua de dilución, se añade 1 ml de las soluciones (b) a (d) y se enrasa a 1 1 con agua de dilución.
II.2.3. Preparación y acondicionamiento previo del inóculo
El inóculo puede obtenerse de diversas fuentes: lodo activado, aguas residuales efluentes, aguas superficiales, suelos, o de una combinación de ellos.
Véanse los puntos I.6.4, I.6.4.I, I.6.4.2 y I.6.5.
II.2.4. Preparación de los matraces
Como ejemplo, se introducen alícuotas de 800 ml de medio mineral en matraces cónicos de 2 litros y se añaden volúmenes suficientes de las soluciones madre de las sustancias problema y de referencia a los distintos matraces para obtener una concentración de sustancia equivalente a 10-40 mg COD/l. Controlar los valores del pH y ajustar a 7,4 si es necesario. Se inoculan los matraces con lodo activado u otra fuente de inóculo (véase el punto I.6.4), para obtener una concentración final que no exceda de 30 mg de sólidos en suspensión/1. También hay que preparar controles del inóculo en el medio mineral pero sin sustancia problema ni de referencia.
En caso necesario, puede utilizarse un recipiente para comprobar el posible efecto inhibidor de la sustancia problema inoculando una solución que contenga, en el medio mineral, concentraciones comparables tanto de la sustancia problema como de la sustancia de referencia.
También, en caso necesario, puede prepararse otro matraz estéril para observar si la sustancia problema se degrada abióticamente. Para ello, se utiliza una solución de la sustancia problema sin inoculo (véase el punto 1.6.6).
Además, si se sospecha que la sustancia problema puede adsorberse de forma significativa en las paredes o fondo del recipiente utilizado para la realización del ensayo, en los lodos, etc., hay que hacer una evaluación previa para determinar la importancia de la adsorción y, en consecuencia, la idoneidad de la prueba para esa sustancia (véase el cuadro 1). Preparar un matraz conteniendo la sustancia problema, el inoculo y el agente esterilizante.
Todos los matraces se enrasan a 1 1 con medio mineral y, tras mezclar, se toma una muestra de cada matraz para determinar la concentración inicial de COD (véase el anexo II.4). Por ejemplo, se tapa la boca de los matraces con papel de aluminio, de forma que se permita la libre circulación de aire entre el matraz y la atmósfera ambiente. A continuación se ponen los recipientes en el agitador para iniciar el ensayo.
II.2.5. Número de matraces en un ensayo normal
Matraces 1 y 2: suspensión de ensayo
Matraces 3 y 4: con inóculo solo (blanco con inóculo)
Matraz 5: control de procedimiento
Preferentemente y cuando sea necesario:
Matraz 6: control estéril abiótico
Matraz 7: control de adsorción
Matraz 8: control de toxicidad.
Véase también el punto 1.6.7.
II.2.6. Realización del ensayo
A lo largo de todo el ensayo, hay que determinar la concentración de COD en cada matraz por duplicado a intervalos de tiempo conocidos con la suficiente frecuencia para poder determinar el principio del período de observación de 10 días y la pérdida porcentual al final de dicho período de 10 días. Debe limitarse al mínimo el volumen de suspensión problema tomado para cada determinación.
Antes de tomar las muestras, hay que compensar las pérdidas por evaporación de los matraces añadiendo agua de dilución (véase el anexo I.6.1) en la cantidad necesaria. Antes de tomar las muestras hay que homogeneizar bien el medio de cultivo y asegurarse de que el material que se adhiere a las paredes de los recipientes se disuelve o resuspende. Inmediatamente después de tomar la muestra, esta se filtra por membrana o se centrifuga (véase el anexo II.4). La muestra filtrada o centrifugada se analiza el mismo día; en caso contrario, se conserva a 2-4 oC durante un máximo de 48 horas, o por debajo de - 18 oC durante un período mayor.
II.3. DATOS E INFORME
II.3.1. Tratamiento de los resultados
Calcular la degradación porcentual al tiempo t según se indica en el punto I.7.1. (determinación de COD) y, de forma optativa, en el punto I.7.2 (análisis específico).
Todos los resultados deben registrarse en las fichas de recogida de datos presentadas.
II.3.2. Validez de los resultados.
Véase el punto I.5.2.
II.3.3. Informe
Véase el punto I.8.
II.4. FICHA DE RECOGIDA DE DATOS
A continuación se da un ejemplo de la ficha de recogida de datos.
ENSAYO DE PÉRDIDA DE COD
|
1. |
LABORATORIO |
|
2. |
FECHA DE COMIENZO DE LA PRUEBA |
3. SUSTANCIA A EXAMINAR
Nombre:…
Concentración de la solución madre: … mg/l en sustancia química
Concentración inicial en el medio, to: … mg/l en sustancia química
4. INÓCULO
Fuente:…
Tratamiento realizado:
Acondicionamiento previo en su caso:…
Concentración de sólidos en suspensión en la mezcla de ensayo: … mg/l
5. DETERMINACIONES DE CARBONO
Analizador de carbono:
|
|
Matraz no |
|
COD tras n días (mg/l) |
||||
|
0 |
n1 |
n2 |
n3 |
nx |
|||
|
Substancia problema más inóculo |
1 |
a1 |
|
|
|
|
|
|
a2 |
|
|
|
|
|
||
|
a, media Ca(t) |
|
|
|
|
|
||
|
2 |
b2 |
|
|
|
|
|
|
|
b2 |
|
|
|
|
|
||
|
b, media Cb(t) |
|
|
|
|
|
||
|
Ensayo en blanco con inóculo pero sin substancia problema |
3 |
c2 |
|
|
|
|
|
|
c2 |
|
|
|
|
|
||
|
c, media Cb(t) |
|
|
|
|
|
||
|
4 |
d2 |
|
|
|
|
|
|
|
d2 |
|
|
|
|
|
||
|
d, media Cb(t) |
|
|
|
|
|
||
|
|
|
|
|
|
|
6. EVALUACIÓN DE LOS RESULTADOS OBTENIDOS
|
Matraz no |
|
% de degradación tras n días |
||||
|
0 |
n1 |
n2 |
n3 |
n4 |
||
|
1 |
|
0 |
|
|
|
|
|
2 |
|
0 |
|
|
|
|
|
Media (3) |
|
0 |
|
|
|
|
Nota: Pueden utilizarse fórmulas similares para la sustancia de referencia y los controles de toxicidad.
7. CONTROL ABIóTICO (optativo)
|
|
Tiempo (días) |
|
|
0 |
t |
|
|
conc. de COD (mg/l) en el control estéril |
Cs(o) |
|

8. ANÁLISIS QUÍMICO ESPECÍFICO (optativo)
|
|
Cantidad residual de la substancia química al final de la prueba (mg/l) |
% degradacíon |
|
Control estéril |
Sb |
|
|
Prueba con el media inoculado |
Sa |
|
PARTE III. ENSAYO DE DETECCIÓN DE LA OCDE MODIFICADO (Método C.4-B)
III. 1. PRINCIPIO DEL MÉTODO
Un volumen medido de medio mineral con una concentración conocida de la sustancia problema (10-40 mg COD/l) como única fuente nominal de carbono orgánico se inocula con 0,5 ml de efluente por litro de medio. La mezcla se airea en la oscuridad o bajo luz difusa a 22 ± 2 oC.
La degradación se va siguiendo mediante análisis del COD a intervalos frecuentes a lo largo de un período de 28 días. El grado de biodegradación se calcula expresando la concentración de COD eliminado (corregida con los valores obtenidos en el blanco con inoculo) como porcentaje de la concentración presente inicialmente. El grado de biodegradación primaria también puede calcularse a partir de análisis químicos suplementarios realizados al principio y al final de la incubación.
III.2. DESCRIPCIÓN DEL MÉTODO
III.2.1. Equipo
|
a) |
Matraces cónicos, por ejemplo de 250 ml a 2 l, según el volumen necesario para el análisis de COD. |
|
b) |
Agitador para matraces cónicos, bien con control automático de temperatura o bien situado en una sala de temperatura constante. La agitación debe ser suficiente como para mantener condiciones aerobias en todos los matraces. |
|
c) |
Equipo de filtración con membranas adecuadas. |
|
d) |
Analizador de COD. |
|
e) |
Equipo para determinar el oxígeno disuelto. |
|
f) |
Centrífuga. |
III.2.2. Preparación del medio mineral
Para la preparación de la solución madre, véase el punto I.6.2.
Se mezclan 10 ml de la solución (a) con 800 ml de agua de dilución, se añade 1 ml de las soluciones (b) a (d) y se enrasa a 1 1 con agua de dilución.
Este método utiliza tan solo 0,5 ml de efluente/l como inoculo y, por tanto, puede ser necesario enriquecer el medio con oligoelementos y factores de crecimiento. Esto se consigue añadiendo 1 ml de cada una de las siguientes soluciones por litro de medio final:
Solución de oligoelementos:
|
Sulfato de manganeso tetrahidratado, MnSO4. 4H2 |
39,9 mg |
|
Acido bórico, H3BO3 |
57,2 mg |
|
Sulfato de zinc heptahidratado, ZnSO4. 7H2 |
42,8 mg |
|
Heptamolibdato de amonio (NH4)6 Mo7O24 |
34,7 mg |
|
Quelato de hierro (FeCl3 ácido etilendiamínotetraacético) |
100,0 mg |
|
Disolver en agua de dilución y llevar a 1 000 ml. |
|
|
Solución de vitaminas: |
|
|
Extracto de levadura |
15,0 mg |
Disolver el extracto de levadura en 100 ml de agua. Esterilizar mediante filtración por membrana de 0,2 micrómetros o bien preparar extemporáneamente.
III.2.3. Preparación y acondicionamiento previo del inóculo
El inóculo está derivado de un efluente secundario de una planta depuradora o de una instalación de laboratorio que reciba de manera primordial aguas residuales domésticas. Véanse los puntos I.6.4.2. y I.6.5.
Se usan 0,5 ml por litro de medio mineral.
III.2.4. Preparación de los matraces
Se introducen porciones de, por ejemplo, 800 ml de medio minera] en matraces cónicos de 2 litros y se añaden volúmenes suficientes de las soluciones madre de las sustancias problema y de referencia a los distintos matraces para obtener una concentración de sustancia equivalente a 10-40 rng COD/l. Revise el valor pH y ajústelo si es necesario, a 7,4. Se inoculan los matraces con efluente de aguas residuales en la proporción de 0,5 ml/l (véase el punto I.6.4.2). También deben prepararse controles del inóculo con el medio mineral pero sin sustancia problema ni de referencia.
En caso necesario, puede utilizarse un matraz para comprobar el posible efecto inhibidor de la sustancia problema inoculando una solución que contenga, en medio mineral, concentraciones comparables tanto de la sustancia problema como de una sustancia de referencia.
También, en caso necesario, puede prepararse otro matraz estéril para comprobar si la sustancia problema se degrada abioticamente utilizando una solución de esta sustancia sin inóculo (véase el punto I.6.6).
Además, si se sospecha que la sustancia problema puede adsorberse de forma significativa en el recipiente utilizado para el ensayo, en los lodos, etc., debe hacerse una prueba previa para determinar el alcance probable de la absorción y, en consecuencia, la adecuación de la prueba para esa sustancia concreta (véase el cuadro 1). Prepárese un matraz conteniendo la sustancia problema, el inóculo y un agente esterilizante.
Todos los matraces se enrasan a 1 1 con medio mineral y, después de agitar, se toma una muestra de cada matraz para determinar la concentración inicial de COD (véase el anexo II.4). Se tapan las bocas de los matraces con papel de aluminio, por ejemplo, de forma que se permita la libre circulación de aire entre el matraz y la atmósfera circundante. A continuación se ponen los recipientes en el agitador para iniciar el ensayo.
III.2.5. Número de matraces en un ensayo normal
Matraces 1 y 2: suspensión de ensayo
Matraces 3 y 4: con inoculo solo (blanco con inóculo)
Matraz 5: control de procedimiento
Preferentemente y cuando sea necesario:
Matraz 6: control estéril abiótico
Matraz 7: control de adsorción
Matraz 8: control de toxicidad.
Véase también el punto I.6.7.
III.2.6. Realización del ensayo
A lo largo del ensayo hay que determinar las concentraciones de COD en cada matraz por duplicado a intervalos de tiempo conocidos, con la suficiente frecuencia como para poder determinar el inicio del período de observación de 10 días y el porcentaje de eliminación al final de dicho período de 10 días. Para cada determinación debe tomarse solo el volumen mínimo necesario de suspensión problema.
Antes de tomar las muestras, hay que compensar las pérdidas que se han producido por evaporación del medio de ensayo en los matraces, añadiendo la cantidad necesaria de agua de dilución (I.6.1). Antes de tomar las muestras hay que agitar bien el medio de cultivo y asegurarse de que el material que se hubiera adherido a las paredes de los recipientes se disuelve o se resuspende. Se filtra por membrana o se centrifuga (véase el anexo II.4) inmediatamente después de haber tomado la muestra. Las muestras filtradas o centrifugadas se analizan el mismo día; en caso contrario, se conservan a 2-4 oC durante 48 horas como máximo o por debajo de - 18 oC durante un período más prolongado
III.3. DATOS E INFORME
III.3.1. Tratamiento de los resultados
Se calcula el porcentaje de degradación en el tiempo t según se indica en I.7.1 (determinación de COD) y, de forma optativa, en I.7.2 (análisis específico).
Todos los resultados deben registrarse en las fichas de recogida de datos indicadas.
III.3.2. Validez de los resultados
Véase el punto I.5.2.
III.3.3. Informe
Véase el punto I.8.
III.4. FICHA DE RECOGIDA DE DATOS
A continuación se ofrece un ejemplo de ficha de recogida de datos.
ENSAYO DE DETECCIÓN DE LA OECD MODIFICADO
|
1. |
LABORATORIO |
|
2. |
FECHA DE COMIENZO DE LA PRUEBA |
3. SUSTANCIA A EXAMINAR
Nombre:
Concentración de la solución madre: … mg/l en sustancia química
Concentración inicial en el medio, t0: … mg/l en sustancia química
4. INÓCULO
Fuente:
Tratamiento realizado:
Acondicionamiento previo en su caso:
Concentración de sólidos en suspensión en la mezcla de reacción: mg/1
5. DETERMINACIONES DE CARBONO
Analizador de carbono:
|
|
Matraz no |
|
COD tras n días (mg/l) |
||||
|
0 |
n1 |
n2 |
n3 |
nx |
|||
|
Substancia problema más inóculo |
1 |
a1 |
|
|
|
|
|
|
a2 |
|
|
|
|
|
||
|
a, media Ca(t) |
|
|
|
|
|
||
|
2 |
b2 |
|
|
|
|
|
|
|
b2 |
|
|
|
|
|
||
|
b, media Cb(t) |
|
|
|
|
|
||
|
Blanco con inóculo pero sin substancia problema |
3 |
c2 |
|
|
|
|
|
|
c2 |
|
|
|
|
|
||
|
c, media Cb(t) |
|
|
|
|
|
||
|
4 |
d2 |
|
|
|
|
|
|
|
d2 |
|
|
|
|
|
||
|
d, media Cb(t) |
|
|
|
|
|
||
|
|
|
|
|
|
|
6. EVALUACIÓN DE LOS RESULTADOS OBTENIDOS
|
Matraz |
|
% de degradación trasndías |
||||
|
0 |
n1 |
n2 |
n3 |
n4 |
||
|
1 |
|
0 |
|
|
|
|
|
2 |
|
0 |
|
|
|
|
|
media (4) |
|
0 |
|
|
|
|
Nota: Pueden utilizarse fórmulas similares para la sustancia de referencia y los controles de toxicidad.
7. CONTROL ABIÓTICO (optativo)
|
|
Tiempo (dias) |
|
|
0 |
t |
|
|
conc. de COD (mg/l) en el control estéril |
Cs(o) |
Cs(t) |

8. ANALISIS QUÍMICO ESPECÍFICO (optativo)
|
|
Cantidad residual de la substancia química al final de la prueba |
% degradacíib primaria |
|
Control estéril |
Sb |
|
|
Prueba con el medio inoculado |
Sa |
|
PARTE IV. ENSAYO DE DESPRENDIMIENTO DE CO2 (Método C.4-C)
IV.1. PRINCIPIO DEL MÉTODO
Se hace pasar una corriente de aire exento de dióxido de carbono a velocidad controlada, en la oscuridad o bajo luz difusa, a través de un volumen medido de medio mineral inoculado que contiene una concentración conocida de la sustancia problema (10-20 mg COD o COT/l) como única fuente nominal de carbono orgánico. La degradación se observa a lo largo de 28 días determinando el dióxido de carbono producido, que se recoge en hidróxido de sodio o de bario y se mide por valoración del hidróxido residual o como carbono inorgánico. La cantidad de dióxido de carbono producido a partir de la sustancia problema (con las correcciones necesarias según el resultado del blanco con inóculo) se expresa como porcentaje de CO2T. El grado de biodegradación también puede calcularse mediante un análisis complementario de COD realizado al principio y al final de la incubación.
IV.2. DESCRIPCIÓN DEL MÉTODO
IV.2.1. Equipo
|
a) |
Matraces, 2-5 litros, provisto cada uno de un tubo de aireación, que llegue practicamente al fondo del recipiente, y de una salida. |
|
b) |
Agitadores magnéticos, cuando se trate de sustancias poco solubles. |
|
c) |
Frascos de absorción de gases. |
|
d) |
Aparato para controlar y medir el flujo de aire. |
|
e) |
Aparato para eliminar el dióxido de carbono en la preparación de aire exento de dióxido de carbono; otra posibilidad es utilizar una mezcla de oxigeno exento de CO2 y de nitrógeno exento de CO2, provenientes de sendas botellas, en las proporciones correctas (20 % O2:80 % N2). |
|
f) |
Equipo para determinar el dióxido de carbono, bien volumétricamente o bien con un analizador de carbono inorgánico. |
|
g) |
Equipo de filtración por membrana (opcional). |
|
h) |
Analizador de COD (opcional). |
IV.2.2. Preparación del medio mineral
Para la preparación de las soluciones madre, véase el puanto I.6.2.
Se mezclan 10 ml de solución (a) con 800 ml de agua de dilución; se añade 1 ml de las soluciones (b) a (d) y se enrasa a 1 1 con agua de dilución.
IV.2.3. Preparación y acondicionamiento previo del inóculo
El inóculo puede obtenerse de diversas fuentes: lodo activado, aguas residuales efluentes, aguas superficiales, suelos, o de una combinación de ellos.
Véanse los puntos I.6.4, I.6.4.1, I.6.4.2 y I.6.5.
IV.2.4. Preparación de los matraces
Los siguientes volúmenes y pesos, dados meramente como ejemplo, indican los valores apropiados para matraces de 5 litros que contengan 3 1 de suspensión. Si se utilizan volúmenes más pequeños hay que modificar los valores en consecuencia, pero hay que asegurarse de que el dióxido de carbono formado puede medirse con precisión.
A cada matraz de 5 litros se añaden 2 400 ml de medio mineral. Se añade a continuación un volumen adecuado del lodo activado ya preparado (véanse los puntos I.6.4.1 y I.6.5) para tener una concentración de sólidos en suspensión que no exceda de 30 mg/l en el volumen final de 3 l de mezcla inoculada. Otra posibilidad es diluir en primer lugar el lodo preparado para obtener una suspensión de 500-1 000 mg/1 en el medio mineral antes de añadir una alícuota al contenido del matraz de 5 litros para obtener una concentración final de 30 mg/l; de esta forma se consigue mayor precisión. También pueden utilizarse otras fuentes de inóculos (véase el punto I.6.4.2). Añadir una alícuota al contenido del matraz de 5 litros para obtener una concentración final de 30 mg/l; de esta forma se consigue mayor precisión. También pueden utilizarse otras fuentes de inóculos (véase el punto I.6.4.2).
Estas mezclas inoculadas se someten a un flujo de aire exento de CO2 durante una noche para purgar el dióxido de carbono del sistema.
Se añade a los matraces, por separado y por duplicado, la sustancia problema y la sustancia de referencia procedentes de sendas soluciones madre, en volúmenes conocidos, como para obtener concentraciones de 10 a 20 mg de COD o COT/l, procedente de las sustancias añadidas; algunos matraces se dejan como controles del inóculo sin añadirles ninguna sustancia. Las sustancias problema poco solubles se añaden directamente, a los matraces teniendo en cuenta una relación de peso o de volumen, o bien se opera según se describe en el anexo III.
En caso necesario, puede utilizarse un matraz para comprobar el posible efecto inhibidor de la sustancia problema añadiendo tanto sustancia problema como sustancia de referencia en las mismas concentraciones que se dan en los otros matraces.
También, en caso necesario, puede utilizarse un matraz estéril para comprobar si la sustancia problema se degrada abioticamente utilizando una solución de la sustancia sin inóculo (véase I.6.6.). Esterilícese mediante la adición de una sustancia tóxica en la concentración apropiada.
Se llevan los volúmenes de las suspensiones de todos los matraces a 3 1 mediante adición de medio mineral previamente aireado con aire exento de CO2. Otra posibilidad es tomar muestras para el análisis del COD (véase el anexo II.4.) o análisis específicos. Se conectan los frascos de absorción a las salidas de aire de los matraces.
Si se utiliza hidróxido de bario, hay que conectar a cada matraz de 5 litros tres frascos de absorción puestos en serie, cada uno con 100 ml de solución de hidróxido de bario 0,0125 M. La solución debe estar exenta de sulfatos y carbonatos precipitados y su concentración debe determinarse justo antes de ser utilizada. Si se utiliza hidróxido de sodio, se conectan dos frascos, de los que el segundo actúa como control para comprobar que todo el dióxido de carbono ha quedado absorbido en el primero. Son adecuados los frascos de absorción provistos de cierres de botellas de suero. A cada frasco se añaden 200 ml de solución de hidróxido de sodio 0,05 M, cantidad suficiente para absorber todo el dióxido de carbono producido si la sustancia química se degrada por completo. La solución de hidróxido de sodio, incluso cuando se prepara extemporáneamente, contiene trazas de carbonatos; esto se corrige deduciendo la cantidad de carbonatos presentes en el blanco.
IV.2.5. Número de matraces en un ensayo normal
Matraces 1 y 2: suspensión de ensayo
Matraces 3 y 4: con inóculo solo (blanco con inóculo)
Matraz 5: control de procedimiento
Preferentemente y cuando sea necesario:
Matraz 6: control estéril abiótico
Matraz 7: control de toxicidad.
Véase también el punto I.6.7.
IV.2.6. Realización del ensayo
El ensayo se inicia haciendo borbotear aire exento de CO2 a través de las suspensiones a la velocidad de 30-100 ml/min. Periódicamente se toman muestras del absorbente de dióxido de carbono para analizar el contenido de CO2. Durante los primeros 10 días se recomienda que los análisis se hagan cada dos o tres días y, después, cada cinco días hasta llegar al día 28, de forma que pueda determinarse el período de observación de 10 días.
El día 28 se toman muestras (opcionalmente) para el análisis de COD o análisis específicos, se mide el pH de las suspensiones y se añade 1 ml de ácido clorhídrico concentrado a cada matraz; se airean los frascos durante una noche para arrastrar el dióxido de carbono presente en las suspensiones problema. El día 29 se hace el último análisis del dióxido de carbono producido.
Los días en que se haga la medición del CO2, se desconecta el absorbente de hidróxido de bario más próximo al matraz y se valora la solución de hidróxido con HC1 0,05 M utilizando fenolftaleína como indicador. Los restantes absorbentes se unen al matraz y se coloca en el extremo libre de la serie un nuevo absorbente con 100 ml de hidróxido de bario 0,0125 M recién preparado. Hay que hacer valoraciones según sea necesario, por ejemplo, cuando se observe la aparición de un precipitado importante en el primer frasco y antes de que aparezca en el segundo, o al menos una vez por semana. Cuando se utiliza NaOH como absorbente, se toma con jeringa una muestra pequeña (según las características del analizador de carbono utilizado) de la solución de hidróxido de sodio del absorbente más próximo al matraz. Se inyecta la muestra en la parte CI del analizador de carbono para analizar directamente el dióxido de carbono producido.
El contenido del segundo frasco solo se analiza al final de la prueba para introducir las correcciones necesarias en caso de que se haya producido arrastre de dióxido de carbono.
IV.3. DATOS E INFORME
IV.3.1. Tratamiento de los resultados
La cantidad de CO2 retenida en un absorbente viene dada por:
mgCO2 = (100 × CB-0,5 × V × CA) × 44
donde:
|
V |
= |
volumen de HC1 utilizado para valorar los 100 ml del absorbente (ml) |
|
CB |
= |
concentración de la solución de hidróxido de bario (M) |
|
CA |
= |
concentración de la solución de ácido clorhídrico (M). |
Si CB es 0,0125 M y CA es 0,05 M, el resultado de la valoración de 100 ml de hidróxido de bario es 50 ml y el peso de CO2 viene dado por:
![]()
Así pues, para pasar el volumen de HC1 valorado a mg de CO2 producido, el factor en este caso es 1,1.
Se calculan los pesos de CO2 producido del inóculo solo y del inóculo más la sustancia problema utilizando los respectivos resultados de valoración y la diferencia es el peso de CO2 producido a partir de la sustancia problema sola.
Por ejemplo, si el inóculo solo da una valoración de 48 ml y el inóculo con la sustancia problema da 45 ml,
CO2 del inóculo = 1,1 × (50-48) = 2,2 mg
CO2 del inóculo con la sustancia problema = 1,1 × (50-45) = 5,5 mg
y, por tanto, el peso de CO2 producido a partir de la sustancia problema es 3,3 mg.
El porcentaje de biodegradación se calcula con la fórmula siguiente:

o bien

3,67 es el factor de conversión (44/12) para pasar de carbono a dióxido de carbono.
El porcentaje de degradación tras un intervalo cualquiera de tiempo se obtiene sumando los valores porcentuales de CO2T calculados para cada uno de los días hasta el momento en que se haya medido.
En el caso de absorbentes de hidróxido de sodio, la cantidad de dióxido de carbono producido, expresado como CI (mg) se calcula multiplicando la concentración de CI en el absorbente por el volumen de este.
. El porcentaje de degradación se calcula con la fórmula siguiente:

La pérdida de COD se calcula opcionalmente según se describe en 1.7. Se registran estos resultados y todos los demás en las fichas de datos mencionadas.
IV.3.2. Validez de los resultados
El contenido en CI de la suspensión de sustancia problema en el medio mineral al inicio de la prueba debe ser inferior al 5 % del CT, y el desprendimiento total de CO2 en el blanco con inóculo al final de la prueba no debe exceder normalmente de 40 mg/l por término medio. Si se obtienen valores superiores a 70 mg CO2/l, es necesario revisar críticamente los datos y la técnica experimental.
Véase también el punto I.5.2.
IV.3.3. Informe
Véase el punto I.8.
IV.4. FICHA DE RECOGIDA DE DATOS
A continuación se presenta un ejemplo de ficha de recogida de datos.
ENSAYO DE DESPRENDIMIENTO DE DIÓXIDO DE CARBONO
|
1. |
LABORATORIO |
|
2. |
FECHA DE COMIENZO DE LA PRUEBA |
3. SUSTANCIA A EXAMINAR
Nombre:…
Concentración de la solución madre: … mg/l en sustancia química
Concentración inicial en el medio: … mg/l en sustancia química
Carbono total añadido al matraz: … mg C
CO2T: … mg CO2
4. INOCULO
Fuente: …
Tratamiento realizado: …
Acondicionamiento previo en su caso: …
Concentración de sólidos en suspensión en la mezcla de reacción: … mg/l
|
Tiemp (día) |
CO2 formado (mg) |
CO2 formado blanco (mg) |
C02 formado acumulado (mg) (ensayo menos blanco) |
% CO2
|
|||||
|
1 2 |
media |
3 4 |
media |
1 |
2 |
1 |
2 |
media |
|
|
0 |
|
|
|
|
|
|
|
|
|
|
n1 |
|
|
|
|
|
|
|
|
|
|
n2 |
|
|
|
|
|
|
|
|
|
|
n3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
28 |
|
|
|
|
|
|
|
|
|
Nota: Pueden utilizarse otros formatos similares para la sustancia de referencia y los controles de toxicidad
5. PRODUCCION DE DIOXIDO DE CARBONO Y DEGRADABILIDAD:
Método: Ba(OH)2/NaOH/otro
6. ANALISIS DE CARBONO (opcional)
Analizador de carbono:
|
Tiempo (día) |
blanco mg/1 |
sustancia problema mg/1 |
|
0 |
Cb(o) |
C0 |
|
28 (5) |
Cb(t) |
Ct |

7. DEGRADACIÓN ABIÓTICA (opcional)

PARTE V. RESPIROMETRÍA MANOMÉTRICA (Método C.4-D)
V.I. PRINCIPIO DEL MÉTODO
Se somete a agitación en un frasco cerrado a temperatura constante (± 1 oC o más exacto), durante 28 días, un volumen medido de medio mineral inoculado que contenga una concentración conocida de la sustancia química de ensayo (100 mg/l de la sustancia de ensayo, para dar al menos 50-100 mg DTO/l) como única fuente nominal de carbono orgánico. Se determina el consumo de oxígeno midiendo la cantidad de oxígeno (producido por electrólisis) requerido para mantener un volumen constante de gas en el frasco del respirómetro, o a partir de los cambios de volumen o presión (o una combinación de ambas cosas) en el aparato. El dióxido de carbono producido se absorbe en una solución de hidróxido potásico u otro absorbente adecuado. La cantidad de oxígeno consumido por la sustancia problema (corregida según el consumo del blanco con inóculo, realizada en paralelo) se expresa como porcentaje de DTO o DQO. Opcionalmente, se puede calcular también la biodegradación primaria mediante un análisis adicional específico realizado al comienzo y al final de la incubación, y la biodegradación última mediante un análisis de COD.
V.2. DESCRIPCIÓN DEL MÉTODO
V.2.1. Equipo
|
a) |
Respirómetro adecuado. |
|
b) |
Control de temperatura, manteniéndolo con un margen de ± 1 oC o más exacto. |
|
c) |
Conjunto de membrana y filtros (opcional). |
|
d) |
Analizador de carbono (opcional). |
V.2.2. Preparación del medio mineral
Para la preparación de las soluciones madre, véase I.6.2.
Mezclar 10 ml de la solución (a) con 800 ml de agua de dilución, añadir 1 ml de las soluciones (b) a (d) y enrasar hasta 1 1 con agua de dilución.
V.2.3. Preparación y acondicionamiento previo del inóculo
El inóculo puede obtenerse de diversas fuentes: lodo activado, aguas residuales efluentes, aguas superficiales, suelos, o de una combinación de ellos.
Véanse los puntos I.6.4, I.6.4.1, I.6.4.2 y I.6.5.
V.2.4. Preparación de los frascos
Preparar soluciones de las sustancias problema y de referencia, en lotes separados, en un medio mineral equivalente a una concentración, normalmente, de 100 mg de sustancia/l (que dé al menos 50-100 mg DTO/l) utilizando soluciones madre.
Calcular la DTO sobre la base de la formación de sales de amonio salvo que se prevea nitrificación; en este caso, el cálculo debe basarse en la formación de nitratos (véase el anexo II.2).
Determinar los valores de pH y, si es necesario, ajustarlos a 7,4±0,2.
Las sustancias débilmente solubles deben añadirse en un estadio posterior (véase más abajo).
Si hay que determinar la toxicidad de la sustancia problema, preparar posteriormente otra solución en medio mineral que contenga tanto la sustancia problema como la de referencia a las mismas concentraciones que las utilizadas para el ensayo.
Si se necesita una medida del consumo fisicoquímico de oxígeno, preparar una solución de la sustancia problema, generalmente, a una concentración de 100 mg DTO/1. Esterilizar dicha solución mediante la adición de una sustancia tóxica adecuada (véase el punto I.6.6).
Introducir en frascos, al menos por duplicado, el volumen necesario de soluciones de sustancias problema y de referencia. Añadir a los siguientes frascos solamente medio mineral (para controles de inóculos) y, si es necesario, la solución con mezcla de sustancia problema y de referencia y la solución estéril.
Si la sustancia problema es poco soluble, se añadirá directamente en este estadio basándose en el volumen o en el peso o actuando como se describe en el anexo III. Añadir hidróxido de potasio con cal sodada u otro absorbente a los compartimentos de absorción de CO2.
V.2.5. Número de frascos en un ensayo normal
Matraces 1 y 2: suspensión de ensayo
Matraces 3 y 4: con inóculo solo (blanco con inóculo)
Matraz 5: control de procedimiento
Preferentemente y cuando sea necesario:
Matraz 6: control estéril
Matraz 7: control de toxicidad.
Véase también el punto I.6.7.
V.2.6. Realización del ensayo
Se deja que los recipientes alcancen la temperatura deseada y se inoculan los recipientes adecuados con lodo activado preparado u otra fuente de inóculo para obtener una concentración de sólidos suspendidos no superior a 30 mg/l. Se monta el equipo, se pone en marcha el agitador y se controla la impermeabilidad al aire, comenzando en este momento la medición del consumo de oxígeno. Generalmente no hay que hacer más que tomar las lecturas necesarias y los controles diarios para asegurarse de que se mantienen la temperatura correcta y la agitación adecuada.
Calcular el consumo de oxígeno según las lecturas tomadas a intervalos frecuentes y regulares, utilizando los métodos proporcionados por el fabricante del equipo. Ai final de la incubación, normalmente 28 días, medir el pH del contenido de los frascos, especialmente si el consumo de oxígeno es inferior o superior a la DTONH4 (en aquellos compuestos que contengan nitrógeno).
Si es necesario, se tomarán muestras de los frascos del respirómetro, al comienzo y al final, para analizar el COD o realizar un análisis químico específico (véase el anexo II.4). Cuando se haga la primera toma de muestras, hay que asegurarse de que se conoce el volumen de suspensión problema que queda en el frasco. Cuando la sustancia problema nitrogenada consuma oxígeno, determinar el aumento de la concentración de nitrito y nitrato en 28 días y calcular la corrección teniendo en cuenta el oxígeno consumido por nitrificación (véase el anexo V).
V.3. DATOS E INFORME
V.3.1. Tratamiento de los resultados
Dividir el consumo de oxígeno (mg) de la sustancia problema después de un tiempo dado (corregido según el blanco con inóculo después del mismo tiempo) entre el peso de la sustancia problema utilizada. Esto dará la DBO expresada en mg de oxígeno/mg de sustancia problema, es decir

= mg de O2 por mg de substancia problema
calcular el porcentaje de biodegradación, bien a partir de:

o bien de:

Debe señalarse que estos dos métodos no dan necesariamente el mismo valor. Es preferible usar el primero.
En el caso de sustancias a examinar que contengan nitrógeno, se utiliza la DTO (NH4 o NO3) adecuada según se espere o se sepa que va haber nitrificación o no (véase el anexo II.2). Si hay nitrificación pero no es completa, se calcula una corrección según el oxígeno consumido por nitrificación basándose en los cambios de la concentración de nitrito y nitrato (véase el anexo V).
Cuando se hagan determinaciones opcionales de carbono orgánico o análisis químicos específicos, calcular la degradación porcentual, tal como se describe en el punto I.7.
Registrar todos los resultados en las fichas de datos adjuntas.
V.3.2. Validez de los resultados
El consumo de oxígeno del blanco con inóculo es normalmente de 20-30 mg O2/l y no debe ser superior a 60 mg/l en 28 días. Los valores superiores a 60 mg/l exigirán un examen crítico de los datos y de las técnicas experimentales. Si el valor del pH está fuera del rango 6-8,5 y el consumo de oxígeno por la sustancia problema es inferior al 60 %, debe repetirse el ensayo con menor concentración de la sustancia problema.
Véase también el punto I.5.2.
V.3.3. Informe
Véase el punto I.8.
V.4. FICHA DE RECOGIDA DE DATOS
A continuación se da un ejemplo de una ficha de recogida de datos.
RESPIROMETRÍA MANOMÉTRICA
|
1. |
LABORATORIO |
|
2. |
FECHA DE COMIENZO DE LA PRUEBA |
3. SUSTANCIA A EXAMINAR
Nombre: …
Concentración de la solución madre: … mg/l
Concentración inicial en el medio, Co: … mg/l
Volumen en el matraz de prueba (V): … ml
DTO de sustancia/DQO de sustancia: … mg O2/mg de sustancia a examinar (NH4, NO3)
4. INÓCULO
Fuente: …
Tratamiento realizado: …
Acondicionamiento previo en su caso: …
Concentración de sólidos en suspensión en la mezcla de reacción: … mg/l
5. CONSUMO DE OXÍGENO: BIODEGRADABILIDAD
|
|
Tiempo (dias) |
||||||||||||
|
0 |
|
7 |
|
14 |
|
|
21 |
|
|
28 |
|
||
|
O2 consumido (mg) sust. problema |
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a, media |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O2 consumido (mg) blanco |
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
b, media |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
DBO (mg) corregida |
(a1 — bm) |
|
|
|
|
|
|
|
|
|
|
|
|
|
(a2 — bm) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
DBO por mg de sustancia problema |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
% degradación
|
D1 (a1) |
|
|
|
|
|
|
|
|
|
|
|
|
|
D2 (a2) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Media (6) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
V = volumen de medio en los frascos del ensayo. |
Nota: Pueden utilizarse formatos similares para las sustancias de referencia y los controles de toxicidad.
6. CORRECCIÓN POR NITRIFICACIÓN (véase el anexo V)
|
Día |
0 |
28 |
Diferencia |
||
|
|
|
(N) |
||
|
— |
|
|
||
|
|
|
(N) |
||
|
— |
— |
|
||
|
— |
— |
|
7. ANÁLISIS DE CARBONO (opcional)
Analizador de carbono:
|
Tiempo (da) |
Blanco mg/l |
Sustancia problema mg/l |
|
0 |
(Cblo) |
(Co) |
|
28 (7) |
(Cblt) |
(Ct) |

8. ANÁLISIS QUÍMICO ESPECÍFICO (opcional)
|
Sb |
= |
concentración en el control fisicoquímico (estéril) a los 28 días. |
|
Sa |
= |
concentración en el frasco inoculado a los 28 días. |

9. DEGRADACIÓN ABIÓTICA (opcional)
|
a |
= |
consumo de oxígeno en frascos estériles después de 28 días, mg. |

(véanse las secciones 1 y 3)

PARTE VI. ENSAYO DEL FRASCO CERRADO (Método C.4-E)
VI. 1. PRINCIPIO DEL MÉTODO
Se prepara una solución de la sustancia problema en medio mineral, generalmente con una concentración de 2-5 mg/l. Esta solución se inocula con un número relativamente pequeño de microorganismos de una población mixta y se mantiene en frascos cerrados completamente llenos, en la oscuridad y a temperatura constante. La degradación se sigue mediante el análisis del oxígeno disuelto durante un período de 28 días. La cantidad de oxígeno consumido por la sustancia problema, corregido según el consumo de oxígeno por parte del blanco con inóculo realizado en paralelo, se expresa como porcentaje de DTO o DQO.
VI.2. DESCRIPCIÓN DEL MÉTODO
VI.2.1. Equipo
|
a) |
Frascos de DBO, con tapones de cristal de, por ejemplo, 250-300 ml. |
|
b) |
Baño maría o incubadora, para mantener los frascos a temperatura constante (± 1 oC o más exacto) en la oscuridad. |
|
c) |
Frascos de cristal grandes (2-5 1) para la preparación de medios y para rellenar los frascos de DBO. |
|
d) |
Electrodo de oxígeno y oxímetro o equipamiento y reactivos para la valoración según Winkler, |
VI.2.2. Preparación del medio mineral
Para la preparación de la solución madre, véase el punto I.6.2.
Mezclar 1 ml de las soluciones (a) a (d) y rellenar hasta 1 l con agua de dilución.
VI.2.3. Preparación del inóculo
El inóculo se deriva normalmente de un efluente secundario de una planta depuradora o de una instalación de laboratorio que reciba predominantemente aguas residuales domésticas. Otra fuente posible de inóculo es el agua superficial. Utilizar normalmente desde una gota (0,05 ml) hasta 5 ml de filtrado por litro de medio; se pueden necesitar varias pruebas para descubrir el volumen óptimo para un efluente dado. (Véanse los puntos I.6.4.2 y I.6.5)
VI.2.4. Preparación de los frascos
Se airea intensamente el medio mineral durante 20 minutos por lo menos. Realizar cada serie de ensayos con el mismo medio mineral. Generalmente, el medio está listo para su uso después de dejarlo reposar 20 horas, a la temperatura del ensayo. Determinar la concentración de oxígeno disuelto como control; debe dar un valor de unos 9 mg/l a 20 oC. Todas las operaciones de transferencia y llenado de medio saturado de aire y sin burbujas, se harán, por ejemplo, mediante sifones.
Se preparan grupos paralelos de frascos de DBO para la determinación de sustancias problema y de referencia en series experimentales simultáneas. Reunir un número suficiente de frascos de DBO, incluyendo blancos con inóculo, para permitir hacer, al menos por duplicado, mediciones de consumo de oxígeno a los intervalos deseados, por ejemplo, a los 0, 7, 14, 21 y 28 días. Pueden ser necesarios más frascos si se quiere determinar el período de observación de 10 días.
Se añade medio mineral bien aireado a frascos grandes de manera que estén llenos aproximadamente hasta un tercio de su capacidad. A continuación se añade suficiente cantidad de las soluciones madre de las sustancias problema y de las sustancias de referencia a frascos grandes por separado de forma que la concentración final de las sustancias químicas no exceda normalmente de 10 mg/l. No se añadirá ninguna sustancia al blanco de control contenido en otro frasco grande.
La concentración de oxígeno disuelto no debe bajar por debajo de 0,5 mg/l en los frascos de DBO, para poder garantizar la no limitación de la actividad del inóculo. Esto limita la concentración de la sustancia problema a unos 2 mg/l. Sin embargo, en caso de compuestos poco degradables y de aquéllos que tienen baja DTO, puede usarse una concentración de 5-10 mg/l. En algunos casos, es aconsejable realizar series paralelas de sustancias problema a dos concentraciones diferentes, por ejemplo, 2 y 5 mg/l. Normalmente, se calculará la DTO basándose en la formación de sales de amonio pero, si se espera o se sabe que va a haber nitrificación, se calculará basándose en la formación de nitrato (DTONO3: véase el anexo II.2). No obstante, si hay nitrificación pero no es completa, se corregirá de acuerdo con los cambios en la concentración de nitrito y nitrato, determinada por análisis (véase el anexo V).
Si se va a investigar la toxicidad de la sustancia problema (en el caso, por ejemplo, de que se haya encontrado previamente un valor de biodegradabilidad bajo), será necesaria otra serie de frascos.
Se prepara otro frasco grande que contenga medio mineral aireado (alrededor de un tercio de su volumen) más la sustancia problema y la de referencia a concentraciones finales normalmente iguales a las de los otros frascos grandes.
Se inoculan las soluciones en los frascos grandes con efluente secundario (desde una gota, alrededor de 0,05 ml, hasta 5 ml/l) o con otra fuente como agua de río (véase el punto I.6.4.2). Finalmente se llevan las soluciones al volumen deseado con medio mineral aireado utilizando un tubo que llegue hasta el fondo del frasco para conseguir una mezcla adecuada.
VI.2.5. Número de frascos en una serie típica
En una serie típica se usan los siguientes frascos:
|
— |
al menos 10 que contengan la sustancia problema y el inóculo (suspensión de ensayo), |
|
— |
al menos 10 que solo contengan el inóculo (blanco con inóculo), |
|
— |
al menos 10 que contengan la sustancia de referencia y el inóculo (control de procedimiento), |
|
— |
y, cuando sea necesario, 6 frascos que contengan la sustancia problema, la sustancia de referencia y el inóculo (control de toxicidad). Sin embargo, puede necesitarse al menos el doble de frascos si se desea garantizar la posibilidad de determinar el período de observación de 10 días. |
VI.2.6. Realización del ensayo
Se introduce inmediatamente cada solución preparada en el respectivo grupo de frascos de DBO mediante un tubo desde el cuarto inferior (no el fondo) del frasco grande apropiado, de forma que se rellenen completamente todos los frascos de DBO. Se golpea ligeramente para eliminar toda burbuja de aire. Se analizan inmediatamente los frascos para determinar el oxigeno disuelto a tiempo cero, mediante los métodos de Winkler o de electrodo. Pueden preservarse los contenidos de los frascos, para su análisis ulterior, mediante el método de Winkler añadiendo sulfato de manganeso (II) e hidróxido sódico (el primer reactivo del método Winkler); estos frascos, que contienen el oxígeno fijado como óxido hidratado de manganeso (III) (precipitado marrón), se pueden almacenar en la oscuridad y a 10-20 oC durante no más de 24 horas antes de continuar con el análisis según el método de Winkler. Se tapan los restantes frascos replicados asegurándose de que no quedan burbujas dentro de ellos y se incuban a 20 oC en la oscuridad. Cada serie debe acompañarse de una serie paralela completa para la determinación en el blanco inoculado. Retirar al menos frascos duplicados de todas las series para el análisis del oxígeno disuelto a intervalos de tiempo (al menos semanalmente) durante los 28 días de incubación.
Las muestras semanales permitirán la valoración del porcentaje de eliminación en un período de observación de 14 días, mientras que las muestras tomadas cada 3 o 4 días permitirán determinar el período de observación de 10 días, lo que requerirá aproximadamente el doble de frascos.
En el caso de las sustancias que contengan nitrógeno, deberán hacerse correcciones del consumo de oxígeno si tiene lugar cualquier tipo de nitrificación. Con este fin, se usará el método del electrodo de O2 a fin de determinar la concentración de oxígeno disuelto y sacar una muestra del frasco de DBO para análisis de nitrito y nitrato. El oxígeno utilizado se calculará a partir del aumento de concentración de nitrito y nitrato (véase el anexo V).
VI.3. DATOS E INFORME
VI.3.1. Tratamiento de los resultados
Se calcula en primer lugar la DBO obtenida tras cada período de tiempo sustrayendo la depleción de oxígeno (mg O2/l) del blanco con inóculo de la que presenta la sustancia problema. Se divide esta depleción corregida entre la concentración (mg/l) de la sustancia problema a fin de obtener la DBO específica en mg de oxígeno por mg de sustancia problema, Se calcula la biodegradabilidad porcentual dividiendo la DBO específica entre la DTO específica (calculada de acuerdo con el anexo II.2) o la DQO (determinada por análisis, véase el anexo II.3); así pues:

= mg de O2 por mg de sustancia problema

o

En el caso de sustancias que contengan nitrógeno, se utiliza la DTO (NH4 o NO3) adecuada según se espere o se sepa que va haber nitrificación o no (véase el anexo II.2), Si hay nitrificación pero no es completa, se calcula una corrección según el oxígeno consumido por nitrificación basándose en los cambios de la concentración de nitrito y nitrato (véase el anexo V).
VI.3.2. Validez de los resultados
La depleción de oxígeno en el blanco con inóculo no debe sobrepasar 1,5 mg de oxígeno disuelto/l después de 28 días. Si hay valores superiores a este, deberá hacerse una revisión de las técnicas experimentales. La concentración residual de oxígeno en los frascos del ensayo no deberá bajar por debajo de 0,5 mg/l en ningún momento. Estos valores tan bajos de oxígeno solo son válidos si el método de determinación del oxigeno disuelto es capaz de medir dichos niveles con exactitud.
Véase también el punto I.5.2
VI.3.3. Informe
Véase el punto I.8.
VI.4. FICHA DE RECOGIDA DE DATOS
A continuación se da un ejemplo de ficha de recogida de datos.
ENSAYO DEL FRASCO CERRADO
|
1. |
LABORATORIO |
|
2. |
FECHA DE COMIENZO DE LA PRUEBA |
3. SUSTANCIA A EXAMINAR
Nombre:
Concentración de la solución madre: … mg/l
Concentración inicial en el frasco: … mg/l
DTO/DQO: mg O2/mg de sustancia a examinar
4. INÓCULO
Fuente:
Tratamiento realizado:
Acondicionamiento previo en su caso:
Concentración de sólidos en suspensión en la mezcla de reacción: … mg/l
5. DETERMINACIÓN DEL OD
Método: Winkler/electrodo
Análisis de los frascos
|
Tiempo de incubación (d) |
OD (mg/l) |
|||||
|
0 |
n1 |
n2 |
|
|||
|
Blanco sin sustancia problema |
1 |
C1 |
|
|
|
|
|
2 |
C2 |
|
|
|
|
|
|
Media |
|
|
|
|
|
|
|
Substancia problema |
1 |
a1 |
|
|
|
|
|
|
2 |
a2 |
|
|
|
|
|
Media |
|
|
|
|
|
Nota: Pueden utilizarse formatos similares para las sustancias de referencia y los controles de toxicidad.
6. CORRECCIÓN POR NITRIFICACIÓN (véase el anexo V)
|
Tiempo de incubación (d) |
0 |
n1 |
n2 |
n3 |
||
|
|
|
|
|
||
|
— |
|
|
|
||
|
— |
|
|
|
||
|
|
|
|
|
||
|
— |
|
|
|
||
|
— |
|
|
|
||
|
— |
|
|
|
7. DEPLECIÓN DE OD: % DE DEGRADACIÓN
|
|
Depleción tras n días (mg/l) |
|||
|
n1 |
n2 |
n3 |
|
|
|
Frasco 1: (mto - mtx) — (mbo - mbx) |
|
|
|
|
|
Frasco 2: (mto - mtx) — (mbo - mbx) |
|
|
|
|
|
Frasco1:
|
|
|
|
|
|
Frasco 2:
|
|
|
|
|
| % D media (8) =
|
|
|
|
|
|
mto |
= |
valor a tiempo 0 en el frasco de la prueba |
|
mtx |
= |
valor a tiempo t en el frasco de la prueba |
|
mbo |
= |
valor medio del blanco a tiempo 0 |
|
mbx |
= |
valor medio del blanco a tiempo x |
Se aplicará también una corrección según la nitrificación basándose en iii + vi de la sección 6.
8. DEPLECIÓN DE OD EN EL BLANCO
Consumo de oxígeno por el blanco: (mbo - mb28) mg/l. Este consumo es importante para la validez del ensayo. Debe ser menor de 1,5 mg/l.
PARTE VII. ENSAYO DEL M.I.T.I. (Método C.4-F)
VII.1. PRINCIPIO DEL MÉTODO
Se mide automáticamente durante un período de 28 días, en un respirómetro cerrado, a 25 ± 1 oC y en la oscuridad, el consumo de oxígeno en una solución o suspensión agitada, de la sustancia problema en un medio mineral, inoculado con un inóculo especial de microorganismos no adaptados. El dióxido de carbono producido es absorbido por cal sodada. La biodegradabilidad se expresa como porcentaje de consumo de oxígeno (corregido según el consumo del blanco) sobre el consumo teórico (DTO). El porcentaje de biodegradabilidad primaria se calcula también basándose en los análisis químicos específicos adicionales hechos al comienzo y al final de la incubación y, opcionalmente, por análisis de COD.
VII.2. DESCRIPCIÓN DEL MÉTODO
VII.2.1. Equipo
|
a) |
Medidor electrolítico automático de DBO o respirómetro normalmente equipado con seis frascos, de 300 ml cada uno y que llevan cápsulas para el absorbente de CO2. |
|
b) |
Temperatura ambiente constante o baño maría a 25 oC ± 1 oC o más exacta. |
|
c) |
Conjunto de membrana y filtros (opcional). |
|
d) |
Analizador de carbono (opcional). |
VII.2.2. Preparación del medio mineral
Se prepararán las siguientes soluciones madre, utilizando reactivos de calidad analítica y agua (véase el punto I.6.1):
|
(a) |
Ortofosfato diácido de potasio, KH2PO4 |
8,50 g |
|
Ortofosfato monoácido de potasio, K2HPO4 |
21,75 g |
|
|
Ortofosfato monoácido de sodio dodecahidratado Na2HPO4.12 H2O |
44,60 g |
|
|
Cloruro amónico, NH4Cl |
1,70 g |
|
|
Disolver en agua y enrasar hasta 1 litro |
|
|
|
El pH de la solución debe ser de 7,2 |
|
|
|
(b) |
Sulfato de magnesio heptahidratado, MgSO4- 7 H2O |
22,50 g |
|
Disolver en agua y enrasar a 1 litro |
|
|
|
(c) |
Cloruro cálcido anhidro, CaCl2 |
27,50 g |
|
Disolver en agua y enrasar a 1 litro |
|
|
|
(d) |
Cloruro de hierro (III) hexahidratado, FeCl3.6 H2O |
0,25 g |
|
Disolver en agua y enrasar a 1 litro |
|
Se toman 3 ml de cada solución (a), (b), (c) y (d) y se enrasa a 1 litro.
VII.2.3. Preparación del inóculo
Se recogen muestras frescas de no menos de 10 puntos, sobre todo en zonas donde se utilicen y viertan diversos productos químicos. En los sitios como las instalaciones de tratamiento de aguas residuales, tratamiento industrial de aguas residuales, ríos, lagos, mares, se recogerá muestras de 1 litro de lodo, suelo superficial, agua, etc. y se mezclarán cuidadosamente. Despues de eliminar la materia flotante y de dejar sedimentar, el sobrenadante se ajustará a pH 7 ± 1 con hidróxido sódico o ácido fosfórico.
Se utiliza un volumen adecuado del sobrenadante filtrado para rellenar un recipiente de decantación de lodo activado y se airea el líquido durante 23 h 30 min. Treinta minutos después de detener la aireación, se elimina alrededor de un tercio del volumen total de sobrenadante y se añade al material sedimentado un volumen igual de una solución (pH 7) que contenga 0,1 % respectivamente de glucosa, peptona y ortofosfato monopotásico, volviendo a comenzar la aireación. Este procedimiento se repite una vez al día. La unidad de lodo funcionará de acuerdo con las prácticas correctas: los efluentes deben estar limpios, la temperatura debe permanecer a 25 ± 2 oC, el pH debe ser 7 ± 1, el lodo debe estar bien sedimentado, debe haber aireación suficiente para mantener la mezcla en aerobiosis en todo momento, los protozoos deben estar presentes; al menos cada tres meses se hará un ensayo de la actividad del lodo frente a una sustancia de referencia. No se debe utilizar lodo como inóculo hasta al menos un mes después de la operación, pero no más de cuatro meses después. Posteriormente, tomar muestras de al menos 10 sitios a intervalos regulares, una vez cada tres meses.
A fin de mantener la misma actividad en el lodo reciente y en el antiguo, se mezcla el sobrenadante filtrado de un lodo activado en uso con un volumen igual de sobrenadante filtrado de una mezcla reciente recogida de diez fuentes y se cultiva el líquido combinado tal como se ha dicho anteriormente. Se toma el lodo para usarlo como inóculo de 18 a 24 horas después de haber alimentado la unidad.
VII.2.4. Preparación de los frascos
Se preparan los seis frascos siguientes:
No 1: sustancia problema en agua de dilución en concentración de 100 mg/l.
Nos 2, 3 y 4: sustancia problema en medio mineral en concentración de 100 mg/l.
No 5: sustancia de referencia (por ejemplo, anilina) en medio mineral en concentración de 100 mg/l.
No 6: medio mineral solo.
Las sustancias problema poco solubles se añaden directamente calculadas en peso o en volumen o se actúa como se describe en el anexo III, excepto que no se utilizan ni disolventes ni agentes emulsificantes. Se añade el absorbente de CO2 a todos los frascos en las cápsulas especiales. Se ajusta el pH a 7,0 en los frascos nos 2, 3 y 4.
VII.2.5. Realización del ensayo
Se inoculan los frascos nos 2, 3 y 4 (suspensiones problema), no 5 (control de actividad) y no 6 (blanco con inóculo) con un pequeño volumen del inóculo de manera que dé una concentración de 30 mg/l de sólidos suspendidos. No se añade inóculo al frasco no 1 que sirve como control abiótico. Se monta el equipo, se controla la impermeabilidad al aire, se ponen en marcha los agitadores y se comienza la medición del consumo de oxígeno en condiciones de oscuridad. Se controla diariamente la temperatura, el agitador y el registrador culombimétrico de consumo de oxígeno, tomando nota de cualquier cambio de color del contenido de los frascos. El consumo de oxígeno se leerá directamente de los seis frascos por un método apropiado, por ejemplo a partir del registrador gráfico de seis puntos, dando lugar a una curva de DBO. Al final de la incubación, normalmente 28 días, se mide el pH del contenido de los frascos y se determina la concentración de la sustancia problema y/o residual y cualquier intermediario y, en el caso de una sustancia soluble en el agua, la concentración de COD (véase el anexo II.4). Se tendrá un cuidado especial en el caso de sustancias volátiles. Si se supone que va a haber nitrificación, se determina la concentración de nitrato y de nitrito en la medida de lo posible.
VII.3. DATOS E INFORME
VII.3.1. Tratamiento de los resultados
Se divide el consumo de oxígeno (mg) de la sustancia problema después de un tiempo dado, corregido según el consumo del blanco con inóculo después del mismo tiempo, entre el peso de la sustancia problema utilizada. Con ello obtenemos la DBO expresada en mg de oxígeno/mg de sustancia problema, es decir:

= mg O2/mg substancia problema
La biodegradación porcentual se obtiene de la siguiente manera:

En las mezclas, se calcula la DTO basándose en el análisis elemental, como si fuera una sustancia simple. Se utilizará la DTO adecuada (DTONH4 o DTONO3) según haya nitrificación nula o completa (véase el anexo II.2). No obstante, si hay nitrificación pero es incompleta, se corregirá según el oxígeno consumido por la nitrificación, calculado según los cambios en las concentraciones de nitrito y nitrato (véase el anexo V).
Se calcula el porcentaje de biodegración primaria a partir de la pérdida de sustancia química originaria específica (véase el punto I.7.2).

Si ha habido en el frasco No. 1 una pérdida de sustancia problema que dé la medida de la eliminación fisicoquímica, se informará de ello y se utilizará la concentración de la sustancia problema (Sb) después de 28 días en este frasco para calcular el porcentaje de biodegradación.
Cuando se hagan determinaciones de COD opcional), el porcentaje de biodegradación última se calculará con la siguiente fórmula:

como se describe en el punto I.7.1. Si ha habido en el frasco no 1 una pérdida de COD, que mide la eliminación fisicoquímica, se utilizará la concentración de COD en este frasco para calcular el porcentaje de biodegradación.
Se registrarán todos los resultados en la ficha de datos adjunta.
VII.3.2. Validez de los resultados
El consumo de oxígeno del blanco con inóculo es normalmente de 20 a 30 mg O2/l y no debe ser mayor de 60 mg/l en 28 días. Los valores superiores a 60 mg/l exigirán una revisión crítica de los datos y de las técnicas experimentales. Si el pH cae fuera del rango 6-8,5 y el consumo de oxígeno con la sustancia problema es menor del 60 %, debe repetirse el ensayo con una concentración inferior de la sustancia problema.
Véase también el punto I.5.2.
Si el porcentaje de degradación de anilina calculado a partir del consumo de oxígeno no excede del 40 % después de 7 días y del 65 % después de 14 días, el ensayo se considerará no válido.
VII.3.3. Informe
Véase el punto I.8.
VII.4. FICHA DE RECOGIDA DE DATOS
A continuación se da un ejemplo de una ficha de recogida de datos.
PRUEBA DEL MITI (I)
|
1. |
LABORATORIO |
|
2. |
FECHA DE COMIENZO DE LA PRUEBA |
3. SUSTANCIA A EXAMINAR
Nombre: . . .
Concentración de la solución madre: … mg/l, en sustancia
Concentración inicial en el medio, Co: … mg/l, en sustancia
Volumen de la mezcla de reacción, V: … ml
DTO de la sustancia: … mg O2/l
4. INÓCULO
Puntos de muestreo del lodo:
|
|
||||
|
|
||||
|
|
||||
|
|
||||
|
|
Concentración de sólidos en suspensión en el lodo activado tras aclimatación con vertidos sintéticos … mg/l
Volumen de lodo activado por litro de medio final = … ml
Concentración de lodo en el medio final = … mg/l
5. CONSUMO DE OXÍGENO: BIODEGRADABILIDAD
Tipo de respirómetro usado:
|
|
Tiempo (dias) |
||||||
|
0 |
7 |
14 |
21 |
28 |
|||
|
Consumo O2 (mg) de la substancia problema |
a1 |
|
|
|
|
|
|
|
a2 |
|
|
|
|
|
||
|
a3 |
|
|
|
|
|
||
|
Consumo O2 (mg) blanco |
b |
|
|
|
|
|
|
|
Consumo O2 corregido (mg) |
(a1 - b) (a2 - b) (a3 - b) |
|
|
|
|
|
|
|
DBO por mg de sustancia problema |
|
Frasco 1 |
|
|
|
|
|
|
Frasco 2 |
|
|
|
|
|
||
|
Frasco 3 |
|
|
|
|
|
||
|
% degradación
|
|
1 |
|
|
|
|
|
|
2 |
|
|
|
|
|
||
|
3 |
|
|
|
|
|
||
|
media (9) |
|
|
|
|
|
Nota: Pueden utilizarse formatos similares para las sustancias de referencia y los controles de toxicidad.
6. ANÁLISIS DE CARBONO (opcional)
Analizador de carbono:
|
Matraz |
COD |
% COD eliminado |
Media |
|||
|
Medido |
Corregido |
|||||
|
Agua + sustancia a examinar |
a |
|
|
|
|
|
|
Lodo + sustancia a examinar |
b1 |
|
b1-c |
|
|
|
|
Lodo + sustancia a examinar |
b2 |
|
b2-c |
|
|
|
|
Lodo + sustancia a examinar |
b3 |
|
b3-c |
|
|
|
|
Control blanco |
c |
|
— |
|
|
|

7. DATOS DEL ANÁLISIS QUÍMICO ESPECÍFICO
|
|
Cantidad residual de sust. problema al final del ensayo |
% degradación |
|
Ensayo blanco con agua |
Sb |
|
|
Medio inoculado |
Sal |
|
|
Sa2 |
|
|
|
Sa3 |
|
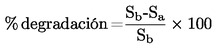
Calcular % de degradación de los frascos a1 a2 y a3 respectivamente.
8. COMENTARIOS
Se adjuntará una curva de DBO en función del tiempo, si se disponede ella.
Anexo I
ABREVIATURAS Y DEFINICIONES
|
OD |
: |
Oxígeno disuelto (mg/l); es la concentración de oxígeno disuelto en una muestra acuosa. |
|
DBO |
: |
Demanda bioquímica de oxígeno (g); es la cantidad de oxígeno consumido por los microorganismos que metabolizan una sustancia problema; se expresa también como gramos de oxígeno consumido por gramo de sustancia problema. (Véase el método C.5.). |
|
DQO |
: |
Demanda química de oxígeno (g); es la cantidad de oxígeno consumido durante la oxidación de una sustancia problema con dicromato en medio ácido caliente. Proporciona una medida de la cantidad de sustancia oxidable presente; se expresa también como gramos de oxígeno consumido por gramo de sustancia problema. (Véase el método C.6.) |
|
COD |
: |
Carbono orgánico disuelto; es el carbono orgánico presente en solución o el que pasa a través de un filtro de 0,45 micrómetros o permanece en el sobrenadante tras centrifugación a 40 000 m.s-2 (±4 000 g) durante 15 minutos. |
|
DTO |
: |
Demanda teórica de oxígeno (mg); es la cantidad total de oxígeno necesario para oxidar completamente una sustancia; se calcula basándose en la fórmula molecular (véase el anexo II.2) y se expresa también como mg de oxígeno requeridos por mg de sustancia problema. |
|
CO2T |
: |
Dióxido de carbono teórico (mg); es la cantidad de dióxido de carbono que se calcula que va a producirse a partir del contenido en carbono, conocido o medido, de la sustancia problema cuando se mineraliza completamente; se expresa también como mg de dióxido de carbono producido por mg de sustancia de ensayo. |
|
COT |
: |
Carbono orgánico total; es la suma total del carbono orgánico en solución y en suspensión en una muestra. |
|
CI |
: |
Carbono inorgánico, |
|
CT |
: |
Carbono total; es el total del carbono orgánico e inorgánico presente en una muestra. |
Biodegradación primaria:
Es la alteración de la estructura química de una sustancia, producida por acción biológica y que da lugar a la pérdida de las propiedades específicas de esa sustancia.
Biodegradación última (aerobia):
Es el nivel de degradación al que llega la sustancia de ensayo cuando es totalmente utilizada por microorganismos dando lugar a la producción de dióxido de carbono, agua, sales minerales y nuevos constituyentes celulares microbianos (biomasa).
Fácilmente biodegradable:
Categoría arbitraria de sustancias químicas que han pasado determinados ensayos específicos de clasificación en cuanto a la biodegradabilidad última; estos ensayos son tan estrictos que es asumido que, según ellos, estos compuestos deben degradarse rápida y completamente en medio ambiente acuático en condiciones de aerobiosis.
Intrínsecamente biodegradable:
Categoría de sustancias químicas en las que hay evidencia inequívoca de biodegradación (primaria o última) en un ensayo de biodegradabilidad reconocido.
Tratabilidad:
Es la capacidad de las sustancias de ser eliminadas durante el tratamiento biológico de los residuos acuosos sin afectar negativamente ai funcionamiento normal de los procesos de tratamiento. Generalmente, los compuestos fácilmente biodegradables son tratables, pero no todos los compuestos intrínsecamente biodegradables son tratables. Pueden darse también procesos abióticos.
Tiempo de latencia
En una prueba de pérdida, es el tiempo transcurrrido desde la inoculación hasta que el porcentaje de degradación aumenta hasta al menos el 10 %. El tiempo de latencia suele ser muy variable y poco reproducible.
Tiempo de degradación
Es el tiempo transcurrido desde el final del tiempo de latencia hasta el momento en que se alcanza el 90 % del nivel máximo de degradación.
Período de observación de 10 días
Son los 10 días que siguen inmediatamente al momento en que se alcanza el 10 % de degradación.
Anexo II
CÁLCULO Y DETERMINACIÓN DE LOS PARÁMETROS INDICATIVOS ADECUADOS
Dependiendo del método elegido, se necesitarán determinados parámetros indicativos. La sección siguiente describe el cálculo de esos valores. El uso de estos parámetros se describe en cada uno de los métodos.
1. Contenido en carbono
El contenido en carbono se calcula basándose en la composición elemental conocida o determinada por análisis elemental de la sustancia problema.
2. Demanda teórica de oxígeno (DTO)
La demanda teórica de oxígeno (DTO) puede calcularse si se conoce la composición.elemental o se determina mediante análisis elemental. Para un compuesto:
CcHhCldNnNanaO0PpSs
sin nitrificación,
 mg/mg
mg/mg
o con nitrification
 mg/mg
mg/mg
3. Demanda química de oxígeno (DQO)
La demanda química de oxígeno (DQO) se determina de acuerdo con el método C.6.
4. Carbono orgánico disuelto (COD)
El carbono orgánico disuelto (COD) es, por definición, el carbono orgánico de cualquier sustancia o mezcla de sustancias en agua que pasa a través de un filtro de 0,45 micrómetros.
Se toman muestras de los recipientes de ensayo y se filtran inmediatamente en el equipo de filtración utilizando un filtro de membrana adecuado. Se desprecian los primeros 20 ml de filtrado (puede disminuirse la cantidad si se utilizan filtros más pequeños). Si se inyectan volúmenes de 10 a 20 ml o menores (el volumen depende de la cantidad necesaria para el analizador de carbono), se guardan para el análisis de carbono. La concentración de COD se determina mediante un analizador de carbono orgánico capaz de medir exactamente una concentración de carbono equivalente o inferior al 10 % de la concentración de COD inicial utilizada en el ensayo.
Las muestras filtradas que no puedan analizarse en el mismo día de trabajo pueden conservarse almacenadas en un refrigerador a 2-4 oC durante 48 horas, o por debajo de - 18 oC durante períodos más prolongados.
Comentarios:
Los filtros de membrana vienen a menudo impregnados con agentes tensoactivos para hacerlos hidrófilos. Así pues, el filtro puede contener varios mg de carbono orgánico soluble que puede interferir en las determinaciones de biodegradabilidad. Los agentes tensoactivos y otros compuestos orgánicos solubles se eliminan de los filtros hirviendo éstos en agua desionizada durante tres períodos de 1 hora cada uno. Los filtros pueden almacenarse a continuación en agua durante una semana. Si se utilizan cartuchos de filtro de un solo uso, cada lote debe controlarse para confirmar que no libera carbono orgánico soluble.
Dependiendo del tipo de filtro de membrana, la sustancia puede ser retenida por absorción. Por ello, es aconsejable asegurarse de que la sustancia problema no queda retenida en el filtro.
Puede usarse centrifugación, en lugar de filtración, a 40 000 m.sec-2 (4 000 g) durante 15 minutos para diferenciar el COT del COD. El método no es fiable con una concentración inicial < 10 mg COD/l ya que ni se eliminan todas las bacterias ni se redisuelve el carbono que forma parte del plasma bacteriano.
REFERENCIAS
|
— |
Standard Methods for the Examination of Water and Wastewater, 12th ed, Am. Pub. Hlth. Ass., Am. Wat. Poll. Control Fed., Oxygen Demand, 1965, p 65. |
|
— |
Wagner, R., Von Wasser, 1976, Vol. 46, 139. |
|
— |
DIN-Entwurf 38409 Teil 41 — Deutsche Einheitsverfahren zur Wasser-, Abwasser- und Schlammuntersu chung, Summarische Wirkungs- und Stoffkenngrößen (Gruppe H). Bestimmung des Chemischen Sauerstoff bedarfs (CSB) (H 41), Normenausschuß Wasserwesen (NAW) in DIN Deutsches Institut für Normung e.V. |
|
— |
Gerike, P. The biodegradability testing of poorly water soluble compounds. Chemosphere, 1984, Vol. 13 (1), 169. |
Anexo III
EVALUACIÓN DE LA BIODEGRADABILIDAD DE LAS SUSTANCIAS POCO SOLUBLES
Debe prestarse una atención especial, en los ensayos de biodegradabilidad de sustancias poco solubles, a los aspectos siguientes.
Aunque los líquidos homogéneos rara vez presentarán problemas de muestreo, se recomienda homogeneizar los materiales sólidos por medios apropiados para evitar errores debidos a la no homogeneidad. Debe prestarse una atención especial cuando se necesiten muestras representativas de unos miligramos tomadas de mezclas de sustancias químicas o de sustancias con grandes cantidades de impurezas.
Pueden utilizarse durante los ensayos diversas formas de agitación. Debe tenerse cuidado de usar solo la agitación suficiente para mantener la dispersión de la sustancia, evitando el sobrecalentamiento, la formación excesiva de espuma y las fuerzas de cizallamiento excesivas.
Puede utilizarse un emulsificante que dé una dispersión estable de la sustancia química. Debe ser atóxico para las bacterias y no debe biodegradarse ni producir espuma en las condiciones del ensayo.
Se aplicarán a los solventes los mismos criterios que a los emulsificantes.
No se recomienda utilizar vehículos sólidos para los ensayos de sustancias sólidas, pero pueden ser adecuados en el caso de sustancias oleosas.
Cuando se utilicen sustancias auxiliares corno emulsificantes, solventes y portadores, debe realizarse un ensayo en blanco con las sustancias auxiliares.
Puede utilizarse para estudiar la biodegradabilidad de las sustancias poco solubles cualquiera de los tres ensayos respirométricos CO2, DBO o MITI.
REFERENCIAS
|
— |
de Morsier, A. et al. Biodegradation tests for poorly- soluble compounds. Chemosphere, 1987, Vol. 16, 833. |
|
— |
Gerike, P. The Biodegradabiliry testing of poorly water soluble compounds. Chemosphere, 1984, Vol. 13, 169. |
Anexo IV
EVALUACIÓN DE LA BIODEGRADABILIDAD DE SUSTANCIAS QUÍMICAS QUE PUEDEN SER TÓXICAS PARA EL INÓCULO
Cuando se someta una sustancia química a ensayos de biodegradabilidad fácil y se vea que no es biodegradable, se recomienda el procedimiento siguiente si se desea distinguir si la sustancia es inhibidora o es inerte (Reynolds et al.; 1987).
Deben utilizarse inóculos similares o idénticos para los ensayos de toxicidad y de biodegradación.
Cuando se considere necesario disponer de un medio para valorar la toxicidad de las sustancias químicas estudiadas en los ensayos de biodegradabilidad fácil, puede ser apropiado aplicar uno o varios de los métodos siguientes: inhibición de la tasa de respiración del lodo (ensayo de inhibición de respiración del lodo activado — Directiva 88/302/CEE), DBO o inhibición del crecimiento.
Si hay que evitar la inhibición debida a la toxicidad, se sugiere que las concentraciones de la sustancia problema utilizadas en las pruebas de biodegradabilidad fácil sean inferiores a 1/10 de la CE50 (o inferiores a los valores de la CE20) obtenida en los ensayos de toxicidad. No es probable que sustancias con una CE50 superior a 300 mg/l tengan efectos tóxicos en los ensayos de biodegradabilidad fácil.
Los valores EC50 inferiores a 20 mg/l pueden presentar problemas en las pruebas subsiguientes. Se emplearán bajas concentraciones de ensayo, haciendo necesario el uso de pruebas sensibles y estrictas como el ensayo del frasco cerrado o el uso de material marcado con 14C. Como alternativa, un inóculo aclimatado puede permitir el uso de concentraciones más elevadas de la sustancia problema. En este último caso, sin embargo, se pierde el criterio específico del ensayo de biodegradabilidad fácil.
REFERENCIAS
Reynolds, L. et al. Evaluation of the toxicity of substances to be assessed for biodegradability. Chemosphere, 1987, Vol. 16, 2259.
Anexo V
CORRECCIÓN DEL CONSUMO DE OXÍGENO POR INTERFERENCIA DE LA NITRIFICACIÓN
Si la sustancia problema no contiene nitrógeno, los errores debidos a no tener en cuenta la nitrificación, en el cálculo del consumo de oxígeno, son despreciables (no superiores al 5 %), incluso si la oxidación del N amónico en el medio se da irregularmente en los distintos recipientes de ensayo y blanco. No obstante, en el caso de sustancias que contienen N, pueden darse errores graves.
Si ha habido nitrificación pero no es completa, el consumo de oxígeno que se ha observado en la mezcla de reacción puede corregirse según la cantidad de oxígeno utilizado para oxidar el amonio a nitrito y nitrato, sí se determinan los cambios en la concentración del nitrito y del nitrato durante la incubación mediante las ecuaciones siguientes:
|
2 NH4Cl + 3 O2 = 2 HNO2 + 2 HC1 + 2 H2O |
(1) |
|
2 HNO2 + O2 = 2 HNO3 |
(2) |
|
Globalmente: |
|
|
2 NH4Cl + 4 O2 = 2 HNO3 + 2 HC1 + 2 H2O |
(3) |
En la ecuación (1) el consumo de oxigeno por 28 g de nitrógeno contenidos en cloruro amónico (NH4Cl) al ser oxidado a nitrito es de 96 g, es decir, un factor de 3,43 (96/28). Del mismo modo, en la ecuación (3) el consumo de oxígeno por 28 g de nitrógeno al ser oxidado a nitrato es de 128 g, es decir, un factor de 4,57 (128/28).
Como las reacciones son secuenciales, desencadenadas por especies bacterianas diferentes bien determinadas, es posible que la concentración de nitrito aumente o disminuya; en este último caso, se formará una concentración equivalente de nitrato. Así pues, el oxígeno consumido en la formación de nitrato es 4,57 multiplicado por el aumento de la concentración de nitrato, mientras que el oxígeno relacionado con la formación de nitrito es 3,43 multiplicado por el aumento en la concentración de nitrito o bien con la disminución de su concentración, la pérdida de oxígeno es -3,43 multiplicado por la disminución de la concentración.
Es decir:
|
O2 consumido en la formación de nitrato-N = 4,57 × aumento en la concentración de nitrato Y |
(4) |
|
O2 consumido en la formación de nitrito-N = 3,43 × aumento en la concentración de nitrito |
(5) |
|
y |
|
|
O2 perdido en la desaparición de nitrito-N = -3,43 × disminución en la concentración de nitrito de forma que |
(6) |
|
consumo de O2 debido a la nitrificación = ±3,43 × cambio en concentración de nitrito-N +4,57 × aumento en concentración de nitrato |
(7) |
|
y, por consiguiente, |
|
|
consumo de O2 debido a oxidación del C = consumo total observado menos consumo debido a nitrificación |
(8) |
Si solo se determina el N oxidado total, puede considerarse que el consumo de oxígeno debido a la nitrificación es, como primera aproximación, 4,57 × aumento del N oxidado.
El valor corregido del consumo de oxígeno debido a la oxidacion del C se compara entonces con la DTO NH4 tal como se calcula en el anexo II.
C.5. DEGRADACIÓN: DEMANDA BIOQUÍMICA DE OXÍGENO
1. MÉTODO
1.1. INTRODUCCIÓN
El objetivo del ensayo es medir la demanda bioquímica de oxígeno (DBO) de las sustancias orgánicas, sólidas o líquidas.
Los resultados que se obtienen con este método de ensayo son representativos, sobre todo, cuando se aplica a los compuestos hidrosolubles; no obstante, al menos en principio, el método puede hacerse extensible a los compuestos volátiles y los compuestos de baja hidrosolubilidad.
El método solo es aplicable a las sustancias orgánicas que no produzcan efectos inhibidores sobre las bacterias a la concentración de ensayo. Si la sustancia de ensayo no es soluble a la concentración de ensayo, se puede recurrir a procedimientos especiales, tales como la dispersión por ultrasonidos, a fin de lograr una dispersión satisfactoria de la sustancia de ensayo.
Es conveniente disponer de información sobre la toxicidad de la sustancia para poder interpretar los resultados bajos y para elegir la concentración de ensayo adecuada.
1.2. DEFINICIONES Y UNIDADES
La DBO se define como la masa de oxígeno disuelto necesario para asegurar, en condiciones determinadas, la oxidación bioquímica de un volumen dado de una solución de la sustancia sometida a ensayo.
Los resultados se expresan en gramos de DBO por gramo de sustancia de prueba.
1.3. SUSTANCIAS DE REFERENCIA
Es conveniente la utilización de una sustancia de referencia apropiada para verificar la actividad del inóculo.
1.4. PRINCIPIO DEL MÉTODO DE ENSAYO
Se siembra con microorganismos y se incuba en la oscuridad a una temperatura ambiente constante dada, una cantidad determinada de sustancia, disuelta o dispersa en un medio apropiado y aireado.
La DBO se determina por la diferencia entre el contenido en oxígeno disuelto al principio y al final del ensayo. La duración mínima del ensayo es de cinco días y la máxima, de 28.
Debe efectuarse un ensayo paralelo en blanco, sin sustancia de ensayo.
1.5. CRITERIOS DE CALIDAD
La determinación de la DBO no puede considerarse como una determinación válida de la biodegradabilidad de la sustancia. Esta prueba solo puede considerarse como prueba exploratoria.
1.6. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
Se prepara una solución o una dispersión preliminar de la sustancia, con el fin de obtener una concentración de DBO compatible con el método utilizado. La DBO se determina entonces aplicando un método nacional o internacional normalizado que sea apropiado.
2. RESULTADOS Y EVALUACIÓN
La DBO contenida en la solución preliminar se calcula según el método normalizado elegido y se expresa en gramos de DBO por gramo de sustancia de ensayo.
3. INFORME
Debe indicarse el método utilizado.
La demanda bioquímica de oxígeno debe ser la media de al menos tres mediciones válidas.
Para facilitar la interpretación de los resultados deben incluirse todos los datos y observaciones que sean pertinentes, especialmente en lo referente a impurezas, estado físico, efectos tóxicos y propiedades específicas de la sustancia que puedan afectar a los resultados.
Debe mencionarse si se ha utilizado un aditivo inhibidor de la nitrificación biológica.
4. REFERENCIAS
Lista de métodos normalizados, por ejemplo:
|
|
NF T 90-103: Determination of the Biochemical Oxygen Demand |
|
|
NBN 407: Biochemical Oxygen Demand |
|
|
NBN 3235 5.4: Bepaling van het biochemisch zuurstofverbruik (BZV) |
|
|
The Determination of Biochemical Oxygen Demand, Methods for the Examination of Water and Associates Materials, HMSO, Londres. |
|
|
ISO 5815: Determination of biochemical oxygen demand after n days. |
C.6. DEGRADACIÓN: DEMANDA QUÍMICA DE OXÍGENO
1. MÉTODO
1.1. INTRODUCCIÓN
El objetivo del ensayo es medir la demanda química de oxígeno (DQO) de las sustancias orgánicas, sólidas o líquidas, mediante la aplicación de cualquier procedimiento normalizado, en condiciones de laboratorio determinadas.
Es conveniente disponer de información sobre la fórmula de la sustancia para facilitar la realización del ensayo y la interpretación de los resultados obtenidos (por ejemplo, sales halógenas, sales ferrosas de compuestos orgánicos, compuestos organoclorados).
1.2. DEFINICIONES Y UNIDADES
La demanda química de oxígeno es la medida de la oxidabilidad de una sustancia, definida como la cantidad equivalente en oxígeno de un reactivo oxidante consumida por la sustancia en condiciones de laboratorio determinadas.
El resultado se expresa en gramos de DQO por gramo de sustancia de ensayo.
1.3. SUSTANCIAS DE REFERENCIA
No es preciso emplear sustancias de referencia cada vez que se analiza una nueva sustancia. Dichas sustancias de referencia sirven, principalmente, para calibrar periódicamente el método y permitir la comparación de resultados cuando se apliquen métodos distintos.
1.4. PRINCIPIO DEL MÉTODO DE ENSAYO
Una cantidad determinada de sustancia, disuelta o dispersa en agua, se oxida con dicromato de potasio en presencia de ácido sulfúrico concentrado, con catalizador de sulfato de plata, en caliente y con reflujo durante dos horas. El dicromato residual se valora con ayuda de sulfato ferroso amónico titulado.
En el caso de las sustancias con cloro, se debe añadir sulfato mercúrico (las soluciones que contienen sales de mercurio deben ser tratadas, después de su utilización, pra evitar que pase mercurio al medio ambiente) para atenuar la interferencia de los cloruros.
1.5. CRITERIOS DE CALIDAD
Dada la arbitrariedad del método de determinación de la DQO, ésta debe considerarse como un «indicador de oxidabilidad» y se utiliza en calidad de tal como método práctico de evaluación de la materia orgánica.
Los cloruros pueden interferir en este ensayo; las sustancias inorgánicas reductoras u oxidantes pueden interferir igualmente con la determinación de la DQO.
Algunos compuestos cíclicos y muchas sustancias volátiles (p.ej., ácidos grasos de cadena corta) no se oxidan totalmente durante el ensayo.
1.6. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
Se prepara una solución o una dispersión preliminar de la sustancia, para conseguir una DQO de 250 a 600 mg por litro.
Notas:
Cuando se trata de sustancias poco solubles y que no se dispersan, se puede pesar una cantidad de sustancia finamente pulverizada, o en estado líquido, correspondiente a unos 5 mg de DQO, e introducirla en el aparato experimental añadiendo agua.
La demanda química de oxígeno (DQO) se determina a menudo mejor, sobre todo en el caso de sustancias poco solubles, con una variante del método, es decir, en un sistema cerrado con ecualizador de presión (H. Kellcenberg, 1975). Con esta modificación puede conseguirse cuantificar sustancias que se determinan con mucha dificultad cuando se usa el método convencional (por ejemplo, ácido acético). Sin embargo, el método falla igualmente en el caso de la piridina. Si se alcanza una concentración de dicromato potásico de 0,25 N (0,0416 M), como se prescribe en la referencia (1), se facilita la adición directa de 5-10 mg de sustancia, lo que resulta esencia] para la determinación de la DQO de las sustancias poco hidrosolubles [referencia (2)].
Por lo demás, la DQO se determina a continuación aplicando cualquier método nacional o internacional normalizado que sea adecuado.
2. RESULTADOS Y EVALUACIÓN
Se calcula la DQO contenida en el frasco experimental según el método normalizado elegido y se expresa en gramos de DQO por gramo de sustancia de ensayo.
3. INFORME
Debe indicarse el método utilizado.
La demanda química de oxigeno debe ser la media de al menos tres mediciones. Se incluirán iodos los datos y observaciones que puedan facilitai la interpretación de los resultados, especialmente los referentes a impurezas estado físico y propiedades especificas de la sustancia (si se conocen) que puedan afectar a los resultados.
Debe mencionarse si se na usado sulfato mercurico para reducir la interferencia de los cloturos.
4. BIBLIOGRAFÍA
|
(1) |
Kelkenberg, H., Z. von Wasser und Abwasssrforschung, 1975, vol. 8, 146. |
|
(2) |
Gerike, P. The biodegradibility testing of poorly water soluble compounds. Chemosphere, 1984, vol. 13, 169. Lista de métodos normalizados, por ejemplo:
|
C.7. DEGRADACIÓN — DEGRADACIÓN ABIÓTICA: HIDRÓLISIS EN FUNCIÓN DEL pH
1. MÉTODO
El presente método de ensayo es equivalente a las directrices de ensayo de la OCDE TG 111 (2004).
1.1. INTRODUCCIÓN
Las sustancias químicas pueden llegar a las aguas superficiales por vías como la aplicación directa, los aerosoles erráticos, la escorrentía, el drenaje, la eliminación de residuos, los efluentes industriales, domésticos o agrícolas y la deposición atmosférica, y pueden transformarse en dichas aguas mediante procesos químicos (por ejemplo, hidrólisis, oxidación), fotoquímicos y microbianos. Las presentes directrices describen un método de ensayo de laboratorio para evaluar la transformación hidrolítica abiótica de sustancias químicas en sistemas acuáticos a valores de pH que se encuentran normalmente en el medio ambiente (pH de 4 a 9) y se basa en otras directrices anteriores [(1) (2) (3) (4) (5) (6) (7)].
Con los experimentos se pretende determinar: i) la velocidad de hidrólisis de la sustancia problema en función del pH, y ii) la identidad o naturaleza y las velocidades de formación y destrucción de los productos de hidrólisis a los que pueden estar expuestos los organismos. Tales estudios pueden ser necesarios en relación con sustancias que se aplican directamente al agua o que pueden llegar al medio ambiente por las otras vías antes citadas.
1.2. DEFINICIONES Y UNIDADES
Véase el anexo 2.
1.3. APLICABILIDAD DEL MÉTODO
El método es aplicable en general a todas las sustancias químicas (tanto marcadas como sin marcar) para las que se disponga de un método analítico de suficiente exactitud y sensibilidad. Es aplicable a compuestos ligeramente volátiles y no volátiles que tengan la suficiente hidrosolubilidad. El ensayo no debe aplicarse a sustancias químicas que sean muy volátiles a partir del agua (por ejemplo, fumigantes o disolventes orgánicos) y, por tanto, no se puedan mantener en solución en las condiciones experimentales del ensayo. Puede ser difícil efectuar el ensayo con sustancias muy poco hidrosolubles (8).
1.4. PRINCIPIO DEL MÉTODO DE ENSAYO
Se tratan con la sustancia problema soluciones acuosas estériles amortiguadoras con diferentes valores de pH (pH de 4, 7 y 9), y se incuban en la oscuridad en condiciones de laboratorio controladas (a temperaturas constantes). A intervalos determinados, se analiza el contenido de sustancia problema y de productos de su hidrólisis en las soluciones amortiguadoras. Si se utiliza una sustancia problema marcada (por ejemplo, 14C) es más fácil calcular el balance de masa.
Este método de ensayo está diseñado según un enfoque secuencial que se muestra y explica en el anexo 1. A cada etapa de la secuencia se pasa en función de los resultados de la etapa anterior.
1.5. INFORMACIÓN SOBRE LA SUSTANCIA DE ENSAYO
Para medir la velocidad de hidrólisis puede utilizarse una sustancia problema marcada o sin marcar. Generalmente se prefiere utilizar material marcado para estudiar la ruta de la hidrólisis y para establecer el balance de masa; sin embargo, el marcado puede no ser absolutamente necesario en casos especiales. Se recomienda el marcado con 14C, pero también puede resultar de utilidad el uso de otros isótopos, tales como 13C, 15N o 3H. En la medida de lo posible, el marcador debe ubicarse en la parte o partes más estables de la molécula. Por ejemplo, si la sustancia problema contiene un solo anillo, es preciso marcar ese anillo; si contiene dos o más anillos, puede resultar necesario efectuar estudios separados para evaluar el destino de cada anillo marcado y obtener información adecuada sobre la formación de los productos de hidrólisis. La pureza de la sustancia problema debe ser de al menos del 95 %.
Antes de llevar a cabo un ensayo de hidrólisis, debe disponerse de la información siguiente sobre la sustancia problema:
|
a) |
hidrosolubilidad (método de ensayo A.6); |
|
b) |
solubilidad en disolventes orgánicos; |
|
c) |
presión de vapor (método de ensayo A.4) o constante de la ley de Henry; |
|
d) |
coeficiente de reparto n-octanol/agua (método de ensayo A.8); |
|
e) |
constante de disociación (pKa) (directrices OCDE 112) (9); |
|
f) |
velocidad de fototransformación directa e indirecta en agua, en su caso. |
Debe disponerse de métodos analíticos de cuantificación de la sustancia problema y, cuando sea pertinente, de identificación y cuantificación de los productos de su hidrólisis en soluciones acuosas (véase también el punto 1.7.2).
1.6. SUSTANCIAS DE REFERENCIA
A ser posible, deben utilizarse sustancias de referencia para la identificación y cuantificación de los productos de la hidrólisis mediante métodos espectroscópicos y cromatográficos, u otros métodos con la sensibilidad adecuada.
1.7. CRITERIOS DE CALIDAD
1.7.1. Recuperación
Mediante el análisis de soluciones amortiguadoras, cuando menos con dos réplicas en paralelo, o de sus extractos inmediatamente después de la adición de la sustancia problema se obtiene una primera indicación de la repetibilidad del método analítico y de la uniformidad del procedimiento de aplicación de la sustancia problema. Los valores de recuperación correspondientes a etapas posteriores de los experimentos los dan los balances de masa respectivos (cuando se utiliza material marcado). Las tasas de recuperación tienen que oscilar entre el 90 % y el 110 % de las sustancias tanto marcadas como sin marcar (7). En caso de que sea técnicamente difícil llegar a esta banda, puede aceptarse una recuperación del 70 % de sustancias sin marcar, pero debe justificarse la situación.
1.7.2. Repetibilidad y sensibilidad del método analítico
La repetibilidad del método o métodos analíticos utilizados para cuantificar la sustancia problema y los productos de hidrólisis posteriormente puede comprobarse mediante análisis de réplicas en paralelo de las mismas soluciones amortiguadoras (o de sus extractos) después de que se hayan formado cantidades de productos de hidrólisis suficientes para su cuantificación.
El método analítico debe tener la sensibilidad suficiente para cuantificar concentraciones de sustancia problema iguales o inferiores al 10 % de la concentración inicial. En su caso, los métodos analíticos deben tener también la suficiente sensibilidad para cuantificar cualquier producto de hidrólisis que represente el 10 % o más de la cantidad aplicada (en cualquier momento a lo largo del estudio) hasta el 25 % o menos de su concentración máxima.
1.7.3. Intervalos de confianza de los datos cinéticos de la hidrólisis
Deben calcularse y presentarse los intervalos de confianza de todos los coeficientes de regresión, constantes de velocidad, semividas y cualesquiera otros parámetros cinéticos (por ejemplo, TD50).
1.8. DESCRIPCIÓN DEL MÉTODO
1.8.1. Equipo y material
El estudio debe realizarse en recipientes de vidrio (por ejemplo, tubos de ensayo, frascos pequeños) en condiciones estériles y de oscuridad, en caso necesario, salvo que se disponga de información previa (como el coeficiente de reparto n-octanol/agua) que indique la posibilidad de adherencia de la sustancia problema al vidrio. En tal situación, puede ser necesario considerar la utilización de otros materiales (como el teflón). También puede caber la posibilidad de solucionar el problema de la adherencia al vidrio con uno o más de los métodos siguientes:
|
— |
determinación de la masa de la sustancia problema y productos de hidrólisis que adsorbe el recipiente de ensayo, |
|
— |
empleo de un baño de ultrasonidos, |
|
— |
lavado con disolventes de todo el material de vidrio en cada intervalo de muestreo, |
|
— |
utilización de productos formulados, |
|
— |
uso de una cantidad mayor de cosolvente para añadir la sustancia problema al sistema; si se utiliza un cosolvente, éste no debe hidrolizar la sustancia problema. |
Normalmente es necesario utilizar agitadores de baños de agua con temperatura controlada o incubadores termostáticos para la incubación de las diversas soluciones problema.
Se precisa el equipo normal de laboratorio y, en particular, el siguiente:
|
— |
pHmetro; |
|
— |
instrumentos analíticos tales como equipos de cromatografía (de gases, de líquidos de alta resolución o de capa fina), incluidos los sistemas de detección adecuados para el análisis de sustancias radiomarcadas y sin marcar o un método inverso de dilución isotópica, |
|
— |
instrumentos de identificación (por ejemplo, espectrometría de masas, cromatografía de gases con espectrometría de masas, cromatografía de líquidos de alta resolución con espectrometría de masas, resonancia magnética nuclear, etc.), |
|
— |
contador de centelleo de líquidos, |
|
— |
ampollas de decantación para la extracción líquido-líquido; |
|
— |
instrumental para concentrar soluciones y extractos (por ejemplo, evaporador rotatorio); |
|
— |
equipo de control de la temperatura (por ejemplo, baño de agua). |
Entre los reactivos químicos figuran, por ejemplo:
|
— |
disolventes orgánicos, con grado analítico de pureza, tales como hexano, diclorometano, etc., |
|
— |
líquido de centelleo, |
|
— |
soluciones amortiguadoras (véase el punto 1.8.3). |
Debe esterilizarse todo el material de vidrio, así como el agua de grado reactivo y las soluciones amortiguadoras que se utilicen en los ensayos de hidrólisis.
1.8.2. Aplicación de la sustancia problema
La sustancia problema debe añadirse como solución acuosa a las diferentes soluciones amortiguadoras (véase el anexo 3). Cuando sea necesario para conseguir una disolución adecuada, se permite utilizar pequeñas cantidades de disolventes miscibles con el agua (como acetonitrilo, acetona o etanol) para la aplicación y distribución de la sustancia problema, pero sin sobrepasar en principio el 1 % v/v. Cuando se desee recurrir a una concentración superior de disolventes (por ejemplo, en caso de sustancias problema poco solubles), esta podría autorizarse solo si se demuestra que el disolvente no afecta a la hidrólisis de la sustancia problema.
No se recomienda en principio el uso de productos formulados, ya que no puede excluirse que los componentes de la formulación influyan sobre el proceso de hidrólisis. Sin embargo, cuando se trate de sustancias problema poco hidrosolubles o que se adhieran al vidrio (véase el punto 1.8.1), puede ser una alternativa adecuada el empleo de material formulado.
Debe utilizarse una sola concentración de la sustancia problema, sin pasar de 0,01 M o de la mitad de la concentración de saturación (véase el anexo 1).
1.8.3. Soluciones amortiguadoras
El ensayo de hidrólisis debe efectuarse a pH de 4, 7 y 9. Para conseguirlo, deben prepararse soluciones amortiguadoras utilizando agua y sustancias de grado reactivo. En el anexo 3 se presentan varios sistemas amortiguadores útiles. Debe observarse que el sistema amortiguador utilizado puede afectar a la velocidad de la hidrólisis; en caso de que sea así, debe emplearse otro sistema amortiguador (10).
Debe comprobarse el pH de cada solución amortiguadora con un pHmetro calibrado que tenga una precisión mínima de 0,1 a la temperatura necesaria.
1.8.4. Condiciones del ensayo
1.8.4.1. Temperatura del ensayo
Los experimentos de hidrólisis deben efectuarse a temperatura constante. A efectos de extrapolación, es importante que la variación máxima de la temperatura sea de ±0,5 oC.
Debe realizarse un ensayo preliminar (etapa 1) a la temperatura de 50 oC si no se conoce el comportamiento hidrolítico de la sustancia problema. Deben efectuarse ensayos cinéticos de etapas superiores con un mínimo de tres temperaturas (incluido el ensayo a 50 oC), salvo que la sustancia problema sea estable frente a la hidrólisis según el ensayo de la etapa 1. Se sugiere la banda de temperaturas de 10-70 oC (de preferencia con al menos una temperatura por debajo de 25 oC), que incluye la temperatura de 25 oC consignada en el informe y la mayoría de las temperaturas observadas sobre el terreno.
1.8.4.2. Luz y oxígeno
Todos los ensayos de hidrólisis deben realizarse siguiendo un método adecuado para evitar efectos fotolíticos. Deben tomarse todas las medidas necesarias para evitar la presencia de oxígeno (por ejemplo, burbujeando helio, nitrógeno o argón durante 5 minutos antes de la preparación de la solución).
1.8.4.3. Duración del ensayo
El ensayo preliminar debe efectuarse durante cinco días, mientras que los ensayos de etapas más avanzadas deben mantenerse hasta conseguir la hidrólisis del 90 % de la sustancia problema, o bien durante 30 días si este plazo es más breve.
1.8.5. Realización del ensayo
1.8.5.1. Ensayo preliminar (etapa 1)
El ensayo preliminar se efectúa a 50 ±0,5 oC y a un pH de 4,0, 7,0 y 9,0. Si al cabo de cinco días se observa menos del 10 % de hidrólisis (t 0,5 25 oC > 1 año), se considera que la sustancia problema es hidrolíticamente estable y, en principio, no es necesario hacer más ensayos. Si se sabe que la sustancia es inestable a temperaturas pertinentes para el medio ambiente (11), no es necesario efectuar el ensayo preliminar. El método analítico debe tener la precisión y sensibilidad suficientes para detectar una reducción del 10 % de la concentración inicial.
1.8.5.2. Hidrólisis de sustancias inestables (etapa 2)
El ensayo en etapas superiores debe realizarse a los valores de pH a los que se haya visto que la sustancia problema es inestable según el ensayo preliminar antes citado. Las soluciones amortiguadoras de la sustancia problema deben mantenerse constantemente a las temperaturas seleccionadas. Para hacer un ensayo respecto a una cinética de primer orden, cada solución de reacción debe analizarse a intervalos que proporcionen datos de un mínimo de seis puntos espaciados, normalmente entre el 10 % y el 90 % de hidrólisis de la sustancia problema. Deben tomarse muestras distintas en paralelo (un mínimo de dos réplicas procedentes de recipientes de reacción diferentes) para analizar su contenido en, como mínimo, 6 momentos de muestreo (para tener un mínimo de 12 puntos de datos en paralelo). Se considera inadecuado utilizar una sola solución a granel de la que se van tomando alícuotas individuales de la solución problema tras cada intervalo de muestreo, ya que esto no permite el análisis de la variabilidad de los datos y puede provocar problemas de contaminación de la solución problema. Deben realizarse pruebas de confirmación de la esterilidad al final del ensayo en etapas superiores (es decir, con el 90 % de hidrólisis o a los 30 días). Sin embargo, si no se observa ninguna degradación (es decir, transformación) no es necesario realizar pruebas de esterilidad.
1.8.5.3. Identificación de los productos de la hidrólisis (etapa 3)
Con métodos analíticos adecuados, deben identificarse todos los productos importantes de la hidrólisis, al menos los que representen una proporción > 10 % de la dosis aplicada.
1.8.5.4. Ensayos facultativos
Puede ser necesario efectuar ensayos complementarios con valores de pH distintos de 4, 7 y 9 en relación con sustancias problema hidrolíticamente inestables. Así, puede ser necesario por motivos fisiológicos efectuar un ensayo en condiciones más ácidas (por ejemplo, a un pH de 1,2) utilizando una única temperatura pertinente a efectos fisiológicos (37 oC).
2. DATOS
Las cantidades de sustancia problema y de productos de su hidrólisis, en su caso, deben indicarse como porcentaje de la concentración inicial aplicada y, cuando corresponda, en mg/l respecto a cada intervalo de muestreo y cada pH y temperatura de ensayo. Además, debe darse un balance de masa en porcentaje de la concentración inicial aplicada cuando se haya utilizado una sustancia problema marcada.
Debe proporcionarse una representación gráfica del logaritmo de la concentración de la sustancia problema frente al tiempo. Deben identificarse todos los productos importantes de la hidrólisis, al menos los que constituyan una proporción > 10 % de la dosis aplicada, y sus concentraciones logarítmicas deben representarse de la misma manera que la sustancia original para mostrar sus velocidades de formación y destrucción.
2.1. TRATAMIENTO DE LOS RESULTADOS
Utilizando cálculos según modelos cinéticos adecuados, deben obtenerse determinaciones más exactas de las semividas o valores de TD50. Se consignarán los valores de semivida y TD50 (incluidos los límites de confianza) respecto a cada pH y temperatura, junto con la descripción del modelo utilizado, el orden de la cinética y el coeficiente de determinación (r2). Si procede, se aplicarán los cálculos asimismo a los productos de la hidrólisis.
Si se trata de estudios de velocidad efectuados a varias temperaturas, las constantes de velocidad de hidrólisis de pseudo-primer orden (kobs) deberán describirse en función de la temperatura. Los cálculos se basarán tanto en la separación de kobs en constantes de velocidad en relación con la hidrólisis catalizada por ácidos, la neutra y la catalizada por bases (kH, kneutral y kOH respectivamente) como en la ecuación de Arrhenius:

donde Ai y Bi son constantes de regresión de la ordenada en el origen y de la pendiente, respectivamente, de las rectas mejor ajustadas obtenidas por regresión lineal de ln ki frente a la inversa de la temperatura absoluta en grados Kelvin (T). Mediante el uso de las relaciones de Arrhenius respecto a la hidrólisis catalizada por ácidos, la neutra y la catalizada por bases, es posible calcular constantes de velocidad de pseudo-primer orden y, por tanto, semividas, relativas a otras temperaturas a las que sea difícil efectuar directamente la determinación experimental de una constante de velocidad (10).
2.2. EVALUACIÓN E INTERPRETACIÓN DE LOS RESULTADOS
La mayoría de las reacciones de hidrólisis muestran una velocidad de reacción de primer orden aparente y, por lo tanto, las semividas son independientes de la concentración (véase la ecuación 4 del anexo 2). Esto permite normalmente aplicar a condiciones ambientales (concentraciones < 10-6 M) los resultados de laboratorio determinados a concentraciones de 10-2 a 10-3 M (10). Mabey y Mill (11) han presentado varios ejemplos de concordancia entre velocidades de hidrólisis medidas en agua tanto pura como natural respecto a diversas sustancias, con la condición de que se hubieran medido tanto el pH como la temperatura.
3. INFORMES
3.1. INFORME DEL ENSAYO
El informe del ensayo debe dar, como mínimo, la información siguiente:
Sustancia de ensayo:
|
— |
nombre común, nombre químico, número CAS, fórmula estructural (indicando la posición del marcador si se usa material marcado radiactivamente) y propiedades fisicoquímicas de interés (véase el punto 1.5), |
|
— |
pureza (impurezas) de la sustancia problema, |
|
— |
pureza radioquímica de la sustancia marcada y actividad molar (si procede). |
|
— |
Soluciones amortiguadoras: |
|
— |
fechas y datos de su preparación, |
|
— |
amortiguadores y aguas utilizados, |
|
— |
molaridad y pH de las soluciones amortiguadoras. |
Condiciones de ensayo:
|
— |
fechas de realización de los estudios, |
|
— |
cantidad de la sustancia problema aplicada, |
|
— |
métodos y disolventes (tipo y cantidad) utilizados para la aplicación de la sustancia problema, |
|
— |
volumen incubado de las soluciones amortiguadoras con la sustancia problema, |
|
— |
descripción del sistema de incubación utilizado, |
|
— |
pH y temperatura durante el estudio, |
|
— |
tiempos de muestreo, |
|
— |
métodos de extracción, |
|
— |
métodos de identificación y cuantificación de la sustancia problema y de los productos de su hidrólisis en las soluciones amortiguadoras, |
|
— |
número de réplicas en paralelo. |
Resultados:
|
— |
repetibilidad y sensibilidad de los métodos analíticos utilizados, |
|
— |
tasas de recuperación (en el punto 1.7.1 se dan los porcentajes necesarios para la validez del estudio), |
|
— |
datos de las réplicas en paralelo y medias en forma tabular, |
|
— |
balance de masa durante los estudios y al final de estos (si se utilizan sustancias problema marcadas), |
|
— |
resultados del ensayo preliminar, |
|
— |
evaluación e interpretación de los resultados, |
|
— |
todos los datos y figuras originales. |
La siguiente información es necesaria solo en caso de que se determine la velocidad de hidrólisis:
|
— |
representación gráfica de la concentración frente al tiempo correspondiente a las sustancias problema y, en su caso, a los productos de la hidrólisis a cada valor de pH y temperatura, |
|
— |
cuadros de resultados de la ecuación de Arrhenius para la temperatura de 20 - 25 oC, con los valores de pH, constante de velocidad [h-1 o día-1], semivida o TD50, temperaturas [ oC], incluidos los límites de confianza y coeficientes de correlación (r2) u otra información comparable, |
|
— |
ruta propuesta de hidrólisis. |
---
4. REFERENCIAS
|
(1) |
OECD (1981). Hydrolysis as a Function of pH. OECD Guideline for Testing of Chemicals Nr. 111, adopted 12 May 1981. |
|
(2) |
US-Environmental Protection Agency (1982). 40 CFR 796.3500, Hydrolysis as a Function of pH at 25 oC. Pesticide Assessment Guidelines, Subdivision N. Chemistry: Environmental Fate. |
|
(3) |
Agriculture Canada (1987). Environmental Chemistry and Fate Guidelines for registration of pesticides in Canada. |
|
(4) |
Unión Europea (UE) (1995). Directiva 95/36/CE de la Comisión, por la que se modifica la Directiva 91/414/CEE del Consejo relativa a la comercialización de productos fitosanitarios. anexo V: Destino y comportamiento en el medio ambiente. |
|
(5) |
Dutch Commission for Registration of Pesticides (1991). Application for registration of a pesticide. Section G: Behaviour of the product and its metabolites in soil, water and air. |
|
(6) |
BBA (1980). Merkblatt Nr. 55, Teil I und II: Prüfung des Verhaltens von Pflanzenbehandlungsmitteln im Wasser (October 1980). |
|
(7) |
SETAC (1995). Procedures for Assessing the Environmental Fate and Ecotoxicity of Pesticides. Mark R. Lynch, Ed. |
|
(8) |
OECD (2000). Guidance document on aquatic toxicity testing of difficult substances and mixtures, OECD Environmental Health and Safety Publications Series on Testing and Assessment Nr.23. |
|
(9) |
OECD (1993). Guidelines for the Testing of Chemicals. Paris. OECD (1994-2000): Addenda 6-11 to Guidelines for the Testing of Chemicals. |
|
(10) |
Nelson, H, Laskowski D, Thermes S, and Hendley P. (1997) Recommended changes in pesticide fate study guidelines for improving input to computer models. (Text version of oral presentation at the 14th Annual Meeting of the Society of Environmental Toxicology and Chemistry, Dallas TX, November 1993). |
|
(11) |
Mabey, W. and Mill, T. (1978). Critical review of hydrolysis of organic compounds in water under environmental conditions. J. Phys. Chem. Ref. Data 7, 383-415. |
Anexo 1
Esquema del ensayo de hidrólisis en etapas

Anexo 2
Definiciones y unidades
Siempre deben utilizarse unidades del Sistema Internacional .
Sustancia problema: cualquier sustancia, sea el compuesto original o los correspondientes productos de transformación.
Productos de transformación: todas las sustancias procedentes de reacciones de transformación biótica o abiótica de la sustancia problema.
Productos de hidrólisis: todas las sustancias procedentes de reacciones de transformación hidrolítica de la sustancia problema.
Hidrólisis: reacción de una sustancia problema RX con el agua, con la sustitución final del grupo X por el OH en el centro de reacción:
|
RX + HOH → ROH + HX |
[1] |
La velocidad a la que disminuye la concentración de RX en este proceso simplificado viene dada por:
|
velocidad = k [H2O] [RX] |
reacción de segundo orden |
|
o |
|
|
velocidad = k [RX] |
reacción de primer orden |
según la fase determinante de la velocidad. Como hay un gran exceso de agua respecto a la sustancia problema, este tipo de reacción se suele describir como reacción de pseudo-primer orden, y la constante de velocidad observada viene dada por la fórmula siguiente:
|
kobs = k [H2O] |
[2] |
and can be determined from the expression (12)
|
[3] |
donde
t= tiempo
y Co, Ct= concentraciones de RX a los tiempos 0 y t.
Las unidades de esta constante tienen las dimensiones de (tiempo)-1 y la semivida de la reacción (tiempo para que reaccione el 50 % de RX) viene dada por:
|
|
[4] |
Semivida: (t0,5) tiempo necesario para la hidrólisis del 50 % de una sustancia problema cuando puede describirse la reacción mediante una cinética de primer orden; es independiente de la concentración.
TD 50 (tiempo de desaparición 50): tiempo necesario para que la concentración de la sustancia problema se reduzca en un 50 %; es diferente de la semivida (t0,5) si la reacción no sigue una cinética de primer orden.
Estimación de k a temperatura diferente
Si se conocen las constantes de velocidad correspondientes a dos temperaturas, pueden calcularse las constantes de velocidad a otras temperaturas utilizando la ecuación de Arrhenius:
 o
o 
La representación gráfica de ln k frente a l/T es una línea recta con una pendiente de -E/R
donde:
|
k |
= |
constante de velocidad, medida a temperaturas diferentes |
|
E |
= |
energía de activación [kJ/mol] |
|
T |
= |
temperatura absoluta [K] |
|
R |
= |
R = constante de los gases [8,314 J/mol.K]. |
La energía de activación se calcula por análisis de regresión o con la siguiente ecuación:

donde: T2> T1.
Anexo 3
Sistemas amortiguadores
A. CLARK Y LUBS:
Mezclas amortiguadoras de CLARK y LUBS (13)
|
Composición |
pH |
|
HCl 0,2 N y KCl 0,2 N a 20 oC |
|
|
47,5 ml HCl + 25 ml KCl diluido hasta 100 ml |
1,0 |
|
32,25 ml HCl + 25 ml KCl diluido hasta 100 ml |
1,2 |
|
20,75 ml HCl + 25 ml KCl diluido hasta 100 ml |
1,4 |
|
13,15 ml HCl + 25 ml KCl diluido hasta 100 ml |
1,6 |
|
8,3 ml HCl + 25 ml KCl diluido hasta 100 ml |
1,8 |
|
5,3 ml HCl + 25 ml KCl diluido hasta 100 ml |
2,0 |
|
3,35 ml HCl + 25 ml KCl diluido hasta 100 ml |
2,2 |
|
Biftalato potásico 0,1 M + HCl 0,1 N a 20 oC |
|
|
46,70 ml HCl 0,1 N + 50 ml biftalato hasta 100 ml |
2,2 |
|
39,60 ml HCl 0,1 N + 50 ml biftalato hasta 100 ml |
2,4 |
|
32,95 ml HCl 0,1 N + 50 ml biftalato hasta 100 ml |
2,6 |
|
26,42 ml HCl 0,1 N + 50 ml biftalato hasta 100 ml |
2,8 |
|
20,32 ml HCl 0,1 N + 50 ml biftalato hasta 100 ml |
3,0 |
|
14,70 ml HCl 0,1 N + 50 ml biftalato hasta 100 ml |
3,2 |
|
9,90 ml HCl 0,1 N + 50 ml biftalato hasta 100 ml |
3,4 |
|
5,97 ml HCl 0,1 N + 50 ml biftalato hasta 100 ml |
3,6 |
|
2,63 ml HCl 0,1 N + 50 ml biftalato hasta 100 ml |
3,8 |
|
Biftalato potásico 0,1 M + NaOH 0,1 N a 20 oC |
|
|
0,40 ml NaOH 0,1 N + 50 ml biftalato hasta 100 ml |
4,0 |
|
3,70 ml NaOH 0,1 N + 50 ml biftalato hasta 100 ml |
4,2 |
|
7,50 ml NaOH 0,1 N + 50 ml biftalato hasta 100 ml |
4,4 |
|
12,15 ml NaOH 0,1 N + 50 ml biftalato hasta 100 ml |
4,6 |
|
17,70 ml NaOH 0,1 N + 50 ml biftalato hasta 100 ml |
4,8 |
|
23,85 ml NaOH 0,1 N + 50 ml biftalato hasta 100 ml |
5,0 |
|
29,95 ml NaOH 0,1 N + 50 ml biftalato hasta 100 ml |
5,2 |
|
35,45 ml NaOH 0,1 N + 50 ml biftalato hasta 100 ml |
5,4 |
|
39,85 ml NaOH 0,1 N + 50 ml biftalato hasta 100 ml |
5,6 |
|
43,00 ml NaOH 0,1 N + 50 ml biftalato hasta 100 ml |
5,8 |
|
45,45 ml NaOH 0,1 N + 50 ml biftalato hasta 100 ml |
6,0 |
Mezclas amortiguadoras de CLARK y LUBS (continuación)
|
Fosfato monopotásico 0,1 M + NaOH 0,1 N a 20 oC |
|
|
5,70 ml NaOH 0,1 N + 50 ml fosfato hasta 100 ml |
6,0 |
|
8,60 ml NaOH 0,1 N + 50 ml fosfato hasta 100 ml |
6,2 |
|
12,60 ml NaOH 0,1 N + 50 ml fosfato hasta 100 ml |
6,4 |
|
17,80 ml NaOH 0,1 N + 50 ml fosfato hasta 100 ml |
6,6 |
|
23,45 ml NaOH 0,1 N + 50 ml fosfato hasta 100 ml |
6,8 |
|
29,63 ml NaOH 0,1 N + 50 ml fosfato hasta 100 ml |
7,0 |
|
35,00 ml NaOH 0,1 N + 50 ml fosfato hasta 100 ml |
7,2 |
|
39,50 ml NaOH 0,1 N + 50 ml fosfato hasta 100 ml |
7,4 |
|
42,80 ml NaOH 0,1 N + 50 ml fosfato hasta 100 ml |
7,6 |
|
45,20 ml NaOH 0,1 N + 50 ml fosfato hasta 100 ml |
7,8 |
|
46,80 ml NaOH 0,1 N + 50 ml fosfato hasta 100 ml |
8,0 |
|
H3BO30,1 M en KCl 0,1 M + NaOH 0,1 N a 20 oC |
|
|
2,61 ml NaOH 0,1 N + 50 ml ácido bórico hasta 100 ml |
7,8 |
|
3,97 ml NaOH 0,1 N + 50 ml ácido bórico hasta 100 ml |
8,0 |
|
5,90 ml NaOH 0,1 N + 50 ml ácido bórico hasta 100 ml |
8,2 |
|
8,50 ml NaOH 0,1 N + 50 ml ácido bórico hasta 100 ml |
8,4 |
|
12,00 ml NaOH 0,1 N + 50 ml ácido bórico hasta 100 ml |
8,6 |
|
16,30 ml NaOH 0,1 N + 50 ml ácido bórico hasta 100 ml |
8,8 |
|
21,30 ml NaOH 0,1 N + 50 ml ácido bórico hasta 100 ml |
9,0 |
|
26,70 ml NaOH 0,1 N + 50 ml ácido bórico hasta 100 ml |
9,2 |
|
32,00 ml NaOH 0,1 N + 50 ml ácido bórico hasta 100 ml |
9,4 |
|
36,85 ml NaOH 0,1 N + 50 ml ácido bórico hasta 100 ml |
9,6 |
|
40,80 ml NaOH 0,1 N + 50 ml ácido bórico hasta 100 ml |
9,8 |
|
43,90 ml NaOH 0,1 N + 50 ml ácido bórico hasta 100 ml |
10,0 |
B. KOLTHOFF Y VLEESCHHOUWER:
Amortiguadores de citrato de KOLTHOFF y VLEESCHHOUWER
|
Composición |
pH |
|
Citrato monopotásico 0,1 M y HCl 0,1 N a 18 oC (14) |
|
|
49,7 ml. HCl 0,1 N + 50 ml citrato hasta 100 ml |
2,2 |
|
43,4 ml HCl 0,1 N + 50 ml citrato hasta 100 ml |
2,4 |
|
36,8 ml HCl 0,1 N + 50 ml citrato hasta 100 ml |
2,6 |
|
30,2 ml HCl 0,1 N + 50 ml citrato hasta 100 ml |
2,8 |
|
23,6 ml HCl 0,1 N + 50 ml citrato hasta 100 ml |
3,0 |
|
17,2 ml HCl 0,1 N + 50 ml citrato hasta 100 ml |
3,2 |
|
10,7 ml HCl 0,1 N + 50 ml citrato hasta 100 ml |
3,4 |
|
4,2 ml HCl 0,1 N + 50 ml citrato hasta 100 ml |
3,6 |
|
Citrato monopotásico 0,1 M y NaOH 0,1 N a 18 oC (14) |
|
|
2,0 ml NaOH 0,1 N + 50 ml citrato hasta 100 ml |
3,8 |
|
9,0 ml NaOH 0,1 N + 50 ml citrato hasta 100 ml |
4,0 |
|
16,3 ml NaOH 0,1 N + 50 ml citrato hasta 100 ml |
4,2 |
|
23,7 ml NaOH 0,1 N + 50 ml citrato hasta 100 ml |
4,4 |
|
31,5 ml NaOH 0,1 N + 50 ml citrato hasta 100 ml |
4,6 |
|
39,2 ml NaOH 0,1 N + 50 ml citrato hasta 100 ml |
4,8 |
|
46,7 ml NaOH 0,1 N + 50 ml citrato hasta 100 ml |
5,0 |
|
54,2 ml NaOH 0,1 N + 50 ml citrato hasta 100 ml |
5,2 |
|
61,0 ml NaOH 0,1 N + 50 ml citrato hasta 100 ml |
5,4 |
|
68,0 ml NaOH 0,1 N + 50 ml citrato hasta 100 ml |
5,6 |
|
74,4 ml NaOH 0,1 N + 50 ml citrato hasta 100 ml |
5,8 |
|
81,2 ml NaOH 0,1 N + 50 ml citrato hasta 100 ml |
6,0 |
C. SÖRENSEN:
Mezclas de borato de SÖRENSEN
|
Composición |
Sörensen 18 oC |
Walbum, pH a |
|||
|
ml de bórax |
ml HCl/NaOH |
10 oC |
40 oC |
70 oC |
|
|
Bórax 0,05 M + HCl 0,1 N |
|||||
|
5,25 |
4,75 |
7,62 |
7,64 |
7,55 |
7,47 |
|
5,50 |
4,50 |
7,94 |
7,98 |
7,86 |
7,76 |
|
5,75 |
4,25 |
8,14 |
8,17 |
8,06 |
7,95 |
|
6,00 |
4,00 |
8,29 |
8,32 |
8,19 |
8,08 |
|
6,50 |
3,50 |
8,51 |
8,54 |
8,40 |
8,28 |
|
7,00 |
3,00 |
8,08 |
8,72 |
8,56 |
8,40 |
|
7,50 |
2,50 |
8,80 |
8,84 |
8,67 |
8,50 |
|
8,00 |
2,00 |
8,91 |
8,96 |
8,77 |
8,59 |
|
8,50 |
1,50 |
9,01 |
9,06 |
8,86 |
8,67 |
|
9,00 |
1,00 |
9,09 |
9,14 |
8,94 |
8,74 |
|
9,50 |
0,50 |
9,17 |
9,22 |
9,01 |
8,80 |
|
10,00 |
0,00 |
9,24 |
9,30 |
9,08 |
8,86 |
|
Bórax 0,05 M + NaOH 0,1 N |
|||||
|
10,0 |
0,0 |
9,24 |
9,30 |
9,08 |
8,86 |
|
9,0 |
1,0 |
9,36 |
9,42 |
9,18 |
8,94 |
|
8,0 |
2,0 |
9,50 |
9,57 |
9,30 |
9,02 |
|
7,0 |
3,0 |
9,68 |
9,76 |
9,44 |
9,12 |
|
6,0 |
4,0 |
9,97 |
10,06 |
9,67 |
9,28 |
Mezclas de fosfato de SÖRENSEN
|
Composición |
pH |
|
Fosfato monopotásico 0,0667 M + fosfato disódico 0,0667 M a 20 oC |
|
|
99,2 ml KH2PO4 + 0,8 ml Na2HPO4 |
5,0 |
|
98,4 ml KH2PO4 + 1,6 ml Na2HPO4 |
5,2 |
|
97,3 ml KH2PO4 + 2,7 ml Na2HPO4 |
5,4 |
|
95,5 ml KH2PO4 + 4,5 ml Na2HPO4 |
5,6 |
|
92,8 ml KH2PO4 + 7,2 ml Na2HPO4 |
5,8 |
|
88,9 ml KH2PO4 + 11,1 ml Na2HPO4 |
6,0 |
|
83,0 ml KH2PO4 + 17,0 ml Na2HPO4 |
6,2 |
|
75,4 ml KH2PO4 + 24,6 ml Na2HPO4 |
6,4 |
|
65,3 ml KH2PO4 + 34,7 ml Na2HPO4 |
6,6 |
|
53,4 ml KH2PO4 + 46,6 ml Na2HPO4 |
6,8 |
|
41,3 ml KH2PO4 + 58,7 ml Na2HPO4 |
7,0 |
|
29,6 ml KH2PO4 + 70,4 ml Na2HPO4 |
7,2 |
|
19,7 ml KH2PO4 + 80,3 ml Na2HPO4 |
7,4 |
|
12,8 ml KH2PO4 + 87,2 ml Na2HPO4 |
7,6 |
|
7,4 ml KH2PO4 + 92,6 ml Na2HPO4 |
7,8 |
|
3,7 ml KH2PO4 + 96,3 ml Na2HPO4 |
8,0 |
C.8 TOXICIDAD PARA GUSANOS DE TIERRA-ENSAYO
CON SUELO ARTIFICIAL
1. MÉTODO.
1.1. INTRODUCCIÓN
En este ensayo de laboratorio, la sustancia con la que se va a experimentar se añade a un suelo artificial en el que se colocan gusanos durante 14 días, Después de este período (que, opcionalmente, puede ser de 7 días) se examina el efecto letal de la sustancia sobre los gusanos. Este ensayo provee un método que permite investigar, a relativamente corto plazo, los efectos causados por sustancias químicas sobre los gusanos, por absorción oral o cutánea.
1.2. DEFINICIÓN Y UNIDADES
CL50: Concentración de una sustancia que se considera la causante de la muerte del 50 % de los animales investigados durante el período de ensayo.
1.3. SUSTANCIA DE REFERENCIA
Se utiliza periódicamente una sustancia de referencia como medio para demostrar que la sensibilidad del sistema de ensayo no ha cambiado de manera significativa.
Se recomienda que esta sustancia sea la cloroacetamida de grado analítico.
1.4. PRINCIPIO DEL ENSAYO
Por ser el suelo un medio variable, para este ensayo se utiliza marga artificial definida cuidadosamente. Los gusanos adultos de la especie Eisenia foetida (véase la nota del anexo) se mantienen en una tierra artificial que se trata con diferentes concentraciones de la sustancia de ensayo. El contenido de los recipientes se extiende sobre una bandeja a los 14 días (opcionalmente, pueden ser 7) del comienzo del ensayo y se cuenten los gusanos supervivientes para cada concentración.
1.5. CRITERIOS DE CALIDAD
El ensayo se proyecta de forma que sea lo más reproducible posible con respecto al sustrato y al organismo de ensayo. La mortalidad en los controles no debe exceder del 10 % al final del ensayo, o de lo contrario este no es válido.
1.6. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
1.6.1. Materiales
1.6.1.1. Sustrato de ensayo
Como sustrato básico de ensayo usa un suelo artificial definido.
|
a) |
Sustrato básico (los porcentajes se expresan en términos de peso en seco:
|
|
b) |
Sustrato de ensayo: El sustrato de ensayo contiene el sustrato básico, la sustancia de ensayo y agua desionizada. El contenido de agua oscila entre el 25 y el 42 % del peso en seco del sustrato básico. El contenido de agua de un sustrato se determina secando una muestra a una temperatura de 105 oC, hasta alcanzar un peso constante. El criterio clave consiste en humedecer la tierra artificial hasta un punto en que no quede agua estancada. Se debe tener cuidado al hacer la mezcla, de manera que se obtenga una distribución uniforme de la sustancia de ensayo y del sustrato. Deberá registrarse la manera en que se introduce la sustancia de ensayo en el sustrato. |
|
c) |
Sustrato de control: El sustrato de control contiene el sustrato básico y agua. Si se usa un agente aditivo, el sustrato de control adicional debe contener la misma cantidad de agente aditivo. |
1.6.1.2. Recipientes de ensayo
Se utilizan recipientes de cristal (cubiertos adecuadamente con membranas, tapaderas o película de plástico con agujeros de ventilación) aproximadamente de un litro de capacidad y llenos de una cantidad de sustrato de ensayo húmedo o de control que sea equivalente a un peso en seco de 500 g de sustrato.
1.6.2. Condiciones del ensayo
Los recipientes se deberán mantener en cámaras climatizadas, a una temperatura de 20 (± 2)o C, con luz continua. La intensidad de la luz será de 400 a 800 lux.
El período de ensayo dura 14 días, aunque, opcíonalmente, se puede evaluar la mortalidad a los 7 días del comienzo del ensayo.
1.6.3. Procedimiento de ensayo
Concentraciones del ensayo
Las concentraciones de la sustancia de ensayo se expresan en peso de sustancia por peso en seco de sustrato básico (mg/kg).
Ensayo para determinar la gama de concentraciones
Por medio de este ensayo se determina la gama de concentraciones que ocasiona porcentajes de mortalidad de 0 a 100; con esta información se establece la gama de concentraciones que se vaya a utilizar en el ensayo definitivo.
La sustancia se estudiará a las siguientes concentraciones: 1 000, 100, 10, 1, 0,1 mg de sustancia/kg de sustrato de ensayo (peso en seco).
Cuando vaya a realizarse un ensayo definitivo, bastará con un lote de ensayo para cada concentración y otro para el sustrato de control sin tratar, cada uno de ellos con diez gusanos, para llevar a cabo el ensayo de determinación de la gama de concentraciones.
Ensayo definitivo
Los resultados del ensayo para determinar la gama de concentraciones se usan para elegir cinco concentraciones como mínimo, en serie geométrica, que abarquen la gama de mortalidad del cero al 100 % y que difieran entre sí en un factor constante que no exceda de 1,8.
En los ensayos en que se utilice esta serie de concentraciones se estimará el valor de CL50 y de sus límites de confianza de la manera más precisa posible.
En el ensayo definitivo se utilizan, como mínimo, cuatro lotes de ensayo por concentración y cuatro sustratos de control sin tratar, cada uno de ellos con diez gusanos. Los resultados de estos lotes reproducidos son la media y la desviación típica.
Cuando de dos concentraciones consecutivas, en una proporción de 1,8, solo se obtienen los porcentajes de mortalidad de 0 % y 100 %, estos dos valores son suficientes para indicar la gama en la que se encuentra el valor de CL50.
Mezcla del sustrato de ensayo básico y de la sustancia de ensayo
Siempre que sea posible, el sustrato de ensayo no deberá estar compuesto de ningún otro agente adicional que no sea agua. Inmediatamente antes del comienzo del ensayo, se mezcla una emulsión o dispersión de la sustancia de ensayo en agua desionizada disolvente con el sustrato de ensayo básico, o se rocía dicha emulsión sobre éste de manera uniforme con un rociador cromatográfico fino o con un aparato similar.
Si la sustancia de ensayo no es soluble en agua, puede disolverse en el mínimo volumen posible de un disolvente orgánico apropiado (por ejemplo, hexano, acetona o cloroformo).
Para solubilizar, dispersar o emulsionar la sustancia de ensayo solo pueden utilizarse los agentes que se volatilizan rápidamente. El sustrato de ensayo debe ser ventilado antes de usarlo. Se repondrá la cantidad de agua evaporada. El sustrato de control deberá contener la misma cantidad de agente aditivo.
Si la sustancia de ensayo no fuese soluble, dispersable o emulsionable en disolventes orgánicos, se mezclarán 10 g de una mezcla de arena de cuarzo finamente molido y la cantidad necesaria de sustancia de ensayo para tratar 500 g (peso en seco) de suelo artificial, con 490 g (peso en seco) de sustrato de ensayo.
Por cada lote de ensayo, se coloca en cada recipiente de cristal una cantidad de sustrato de ensayo húmedo equivalente a 500 g de peso en seco, y en la superficie del mismo se colocan diez gusanos, que han sido acondicionados durante 24 horas en un sustrato básico húmedo similar y a los que, después de lavarlos rápidamente, se les ha extraído el exceso de agua con papel de filtro antes de usarlos.
Los recipientes se cubren con tapas de plástico perforado, platos o película para evitar el secado del sustrato, y se guardan bajo las condiciones de ensayo durante 14 días.
Se evaluarán los resultados después de 14 días (u opcionalmente, después de 7 días) del comienzo del ensayo. El sustrato se extiende en una placa de vidrio o de acero inoxidable. Se examinan los gusanos y se determina el número de supervivientes. Los gusanos te consideran muertos si no responden a un suave estímulo mecánico en la parte frontal de su cuerpo.
Cuando el examen se efectúa a los 7 días, el recipiente se vuelve a llenar con el sustrato y los gusanos supervivientes se colocan otra vez en la superficie del mismo.
1.6.4. Organismos de ensayo
Los organismos de ensayo deberán ser adultos de la especie Eisenia foetida (véase la nota en el anexo) (como mínimo con dos meses de edad y con clitelo) y de un peso húmedo de 300 a 600 mg (véase el anexo para el método de reproducción).
2. DATOS
2.1. TRATAMIENTO Y EVALUACIÓN DE LOS RESULTADOS
Las concentraciones de la sustancia ensayada se registran con referencia a los porcentajes correspondientes de gusanos de tierra muertos.
Cuando los datos son adecuados, el valor de CL50 y los límites de fiabilidad(p = 0,05) se determinan por medio de métodos estándar (como el de Litchfield y Wilcoxon, 1949, o un método equivalente). El valor de CL50 debe ser expresado en mg de sustancia de ensayo por kg de sustrato de ensayo (peso en seco).
En los casos en que la pendiente de la curva de concentración es tan pronunciada que no puede calcularse el valor de CL50, se considera suficiente una estimación gráfica de este valor.
Cuando de dos concentraciones consecutivas en una proporción de 1,8 solo se obtienen porcentajes de mortalidad de 0 % y 100 %, estos dos valores son suficientes para indicar la gama en la que se encuentra el valor de CL50.
3. INFORMES
3.1. INFORME DEL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
declaración en la que conste que el ensayo se ha efectuado de conformidad con los criterios de calidad mencionados anteriormente, |
|
— |
tipo de ensayo efectuado (ensayo de determinación de la gama de concentraciones y/o ensayo definitivo), |
|
— |
descripción exacta de las condiciones del ensayo o declaración en la que conste que el ensayo se ha efectuado de conformidad con el método; se informará de cualesquiera diferencias con respecto a dicho método, |
|
— |
descripción exacta de la manera en que se ha mezclado la sustancia de ensayo con el sustrato de ensayo básico, |
|
— |
información acerca de los organismos de ensayo (especie, edad, peso medio y gama de variación, condiciones relativas a reproducción y cría, abastecedor), |
|
— |
método usado en la determinación del valor de CL50, |
|
— |
resultados del ensayo, incluidos todos los datos utilizados, |
|
— |
descripción de los síntomas o de los cambios en el comportamiento de los organismos de ensayo que se hayan observado, |
|
— |
mortalidad en los sustratos de control, |
|
— |
valor de CL50 o la máxima concentración con mortalidad y la mínima con una mortalidad del 100 %, después de 14 días (y opcionalmente después de siete días) del comienzo del ensayo, |
|
— |
trazado de la curva de concentración — respuesta, |
|
— |
resultados obtenidos con la sustancia de referencia, ya sea en relación con el ensayo actual o con ejercicios previos de control de calidad. |
4. REFERENCIAS
|
(1) |
OCDE, París, 1981, Directriz para pruebas 207. Decisión del Consejo C(81) 30 final. |
|
(2) |
Edwards, D. A. and Lofty, J. R., 1977. Biology of Earthworms. London: Chapman and Hall, 331 pp. |
|
(3) |
Bouche, M. B. 1972. Lombriciens de France, Ecologie et Systematique. Publ. Institut National de la Recherche Agronomique, 671 pp. |
|
(4) |
Litchfield, J. T. and Wilcoxon, F., 1949. A simpliefied method of evaluating dose-effect experiments.J. Pharm. Exp. Therap., 96, 99-113. |
|
(5) |
CEE 1983. Development of a standardized laboratory method for assessing the toxictty of chemical substances to earthworms. Report EUR 8714 EN. |
|
(6) |
Umweltbundesamt/Biologische Bundesanstalt für Land- und Forstwirtschaft, Berlin, 1984, Verfahrens vorschlag «Toxizitätstest am Regenwurm Eisern a foetid a in künstlichem Boden», in: Rudolph/Boje: Ökotoxikologie, ecomed, Landsberg, 1986. |
Apéndice
Reproducción y cría de los gusanos con anterioridad at ensayo
A fines de reproducción, se colocan en una caja de cría con sustrato fresco de 30 a 50 gusanos adultos, y se sacan después de 14 días. Estos animales se pueden utilizar para lotes de reproducción adicionales. Los gusanos nacidos de los capullos se utilizan en los ensayos una vez que han alcanzado la madurez (según las condiciones prescritas, después de 2 a 4 meses).
Condiciones de reproducción y cría
|
Cámara climatizada |
: |
a una temperatura de 20 (± 2)o C, preferiblemente con luz continua (intensidad de 400 a 800 lux). |
|
Cajas de reproducción |
: |
recipientes poco profundos apropiados, con un volumen de 10 a 20 litros. |
|
Sustrato |
: |
Eisenia foetida: Se puede criar en los excrementos de varios animales. Como medio de crianza se recomienda utilizar una mezcla con un 50 % en volumen de turba y el otro 50 % de excrementos de vaca o caballo. Este medio debe tener un valor aproximado de pH entre 6 y 7 (regulado con carbonato cálcico) y una conductividad iónica baja (menos de 6 mmhos o una concentración de sal del 0,5 %). El sustrato debe estar húmedo, pero no demasiado mojado. Además del método expuesto anteriormente, se pueden utilizar otros procedimientos con éxito. |
Nota: Existen dos razas de Eisenia foetida, que algunos taxonomistas han suhdividido en especies (Bouche, 1972). Estas dos razas son morfológicamente similares, aunque una de ellas, la Eisenia foetida foetida, exhibe rayas o bandas transversales típicas en los segmentos, mientras que a la otra, la Eisenia foetida andrei, de color rojizo jaspeado, le faltan. Siempre que sea posible se utilizará la Eisenia foetida andrei. Se pueden utilizar otras especies, siempre que se disponga de la metodología necesaria.
C.9. BIODEGRADACIÓN
PRUEBA ZAHNWELLENS
1. MÉTODO
1.1. INTRODUCCIÓN
El objeto de este método es la evaluación de la máxima degradación potencial de sustancias orgánicas solubles en agua y no volátiles, cuando se encuentran expuestas a concentraciones relativamente elevadas de microorganismos en un ensayo estático.
Puede producirse una absorción físico-química sobre los sólidos en suspensión y esto hay que tenerlo en cuenta a la hora de interpretar los resultados (véase el punto 3.2).
Las sustancias que vayan a estudiarse serán utilizadas en concentraciones equivalentes a valores de COD entre los 50 y 400 mg/litro o valores de DQO entre los 100 y 1 000 mg/litro(COD = carbón orgánico disuelto; DQO = demanda química de oxígeno). Estas concentraciones relativamente elevadas tienen la ventaja de ser analíticamente fiables. Los compuestos con propiedades tóxicas pueden retardar o inhibir el proceso de degradación.
En este método, la medida de la concentración de carbón orgánico disuelto, o la demanda de oxígeno químico, se utiliza para calcular la biodegradación máxima de la sustancia objeto del ensayo.
La utilización simultánea de un método analítico específico puede permitir calcular la biodegradación primaria de la sustancia (desaparición de la estructura química madre).
Este método es aplicable solamente a aquellas sustancias orgánicas que, a la concentración utilizada en el ensayo:
|
— |
sean solubles en agua en las condiciones del ensayo, |
|
— |
tengan una presión de vapor insignificante en las condiciones del ensayo, |
|
— |
no provoquen inhibición en las bacterias, |
|
— |
sean absorbidas dentro del sistema del ensayo solo hasta un cierto punto limitado, |
|
— |
no se pierdan a causa de la formación de espuma a partir de la. solución del ensayo. |
La información sobre las proporciones relativas de los principales componentes del material del ensayo será necesaria para la interpretación de los resultados obtenidos, particularmente en los casos en que los resultados sean reducidos o marginales.
La información sobre la toxicidad de la sustancia para los microorganismos resulta conveniente para la interpretación de resultados reducidos y para la selección de las concentraciones apropiadas para el ensayo.
1.2. DEFINICIONES Y UNIDADES
La cantidad de degradación alcanzada al final del ensayo se presenta como la biodegradabilidad en la prueba Zahn — Wellens:

donde:
|
DT |
= |
biodegradación ( %) en el tiempo T |
|
CD |
= |
valores de COD (o DQO) en la mezcla del ensayo medidos tres horas después del comienzo del mismo (mg/l) (COD = Carbón orgánico disueleo; DQO = demanda química de oxígeno) |
|
CT |
= |
valores de COD o DQO e la mezcla del ensayo en el momento en que se toma la muestra (mg/l) |
|
CV |
= |
valores de COD o DQO del blanco en el momento de la toma de la muestra (mg/l) |
|
CVd |
= |
valores de COD y DQO en el blanco, medidos 3 horas después del comienzo de la prueba (mg/l). |
El alcance de la degradación se redondea hasta llegar a completar la unidad de porcentaje más cercana.
El porcentaje de degradación se expresa como el porcentaje de supresión de COD (o DQO) de la sustancia objeto del ensayo.
La diferencia entre los valores medidos tras 3 horas y el valor inical calculado, o preferiblemente medido, puede proporcionar una información útil sobre la eliminación de la sustancia (véase el punto 3.2, «Interpretación de los resultados»).
1.3. SUSTANCIAS DE REFERENCIA
En algunos casos, cuando se están investigando sustancias nuevas, las sustancias de referencia pueden resultar útiles; no obstante, todavía no se pueden recomendar sustancias de referencia específicas.
1.4. PRINCIPIO DEL MÉTODO DE ENSAYO
Se ponen juntos, en un recipiente de vidrio de 1 a 4 litros de capacidad y equipado con un mezclador y un aireador, lodo activado, nutrientes minerales y et material del ensayo como única fuente de carbón en solución acuosa. Se remueve y se airea la mezcla a 20-25 oC, bajo una iluminación difusa o en un cuarto oscuro por un período de hasta 28 días. El proceso de degradación se controla por medio de la determinación de los valores de COD (o DQO) en la solución filtrada a intervalos diarios o a intervalos regulares que resulten apropiados. La relación entre la cantidad de COD (DQO) eliminada tras cada intervalo y el valor 3 horas después del comienzo, se expresa en forma de porcentaje de biodegradación y sirve de medida del alcance de la degradación en ese momento. El resultado se indica en un gráfico en función del tiempo para obtener así la curva de biodegradación.
Cuando se utiliza un método analítico específico, pueden medirse los cambios en la concentración de la molécula madre debidos a la biodegradación (biodegradabilidad primaria),
1.5. CRITERIOS DE CALIDAD
Ha quedado demostrado que la reproducibilidad de esta prueba es satisfactoria en una prueba en varios centros.
La sensibilidad de este método viene determinada en gran medida por la variabilidad del blanco y, en menor medida, por la precisión de la determinación del carbón orgánico disuelto y el nivel del compuesto del ensayo en Ja mezcla liquida.
1.6. DESCRIPCIÓN DEL PROCEDIMIENTO DEL ENSAYO
1.6.1. Preparativos
1.6.1.1. Reactivos
Agua de ensayo: agua potable con un cotenido de carbón orgánico menor de 5 mg/l. La concentración de iones de calcio y magnesio juntos no puede exceder los 2,7 mmoles/l; de otra manera, es necesaria una dilución suficiente con agua desionizada o destilada.
|
Ácido sulfúrico, reactivo analítico (R.A.): |
50 g/l |
|
Solución de hidróxído de sodio R.A.: |
40 g/l |
|
Solución de nutriente mineral: disuélvase en un litro de agua desionizada: |
|
|
cloruro de amonio, NH4Cl, R.A.: |
38,5 g |
|
dihidrógeno fosfato de sodio, NaH2PO4,. 2H2O, R.A.: |
33,4 g |
|
dihidrógeno fosfato de potasio, KH2PO4, R.A.: |
8,5 g |
|
monohidrógeno fosfato de dipotasio, K2HPO4, R.A.: |
21,75 g. |
La mezcla sirve a la vez de elemento nutritivo y de sistema tampón.
1.6.1.2. Aparatos
Recipientes de vidrio de un volumen de 1 a 4 litros (por ejemplo, recipientes cilindricos).
Mezclador con un agitador de vidrio o metal colocado en un eje apropiado. (El agitador debe girar a 5-10 cm por encima del fondo del recipiente). Se puede utilizar en su lugar un agitador magnético con una varilla de 7-10 cm de larga.
Un tubo de vidrio de 2 a 4 mm de diámetro interior para introducir aire. La abertura del tubo debe estar a, más o menos, 1 cm por encima del fondo del recipiente.
Una centrífuga (unos 3 550 g).
Un medidor de pH.
Un medidor de oxígeno disuelto.
Filtros de papel.
Un aparato de filtración por membrana.
Filtros de membrana, tamaño de los poros 0,45 um. Los filtros de membrana son apropiados si es seguro que ni desprenden carbón ni absorben la sustancia en la fase de filtración.
Un equipo analítico para determinar el contenido de carbón orgánico y la demanda química de oxígeno.
1.6.1.3. Preparación del inóculo
Se lava (repetidamente) el lodo activado recogido en una planta de tratamiento biológico, centrifugándolo o dejándolo sedimentarse con agua de iguales características a la que se usa para el ensayo (antes citado).
El lodo activado tiene que estar en condiciones adecuadas. Este fango puede encontrarse en una planta depuradora de aguas residuales que funcione debidamente. Para coger tantas especies o cepas de bacterias diferentes como sea posible, puede ser preferible mezclar inóculos de diferentes fuentes (por ejemplo, diferentes plantas depuradoras, extractos de suelos, aguas de río, etc.). La mezcla debe ser tratada como se describe anteriormente.
Para comprobar la actividad del lodo activado, véase «Control funcional».
1.6.1.4. Preparación de las soluciones del ensayo
Pónganse, en el recipiente que se utiliza para el ensayo, 500 ml de agua de ensayo, 2,5 ml/l de una solución de nutrientes minerales y lodo activado en una cantidad equivalente a 0,2 a 1,0 g/l de materia seca en la mezcla final. Añádase suficiente solución madre de la sustancia a ensayar como para que en la mezcla final se obtenga una concentración de COD de 50 a 400 mg/l. Los valores correspondientes de DQO son 100-1 000 mg/l. Añádase agua de ensayo hasta llegar a un volumen total de 1 a 4 litros. El volumen toral que se elija dependerá del número de muestras que se vayan a tomar para las determinaciones del COD y DQO y las cantidades necesarias para el procedimiento analítico.
Normalmente, puede considerarse satisfactorio un volumen de 2 litros. Se dispone por lo menos un recipiente de control (blanco), paralelamente a cada una de las series de ensayo, que contenga tan solo una solución de lodo activado y la solución de nutrientes minerales, que se completará con agua de ensayo hasta alcanzar un volumen total igual al de los recipientes de ensayo.
1.6.2. Realización del ensayo
Se agitan los recipientes del ensayo con agitadores magnéticos o hélices bajo una iluminación difusa o en un cuarto oscuro a 20-25o C. La aireación se efectúa con aire comprimido limpiado por medio de un filtro de algodón y lana y un frasco lavador si fuera necesario. Hay que asegurarse de que el lodo no sedimenta y de que la concentración de oxígeno no cae por debajo de los 2 mg/l.
Hay que controlar el pH a intervalos regulares (por ejemplo, cada día) y, si fuera necesario, corregirlo ajustándolo a 7-8.
Las pérdidas por evaporación se compensan, justo antes de tomar cada muestra, con agua desionizada o destilada en las cantidades necesarias. Un buen procedimiento es marcar el nivel del líquido sobre el recipiente antes de iniciar el ensayo. Se harán marcas nuevas después de cada toma (sin aireación y sín remover). Las primeras muestras se toman siempre 3 horas después del comienzo del ensayo, para detectar la absorción del material de ensayó por parte del lodo activado.
La eliminación del material de ensayo se controla por medio de las determinaciones de COD y DQO hechas diariamente o a intervalos regulares. Las muestras del recipiente utilizado para el ensayo y del recipiente de control son filtradas con un filtro de papel cuidadosamente lavado. Se desechan los primeros 5 ml de la solución filtrada. Los lodos difíciles de filtrar pueden ser separados previamente centrifugándolos durante 10 minutos. Las determinaciones de COD y DQO deben hacerse por lo menos por duplicado. El ensayo se lleva a cabo durante un período de hasta 28 días.
Nota: Las muestras que todavía estén turbias son filtradas con filtros de membrana. Los filtros de membrana no han de desprender ni absorber material orgánico alguno.
Control funcional del lodo activado
En un recipiente que contenga una sustancia conocida deben llevarse a cabo paralelamente cada serie de pruebas, con vistas a controlar la capacidad funcional del lodo activado. Se ha comprobado que el dietilcnoglicol es de utilidad para este propósito.
Adaptación
Si los análisis se llevan a cabo a intervalos relativamente cortos (por ejemplo, diariamente), se puede reconocer claramente la adaptación por medio de la curva de degradación (véase la figura 2). Así pues, el ensayo no debe iniciarse justo antes del fin de semana.
Si la adaptación tiene lugar al final del período, el ensayo puede prolongarse hasta que finalice la degradación.
Nota: Si fuera necesario un conocimiento más amplio del comportamiento del lodo adaptado, se expone de nuevo el mismo lodo activado al mismo material del ensayo, de acuerdo con el siguiente procedimiento:
Desconéctense el agitador y el aireador y permítase que el lodo activado se sedimente. Vacíese el líquido sobrenadante, añádase agua de ensayo hasta alcanzar los 2 litros, agítese durante 15 minutos y déjese sedimentar de nuevo. Después de haber vaciado de nuevo el líquido sobrenadante, utilícese el lodo que queda para repetir el ensayo con el mismo material, de acuerdo con los puntos 1.6.1.4 y 1.6.2. Se puede aislar el lodo activado centrifugando en vez de dejándolo sedimentar.
El lodo adaptado puede mezclarse con lodo fresco hasta alcanzar un total de 0,2 a 1 g de peso en seco, por litro.
Medios analíticos
Normalmente, se filtran las muestras con un filtro de papel cuidadosamente lavado (para lavarlo, utilícese agua desionizada).
Las muestras que sigan estando turbias se filtrarán con filtros de membrana (0,45 μm).
La concentración de COD se determina por duplicado en las muestras de líquido filtrado (se desechan los primeros 5 ml) por medio del instrumento TOC. Si el líquido filtrado no puede ser analizado el mismo día, tiene que almacenarse en el refrigerador hasta el día siguiente. No se recomienda un almacenaje más prolongado.
La concentración de DQO se determina por duplicado en las muestras de líquido filtrado con un sistema analítico siguiendo el procedimiento descrito en la referencia (2) posterior.
2. DATOS Y EVALUACIÓN
Las concentraciones de COD y DQO se determinan en las muestras, por lo menos por duplicado, de acuerdo con el punto 1.6.2. La degradación en el tiempo T se calcula según la fórmula (con definiciones) dada en el punto 1.2.
El alcance de la degradación se redondea hasta llegar a completar la unidad de porcentaje más cercana. La cantidad de degradación lograda al final del ensayo se presenta como la «biodegradabilidad en la prueba Zahn — Wellens».
Nota: Si se logra una degradación completa antes de que termine el período de duración del ensayo y si este resultado es confirmado por un segundo análisis al día siguiente, el ensayo puede darse por concluido.
3. PRESENTACIÓN DEL INFORME
3.1. INFORME SOBRE EL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
la concentración inicial de la sustancia, |
|
— |
cualquier otra información y los resultados experimentales relativos a la sustancia objeto del ensayo, a la sustancia de referencia, si se utiliza alguna, y al control, |
|
— |
la concentración después de tres horas, |
|
— |
la curva de biodegradación con una descripción, |
|
— |
fecha y lugar en que se tomaron las muestras de los organismos utilizados en el ensayo, status de adaptación, concentración utilizada, etc., |
|
— |
causas científicas de cualquier cambio en el procedimiento del ensayo. |
3.2. INTERPRETACIÓN DE LOS RESULTADOS
La eliminación de COD (o DQO) que tiene lugar gradualmente, durante días o semanas, indica que la sustancia objeto del ensayo se está biodegradando.
No obstante, la absorción físico-química puede, en algunos casos, desempeñar un papel, y esto se pone de manifiesto cuando hay una eliminación completa o parcial desde el principio, dentro de las tres primeras horas, y la diferencia entre las mezclas líquidas sobrenadantes del control y del ensayo se mantienen a un nivel inesperadamente bajo.
Son necesarias pruebas adicionales si se quiere establecer una diferencia entre biodegradación (o biodegradación parcial) y absorción.
Esto puede hacerse de varias maneras, pero lo más conveniente es usar el sobrenadante como inóculo en un ensayo de base fija (una prueba respírométrica preferentemente).
Las sustancias que en este ensayo den una eliminación de COD (o DQO) alta, y no debida a la absorción, deben ser consideradas como potencialmente biodegradables. Una eliminación parcial y no debida a la absorción indica que el producto químico puede sufrir al menos cierta biodegradación. Una eliminación baja o nula de COD (o DQO) puede deberse a que la sustancia objeto del ensayo provoca la inhibición de los microorganismos. Esto puede asimismo ponerse de manifiesto a través de una desintegración y pérdida del lodo, produciendo sobrenadantes turbios. Se debe repetir el ensayo utilizando una concentración inferior de la sustancia objeto de estudio.
La utilización de un método analítico específico para el compuesto, o de una sustancia del ensayo marcada con 14C, puede permitir una mayor sensibilidad. En el caso del compuesto de ensayo marcado con 14 C la obtención de 14CO2 confirmará que se ha producido biódegradación.
Cuando los resultados se presenten en términos de bíodegradación primaria, se debe dar, si fuera posible, una explicación sobre el cambio de estructura química que lleva a la pérdida de respuesta de la sustancia madre utilizada en el ensayo.
La validación del método analítico tiene que presentarse junto con la respuesta encontrada con el medio de ensayo control (blanco).
REFERENCIAS
|
(1) |
OCDE, París, 1981, Directriz para pruebas 302 B. Decisión del Consejo C(81) 30 final. |
|
(2) |
anexo V C.9 Degradación: Demanda química de oxígeno, Directiva 84/449/CEE de la Comisión, Diario Oficial de las Comunidades Europeas No L 251 de 19.9.1984. |
Anexo
EJEMPLO DE EVALUACIÓN
|
Compuesto orgánico: |
Ácido 4-Etoxibenzoico |
|
Concentración teórica de ensayo: |
600 mg/l |
|
COD teórico: |
390 mg/l |
|
inóculo: |
Planta depuradora de aguas residuales . . . |
|
Concentración: |
1 g de material seco/litro |
|
Status de adaptación: |
No adaptado |
|
Análisis: |
Determinación de COD |
|
Cantidad de muestra: |
3 ml |
|
Sustancia de control: |
Dietilenglicol |
|
Toxicidad del compuesta: |
Sin efectos tóxicos por debajo de los 1 000 mg/l. Ensayo utilizado: Ensayo de fermentación con tubos. |
|
Tiempo de ensayo |
Sustancia de control |
Sustancia de ensayo |
|||||
|
COD (15) del control mg/l |
COD (15) mg/l |
COD neto mg/l |
Degradación % |
COD (15) mg/l |
COD neto mg/l |
Degradación ( %) |
|
|
0 |
— |
— |
300,0 |
— |
— |
390,0 |
_ |
|
3h |
4,0 |
298,0 |
294,0 |
2 |
371,6 |
367,6 |
6 |
|
1 d |
6,1 |
288,3 |
282,2 |
6 |
373,3 |
367,2 |
6 |
|
2d |
5,0 |
281,2 |
276,2 |
8 |
360,0 |
355,0 |
9 |
|
5 d |
6,3 |
270,5 |
264,2 |
12 |
193,8 |
187,5 |
52 |
|
6d |
7,4 |
253,3 |
245,9 |
18 |
143,9 |
136,5 |
65 |
|
7d |
11,3 |
212,5 |
201,2 |
33 |
104,5 |
93,2 |
76 |
|
8 d |
7,8 |
142,5 |
134,7 |
55 |
58,9 |
51,1 |
87 |
|
9 d |
7,0 |
35,0 |
28,0 |
91 |
18,1 |
11,1 |
97 |
|
10 d |
18,0 |
37,0 |
19,0 |
94 |
20,0 |
2,0 |
99 |
Figura 1
Ejemplos de curvas de biodegradación

Figura 2
Ejemplos de adaptación del lodo
C.10. BIODEGRADACIÓN
ENSAYO DE SIMULACIÓN CON LODO ACTIVADO
1. MÉTODO
1.1. INTRODUCCIÓN
1.1.1. Comentarios generales
Este método solo es aplicable a aquellas sustancias orgánicas que, a la concentración utilizada en el ensayo:
|
— |
sean tan solubles en agua como sea necesario para la preparación de las soluciones de ensayo, |
|
— |
sometidas a las condiciones del ensayo, tengan una presión de vapor insignificante, |
|
— |
no provoquen inhibición en las bacterias. |
La información sobre las proporciones relativas de los principales componentes del material de ensayo será necesaria para la interpretación de los resultados obtenidos, particularmente en los casos en que los resultados sean' reducidos o marginales.
La información sobre la toxicidad de la sustancia para los microorganismos resulta conveniente para la interpretación de resultados reducidos y para la selección de las concentraciones apropiadas para el ensayo.
1.1.2. Determinación de la bíodegradabilidad máxima (análisis del COD/DQO)
El objeto de este método es determinar la biodegradabilidad máxima a través de la medición de la eliminación de la sustancia y de cualquier metabolito en un modelo de planta de lodo activado con una concentración equivalente a > 12 mg de COD/1 (o aproximadamente 40 mg de DQO/l). 20 mg de COD/l parece ser la óptima.
(COD = carbón orgánico disuelto por litro, DQO = demanda química de oxígeno). El contenido de carbón orgánico (o la demanda química de oxígeno) del material de ensayo tiene que ser determinado.
1.1.3. Determinación de la biodegradabilidad primaria (análisis específico)
El objeto del método es determinar la biodegradabilidad primaria de una sustancia en un modelo de planta de lodo activado, con una concentración de unos 20 mg/l, por medio de un método analítico específico (se puede utilizar una concentración superior o inferior si el método analítico y su toxicidad lo permiten). Esto permite la valoración de la biodegradabilidad primaria de la sustancia (desaparición de la estructura química madre).
El objeto de este método no es la determinación de la mineralización de la sustancia ensayada.
Se tiene que contar con un método analítico adecuado para la determinación de la sustancia.
1.2. DEFINICIONES Y UNIDADES
1.2.1. Análisis del COD/DQO
El grado de eliminación de la sustancia viene dado por:
|
|
[1(a)] |
donde:
|
GE |
= |
grado de eliminación en porcentaje de COD (o DQO) dentro del tiempo de retención medio dado con respecto al material de ensayo |
|
T |
= |
concentración del material de ensayo en el afluente en mg de COD/litro (o mg de DQO/litro) |
|
E |
= |
concentración de COD (o DQO) en el efluente de la unidad de ensayo en mg de COD/litro (o DQP/litro) |
|
Eo |
= |
concentración de COD (o DQO) en el efluente del blanco en mg de COD/litro (o DQO/litro). |
La degradación se expresa, como la eliminación en porcentaje de COD (o DQO) dentro del tiempo de retención dado con respecto al material de ensayo.
1.2.2. Análisis específico
La eliminación porcentual de la sustancia ensayada de su fase acuosa (R2) dentro del tiempo medio de retención dado viene dada por:
|
|
[1(b)] |
donde:
|
CI |
= |
concentración de la sustancia en el afluente de la unidad en la que se realiza el ensayo (mg de sustancia por litro, determinada por medio de un análisis específico) |
|
C O |
= |
concentración de la sustancia en el efluente de la unidad en la que se realiza el ensayo (mg de sustancia por litro, determinada por medio de un análisis específico). |
1.3. SUSTANCIAS DE REFERENCIA
En algunos casos, cuando se está investigando una sustancia nueva, las sustancias de referencia pueden resultar útiles; no obstante, todavía no pueden recomendarse sustancias de referencia específicas.
1.4. PRINCIPIOS DE LOS MÉTODOS DE ENSAYO
Para determinar la biodegradabilidad máxima, se organizan paralelamente dos unidades piloto de lodo activado (unidades de ensayo confirmatorio OCDE o de depósito poroso). Se vierte la sustancia objeto del ensayo en el afluente (aguas residuales domésticas o sintéticas) de una de las unidades, mientras que la otra recibe tan solo aguas residuales. Para la determinación de la biodegradación primaria con un análisis específico en el afluente y el efluente, solo se utiliza una unidad.
Las concentraciones de COD (o DQO) se miden en los efluentes, o se determinan las concentraciones de sustancia por medio de un análisis específico.
El COD debido al material del ensayo no es medido sino simplemente especificado.
Cuando se realizan las mediciones de COG (o DQO), se supone que las diferencias entre las concentraciones medias de los efluentes del ensayo y del control se deben a material del ensayo no degradado.
Cuando se realizan análisis específicos, se pueden medir los cambios en la concentración de la molécula madre (biodegradación primaria).
Las unidades pueden manejarse siguiendo el «método de unidades acopladas», por medio de un procedimiento de transinoculación.
1.5. CRITERIOS DE CALIDAD
La concentración inicial de la sustancia depende del tipo de análisis que se vaya a realizar y su limitación.
1.6. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
1.6.1. Preparación
1.6.1.1. Aparatos
Se necesitan un par de unidades del mismo tipo, excepto cuando se realizan análisis específicos. Se pueden utilizar dos tipos de dispositivos:
La prueba confirmatoria OCDE:
E1 equipo (anexo I) consiste en un recipiente de almacenaje (A) para las aguas residuales sintéticas, una bomba de dosificación (B), un recipiente de aireación (C), un separador (D), una bomba de aire (E) para reciclar el lodo activado y un recipiente (F) para recoger el efluente tratado.
Los recipientes (A) y (F) tienen que ser de vidrio o de un plástico apropiado y deben tener una capacidad de 24 litros por los menos. La bomba (B) tiene que proporcionar una corriente constante de aguas residuales sintéticas en el recipiente de aireación; se puede usar cualquier sistema conveniente, con tal que la corriente de entrada y la concentración queden aseguradas. Durante el funcionamiento normal, se fija la altura del separador de modo que el volumen contenido en el recipiente de aireación sea de 3 litros de mezcla líquida. Se cuelga en el recipiente (C), en el vértice del cono, un cubo de aireación sintetizado (G). La cantidad de aire soplado a través del aireador puede controlarse por medio de un contador de corriente.
La bomba de aire (E) se coloca de manera que el lodo activado del separador sea continua y regularmente reciclado en el recipiente de aireación (C).
El depósito poroso
El depósito poroso está construido a base de láminas de polietileno poroso (2 mm de espesor, tamaño máximo de los poros: 95 μm), que adoptan la forma de cilindros de 14 cm de diámetro con una base cónica a 45o (figuras 1 y 2 del anexo II). El depósito poroso se encuentra en el interior de un recipiente impermeable, hecho de un plástico conveniente, de 15 cm de diámetro y con una salida a una altura de 17,2 cm en la parte cilíndrica, que determina el volumen (3 1) en el depósito. Hay un anillo rígido de soporte, hecho de un plástico conveniente, alrededor de la parte superior del recipiente interior, de modo que hay un espacio de 0,5 cm para el efluente entre el recipiente interior y el exterior.
Los depósitos porosos pueden montarse en la base de un baño maría termostáticamente regulado. Se suministra aire en la base del recipiente interior, en el que se colocan unos difusores apropiados.
Los recipientes (A) y (E) tienen que ser de vidrio o de un plástico conveniente y deben tener una capacidad de 24 litros por lo menos. La bomba (B) tiene que proporcionar una corriente constante de aguas residuales sintéticas al recipiente de aireación; puede usarse cualquier sistema apropiado, con tal que se asegure la corriente de entrada y la concentración.
Se necesitan depósitos porosos interiores de recambio para reemplazar a los que puedan bloquearse; los depósitos bloqueados se limpian por medio de una inmersión de 24 horas en una solución de hipoclorito seguida de un minucioso lavado con agua del grifo.
1.6.1.2. Filtración
Aparatos de filtración por membrana y filtros de membrana con poros de un tamaño de 0,45 μm. Los filtros de membrana son apropiados si es seguro que ni desprenden carbón ni absorben la sustancia en la fase de filtración.
1.6.1.3. Aguas residuales
Se puede usar tanto afluente sintético apropiado como aguas residuales domésticas.
Ejemplo de afluente sintético:
disuélvase en cada litro de agua del grifo:
|
peptona |
160 mg |
|
extracto de carne |
110 mg |
|
urea |
30 mg |
|
NaCl |
7 mg |
|
CaCl2.2H2O |
4 mg |
|
MgS04.7H2O |
2 mg |
|
K2HPO4 |
28 mg. |
Aguas residuales domésticas:
Deben recogerse frescas cada día en una cañería de desagüe de un tanque de sedimentación primaria de una planta de tratamiento que trate aguas residuales domésticas predominantemente.
1.6.1.4. Solución madre del material del ensayo
Debe prepararse una solución del material del ensayo (por ejemplo, 1 %) para ser añadida a la unidad en que se realiza el ensayo. La concentración del material tiene que determinarse, de modo que se conozca el volumen apropiado que hay que añadir a las aguas residuales o directamente a la unidad por medio de una segunda bomba, para obtener la concentración necesaria para el ensayo.
1.6.1.5. Inόculo
Nota: Cuando se utilizan aguas residuales domésticas, no vale la pena utilizar un inόculo de baja concentración bacterial, sino que se puede utilizar lodo activado.
Pueden utilizarse diversos inóculos.
Tres ejemplos de inóculos apropiados son:
|
a) |
inόculo procedente de un efluente secundario: El inόculo debe obtenerse a partir de un efluente secundario de buena calidad recogido en una planta de tratamiento que trate predominantemente aguas residuales domésticas. Hay que mantener el efluente en condiciones aeróbicas durante el tiempo que va desde la recogida de las muestras hasta su uso. Para preparar el inόculo, se filtra la muestra en un filtro poco fino y se desechan los primeros 200 ml. El líquido así filtrado es mantenido en condiciones aeróbicas hasta su uso. El inόculo tiene que ser usado el mismo día que es recogido. Se deben usar por lo menos 3 ml para la inoculación; |
|
b) |
inόculo compuesto: Inόculo procedente de un efluente secundario: Véase la descripción anterior. Inóculo procedente de suelo: Se suspenden 100 g de suelo de jardín (fértil, no estéril) en 1 000 ml de agua potable sin cloro (los suelos con una proporción extremadamente elevada de arcilla, arena o humus son inadecuados). Tras agitar la mezcla, se deja que la suspensión se deposite durante 30 minutos. Se filtra el sobrenadante con un filtro de papel poco fino y se desechan los primeros 200 ml. Se airea el líquido filtrado inmediatamente y hasta su uso. El inόculo tiene que usarse el mismo día en que es recogido. Inόculo precedente de aguas superficiales: Un tercer inόculo parcial se obtiene de aguas superficiales mesosapróbìcas. La muestra se filtra con un papel poco fino y se desechan los primeros 200 ml. El líquido así filtrado es mantenido en condiciones aeróbicas hasta su uso. El inóculo tiene que usarse el mismo día en que es recogido. Se reúnen y mezclan bien volúmenes iguales de las 3 muestras parciales de inóculo y se extrae de esta mezcla la muestra final. Deben usarse por lo menos 3 ml para la inoculación; |
|
c) |
inóculo procedente de iodo activado: Se puede usar como inóculo un volumen (no más de 3 litros) de lodo activado (contenido de sólidos en suspensión de hasta 2,5 g/l) sacado del tanque de aireación de una planta que trate predominantemente aguas residuales domésticas. |
1.6.2. Procedimiento
El ensayo se realiza a temperatura ambiental; esta debe mantenerse entre los 18 y los 25 oC.
Si fuera apropiado, el ensayo puede realizarse a una temperatura inferior (de hasta 10 oC): si la sustancia se degrada, entonces no es necesario ningún trabajo más. Si, sin embargo, la sustancia no se degrada, el ensayo debe llevarse a cabo a una temperatura constante entre los 18 y los 25 oC.
1.6.2.1. Período de rodaje: formación/estabilización del lodo de las unidades
El período de crecimiento/estabilización del lodo activado es el período durante el cual la concentración de los sólidos en suspensión del lodo activado y el funcionamiento de las unidades, progresan hasta llegar a un estado constante en las condiciones de explotación empleadas.
El período de rodaje es el período que va desde el momento en que la sustancia objeto de ensayo es añadida por primera vez hasta el momento en que su eliminación alcanza un trazado constante (valor relativamente constante). Este período no tiene que sobrepasar las seis semanas.
El período de evaluación es un período de tres semanas, tres semanas desde el momento en que la eliminación de la sustancia alcanza un valor relativamente constante, generalmente alto. Para aquellas sustancias que muestran escasa o nula degradación durante las primeras seis semanas, las tres semanas siguientes se consideran como el período de evaluación.
Al principio, lléne(n)se la(s) unidad(es) necesaria(s) para un ensayo con el inóculo mezclado con afluente.
Entonces se ponen en funcionamiento el aireador y la bomba de aireación (E) en el caso de tener unidades de prueba confirmatoria OCDE) y el mecanismo de administración (B).
El afluente sin la sustancia tiene que pasar a través del recipiente de aireación (C) bien a la velocidad de un litro por hora, bien a la velocidad constante de medio litro por hora; esto da un tiempo medio de retención de tres o seis horas respectivamente.
La velocidad de aireación debe regularse de modo que el contenido del recipiente (C) se mantenga constantemente en suspensión, mientras el contenido de oxígeno disuelto es por lo menos de 2 mg/l.
Hay que evitar la formación de espuma de una manera apropiada. No hay que usar agentes antiespuma que inhiban el lodo activado.
El fango que se acumula alrededor de la parte superior del recipiente de aireación (C) [y, en el caso de las unidades de prueba confirmatoria OCDE, en la base del recipiente de sedimentación (D) y en el circuito de circulación] debe ser devuelto a la mezcla liquida al menos una vez al día rascando con un cepillo o de cualquier otro modo que resulte apropiado.
Cuando el fango no se sedimenta, puede aumentarse su densidad añadiendo porciones de 2 ml de una solución al 5 % de cloruro de hierro; repítase si fuera necesario.
El efluente se acumula en un recipiente (E o F) durante 20 a 24 horas, y se toma una muestra después de mezclarlo concienzudamente. Hay que limpiar el recipiente (E o F) cuidadosamente.
A fin de supervisar y controlar la eficiencia del proceso, se mide por lo menos dos veces a la semana la demanda química de oxígeno (DQO) o el carbón orgánico disuelto (COD) del líquido filtrado a partir del efluente acumulado, así como la de] líquido filtrado a partir del efluente (utilizando una membrana de poros de tamaño 0,45 μm). Los primeros 20 ml (aproximadamente) del líquido filtrado se desechan.
La reducción del DQO o COD debe estabilizarse cuando se obtiene una degradación diaria más o menos regular.
El contenido de materia seca del lodo activado que está en el tanque de aireación debe determinarse dos veces a la semana (en g/l). Las unidades pueden operarse de dos maneras: bien se determina el contenido de materia seca del lodo activado dos veces a la semana y, si este es superior a 2,5 g/l, se desecha el exceso de lodo activado; o bien se retiran cada día 500 ml de mezcla líquida, de cada depósito, para conseguir un tiempo medio de retención de 6 días.
Cuando los parámentros [eficiencia del proceso (eliminación de DQO o COD), concentración del lodo, sedimentabilidad del lodo, turbidez de los efluentes, etc.] medidos y calculados de las dos unidades, son suficientemente constantes, se puede introducir la sustancia objeto de ensayo en el afluente de una de las dos unidades (como se indica en el punto 1.6.2.2).
Por otra parte, la sustancia puede asimismo añadirse al principio del período de crecimiento del lodo (punto 1.6.2.1) especialmente cuando el lodo es añadido en tanto que inóculo.
1.6.2.2. Procedimiento de ensayo
Se mantienen las condiciones de explotación del período de rodaje y se añade la suficiente cantidad de solución madre (aproximadamente el 1 %) del material de ensayo afluente de la unidad en que se realiza el mismo, de modo que se obtenga la concentración deseada del material en las aguas residuales (aproximadamente 10 o 20 mg de COD/l o 40 mg de DQO/l). Esto se puede lograr mezclando la solución de reserva con las aguas residuales diariamente o por medio de un sistema de bombeo separado. Esta concentración puede alcanzarse progresivamente. Si la sustancia objeto de ensayo no tiene efectos tóxicos sobre el lodo activado, se pueden probar concentraciones superiores.
Se alimenta el blanco solo con afluente sin sustancias añadidas. Se toman para su análisis volúmenes suficientes de efluentes y se filtran con filtros de membrana (0,45 μm). Se desechan los primeros 20 ml (aproximadamente).
Las muestras filtradas tienen que analizarse el mismo día, sí no, hay que preservarlas por medio de un método apropiado, por ejemplo, usando 0,05 ml de una solución de cloruro de mercurio al 1 % por cada 10 ml de líquido filtrado o almacenándolas a 2 o 4 oC hasta 24 horas, o por debajo de los - 18 oC para períodos más largos.
El período de rodaje, con la adición de la sustancia a ensayar, no debe exceder las 6 semanas y el período de evaluación no debe ser inferior a 3 semanas, esto es, debe haber disponibles unas 14 o 20 determinaciones para calcular el resultado final.
Método de unidades acopladas:
El acoplamiento de las unidades se consigue intercambiando 1,5 litros de mezcla líquida (incluido el lodo) de los recipientes de aireación del lodo activado entre las dos unidades una vez al día. Cuando los materiales de ensayo son fuertemente absorbentes, se sacan solo 1,5 litros de líquido sobrenadante de los recipientes de sedimentación y se vierten en el recipiente de lodo activado de la otra unidad.
1.6.2.3. Análisis
Se pueden realizar dos tipos de análisis para examinar el comportamiento de la sustancia:
COD y DQO:
Las concentraciones de COD se realizan por duplicado con el analizador de carbón y/o los valores de DQO según la referencia bibliográfica (2).
Análisis específico:
Las concentraciones de la sustancia en estudio se determinan por medio de un método analítico apropiado. Cuando sea posible, se debe llevar a cabo la determinación específica de la sustancia absorbida en el lodo.
2. DATOS Y EVALUACIÓN
2.1. MÉTODO DE UNIDADES ACOPLADAS
Cuando se utiliza el «método de unidades acopladas», los grados de eliminación diaria GE % se calculan de acuerdo con el punto 1.2.1.
Estos grados de eliminación diana GE son corregidos con la ecuación [2] para un tiempo medio de retención de 3 horas y la ecuación [3] para 6 horas, dando GEc para la transferencia de material debida al procedimiento de transinoculación.
|
|
[2] |
|
|
[3] |
Se calcula la media de la serie de valores de GEc, así como la desviación estándar de acuerdo con la ecuación [4]:
|
|
[4] |
donde:
|
SGEc |
= |
desviación estándar de la serie de valores de GEc |
|
= |
media de los valores de GEc |
|
n |
= |
número de determinaciones. |
Los valores atípleos de la serie de GEc se eliminan conforme a un procedimiento estadístico adecuado, por ejemplo, Nalimov [referencia (6)], al nivel de probabilidad del 95 %, y se vuelven a calcular la medía y la desviación estándar de la serie de valores de DRc una vez libre de valores atípicos.
El resultado final se calcula entonces con la ecuación [5]:
|
|
[5] |
donde
|
tn _ 1; α |
= |
valor tabular de t para n pares E y Eo, y fiabilidad estadística P (P = 1 — α), según la cual P se fija en un 95 % [referencia (1). |
El resultado se expresa dando la media con límites de tolerancia al nivel de probabilidad del 95 %, la respectiva desviación estándar y el número de valores de la serie DRc, una vez libre de valores atípicos, así como el número de estos, por ejemplo:
|
GEc |
= |
98,6±2,3 % de eliminación de COD |
|
s |
= |
4,65 % de eliminación de COD |
|
n |
= |
18 |
|
x |
= |
número de valores atípicos. |
2.2. MODO DE UNIDADES NO ACOPLADAS
El funcionamiento de las unidades puede controlarse como sigue:

Estas eliminaciones diarias pueden trazarse gráficamente para revelar las tendencias, por ejemplo, a una aclimatación.
2.2.1. Cómo utilizar las determinaciones de DQO/COD
El grado diario de eliminación GE % se calcula según el punto 1.2.1.
Se calcula la media de la serie de valores de GE, así como la desviación estándar de acuerdo con:
|
|
[6] |
donde:
|
SGE |
= |
Desviación estándar de la serie de valores de GEi |
|
|
= |
Media de los valores de GEi |
|
n |
= |
Número de determinaciones. |
Los valores atípicos de la serie de GE se eliminan conforme a un procedimiento estadístico adecuado, p. ej. Nalimov (referencia 6), al nivel de probabilidad del 95 %, y se vuelven a calcular la medía y la desviación estándar de la serie de los valores de GE una ver libre de valores atípicos.
El resultado final se calcula entonces con la ecuación [7]:
|
|
[7] |
donde:
|
tn-1; α |
= |
valor tabular de t para n pares de valores de E y Eo y fiabilidad estadística P (P = 1 — α) según la cual P se fija en un 95 % [referencia (1)]. |
El resultado se expresa dando la media con límites de tolerancia al nivel de probabilidad del 95 %, la respectiva desviación estándar, y el número de valores de la serie GE una vez libre de valores atípicos, así como el número de estos, por ejemplo:
|
GE |
= |
(98,6±2,3 %) de eliminación de COD |
|
s |
= |
4,65 % de eliminación de COD |
|
n |
= |
18 |
|
x |
= |
número de valores atípicos. |
2.2.2. Cómo utilizar un análisis específico
El porcentaje de eliminación de la sustancia objeto de estudio en la fase acuosa (RB) se calcula de acuerdo con el punto 1.2.2.
3. PRESENTACIÓN DEL INFORME
3.1. INFORME SOBRE EL ENSAYO
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
el formulario del anexo III, indicando las condiciones de funcionamiento del ensayo, |
|
— |
qué aparato se eligió (prueba confirmatoria OCDE o depósito poroso), |
|
— |
qué método se ha elegido: método de unidades acopladas o no, |
|
— |
qué aguas residuales: sintéticas o domésticas. En caso de que sean aguas residuales domésticas, fecha y lugar de la muestra, |
|
— |
qué inóculo, con fecha y lugar de la muestra, |
|
— |
una descripción del método analítico si se realizaron análisis específicos, |
|
— |
gráfico de la eliminación de DQO o COD en función del tiempo, incluyendo el período de rodaje y de evaluación, |
|
— |
recuperación analítica de la sustancia en estudio en forma de DQO o COD en la solución madre, |
|
— |
si se realizaron análisis específicos, gráfico del porcentaje de la eliminación de la sustancia de la fase acuosa en función del tiempo (período de rodaje y de evaluación), |
|
— |
la media de eliminación de COD, DQO o de la sustancia y la desviación estándar se calculan a partir de los resultados del período de evaluación, es decir, cuando hay una eliminación constante de material del ensayo o un período de funcionamiento constante, |
|
— |
gráfico de la concentración de lodo activado en función del tiempo |
|
— |
cualquier observación sobre el lodo activado (exceso de lodo desechado, presencia de aglomeraciones, FeCl3,), |
|
— |
concentración de la sustancia utilizada en el ensayo, |
|
— |
cualquier resultado sobre análisis realizados en el lodo, |
|
— |
toda la información y todos los resultados experimentales sobre la sustancia en estudio y la sustancia de referencia, si se utilizó alguna, |
|
— |
causas científicas de cualquier cambio de procedimiento. |
3.2. INTERPRETACIÓN DE LOS RESULTADOS
Una baja eliminación de la sustancia de estudio en la fase acuosa puede deberse a una inhibición de los microorganismos por parte de la sustancia. Esto puede asimismo ponerse de manifiesto a través de una desintegración y pérdida de lodo» produciendo un sobrenadante turbio, y a través de una reducción de la eficiencia en la eliminación de DQO (o COD) de la planta piloto.
La absorción físico-química puede desempeñar a veces un papel. Los análisis realizados en el lodo tras una adecuada desorción pueden revelar diferencias entre la acción biológica de la molécula y la absorción físico-química.
Son necesarias pruebas adicionales si se quiere establecer una distinción entre biodegradación (o biodegradación parcial) y absorción.
Esto puede hacerse de varias maneras, pero la más conveniente es usar el sobrenadante como inóculo en un ensayo de grupo base (una prueba respirométrica preferentemente).
Si se observan eliminaciones elevadas de COD o DQO, entonces ello es debido a la biodegradación, mientras que, en el caso de eliminaciones reducidas, la biodegradación no se puede distinguir de la eliminación, por ejemplo, si un compuesto soluble da muestras de una absorción constante del 98 % y la proporción de excedente de fango retirado es del 10 % diario, una eliminación de hasta el 40 % es posible; con una proporción de excedente de lodo retirado del 30. %, la eliminación debida a la absorción y a la retirada con excedente de lodo puede llegar a ser del 65 % referencia (4)].
Cuando se utilicen análisis específicos, se debe prestar atención a la relación entre la estructura de la sustancia y el análisis específico empleado. En este caso, el fenómeno observado no puede ser interpretado como una mineralización de la sustancia.
4. REFERENCIAS
|
(1) |
OCDE, París, 1981, Directriz para pruebas 303 A. Decisión del Consejo C(81) 30 final. |
|
(2) |
Anexo V C 9 Prueba de degradación — Demanda de oxígeno químico. Directiva 84/449/CEE de la Comisión [Diario Oficial de las Commidades Europeas no L 251 de 19.9.1984). |
|
(3) |
Painter, H. A. y King, E. F., WRC Porous-Pot method for assessing biodegradability. Technical Report TR70. June 1978, Water Research Center Reino Unido. |
|
(4) |
Wierich, P. and Gerike, P., — «The Fate of Soluble, Recalcitrant, and Adsorbing Compounds in Activated Sludge Plants» — Ecotoxicoiogy and Environmental Safety, Vol 5, no 2. June 1981, p. 161a, 171. |
|
(5) |
Directivas 82/242/CEE y 82/243/CEE del Consejo (Diario Oficial de las Commidades Europeas no L109 de 22.4.1982), corrigiendo las Directivas 73/404/CEE y 73/405/CEE: Biodegradabilidad de los detergentes [Diario Oficial de las Comunidades Europeas no L 347 de 17.12.1973). |
|
(6) |
Streuli, H., Fehlerhafte Interpretation und Anwendung von Ausreissertests, insbesondere bei Ringversuchen zur Überprüfung analytisch-chemischer Untersuchungsmethoden, Fesenius-Zeitschrifí für Analytische Chemie, 303 (1980) 406-408. |
---
Apéndice 1
Figura 1
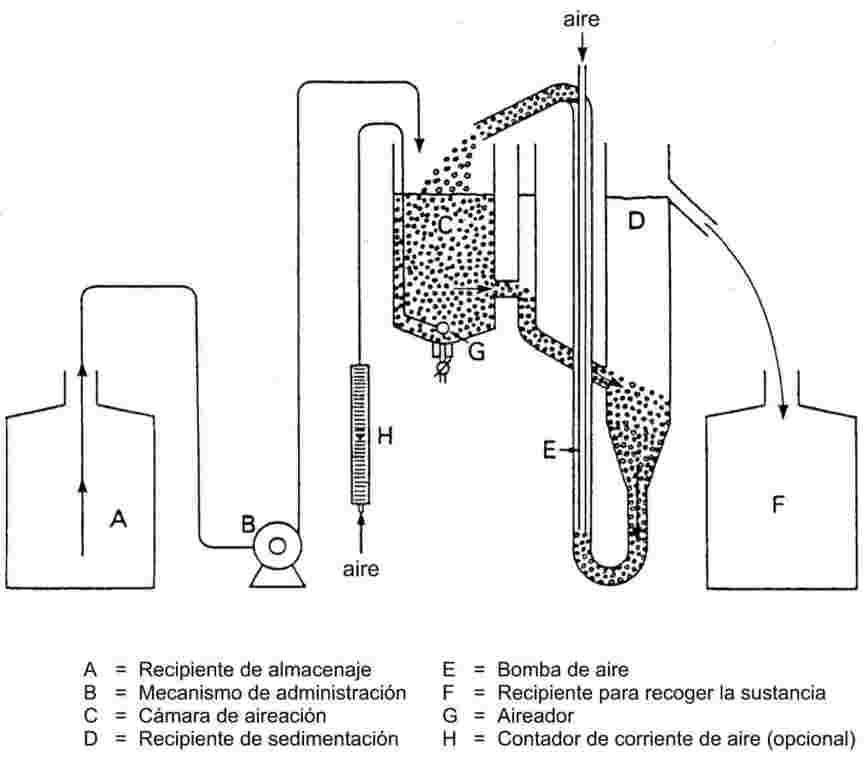
Figura 2

Apéndice 2
Figura 1
Equipo utilizado para valorar la biodegradabílidad

Figura 2
Detalles del recipiente de aireación con depósito poroso de tres litros

Apéndice 3
Condiciones de funcionamiento del ensayo de simulación con lodo activado
Márquese la respuesta en cada grupo
Aparato
|
confirmatorio OCDE |
|
|
depósito poroso |
|
Método de funcionamiento
|
unidad sencilla |
|
|
unidades acopladas |
|
|
unidades no acopladas |
|
Transinoculación
|
ninguna. |
|
|
lodo activado |
|
|
sobrenadante |
|
Tiempo medio de retención
|
3 horas |
|
|
6 horas |
|
Alimento de base
|
aguas residualses domésticas |
|
|
aguas residuales sintéticas |
|
Inóculo
|
efluente secundario |
|
|
compuesto |
|
|
lodo activado |
|
Adición del material de ensayo
|
deside el principio |
|
|
incremento gradual |
|
|
tras la formación del lodo |
|
Análisis
|
especifico |
|
|
DQO |
|
|
COD |
|
C.11. BIODEGRADACIÓN
LODO ACTIVADO: PRUEBA DE INHIBICIÓN DE LA RESPIRACIÓN
MÉTODO
1.1. INTRODUCCIÓN
El método descrito en este anexo sirve para evaluar el efecto que la sustancia objeto de ensayo tiene sobre los microorganismos, midiendo la tasa de respiración en unas condiciones determinadas y en presencia de concentraciones diferentes de la misma.
El propósito de este método consiste en establecer un método de investigación rápido por el que se puedan identificar las sustancias que afectan de manera adversa a las plantas de tratamiento microbiano aeróbico, así como indicar las concentraciones no inhibitorias apropiadas de las sustancias objeto de ensayo que vayan a ser utilizadas en las pruebas de biodegradabilidad.
Una prueba preliminar puede realizarse antes de la prueba definitiva. Esta nos facilitará información acerca del rango de concentraciones que deben utilizarse en la prueba final.
Al proyectar la prueba se incluyen dos muestras control sin la sustancia objeto de ensayo, una al principio y otra al final de la serie de pruebas. Cada lote de lodo activado debe ser revisado utilizando una sustancia de referencia,
Este método es más fácilmente aplicable a sustancias que, debido a su solubilidad en agua y a su baja volatilidad, es probable que permanezcan en el agua.
Para sustancias con solubilidad limitada en el medio de prueba, puede que no sea posible la determinación del valor CE50.
Cuando la sustancia de prueba es propensa a provocar la fosforilación oxídativa, los resultados basados en la disminución de oxígeno pueden conducir a conclusiones erróneas.
Para realizar el ensayo conviene disponer de la siguiente información:
|
— |
solubilidad en agua, |
|
— |
presión de vapor, |
|
— |
fórmula estructural, |
|
— |
pureza de la sustancia objeto del ensayo. |
Recomendación.
El lodo activado puede contener organismos potencialmente patógenos; manéjese con cuidado.
1.2. DEFINICIONES Y UNIDADES
La tasa de respiración es el consumo de oxígeno que tiene lugar en el lodo activado en condiciones aerobias por los microorganismos procedentes de aguas residuales; se expresa generalmente en mg O2 por mg de lodo en una hora.
Para poder calcular el efecto inhibitorio de una sustancia de prueba a una determinada concentración, la tasa de respiración se expresa como porcentaje de la media de las tasas de respiración de las dos muestras control:

donde:
|
Rs |
= |
consumo de oxígeno de la sustancia de prueba a la concentración utilizada en el ensayo |
|
RC1 |
= |
consumo de oxígeno de la muestra control 1 |
|
RC2 |
= |
consumo de oxígeno de la muestra control 2. |
En este método el valor CE50 expresa la concentración de la sustancia de prueba en la que la tasa de respiración es el 50 % de la obtenida en la muestra control, en las condiciones descritas en el método de ensayo.
1.3. SUSTANCIAS DE REFERENCIA
Como sustancia de referencia, se recomienda utilizar 3,5-didorofenol, conocido inhibidor de la respiración, efectuando una prueba para determinar el valor de CE50 en cada lote de lodo activado, a fin de comprobar que la sensibilidad del lodo no es anormal.
1.4. PRINCIPIO DEL MÉTODO
La tasa de respiración de un lodo activado alimentado con una cantidad estándar de agua residual sintética, se determina después de un tiempo de contacto de 30 minutos, o de 3 horas, o de ambos. La tasa de respiración del mismo lodo activado se mide también en presencia de varias concentraciones de la sustancia de prueba y en condiciones idénticas. El efecto inhibitorio de la sustancia de prueba, a una determinada concentración, se expresa como porcentaje de la media de las tasas de respiración de dos muestras control. El valor de CE50 se calcula mediante determinaciones realizadas a diferentes concentraciones.
1.5. CRITERIOS DE CALIDAD
Los resultados de la prueba son válidos si:
|
— |
las tasas de respiración de las muestras de control difieren una de la otra en menos de 15 %, |
|
— |
el valor de CE50 (30 minutos y/o tres horas) del 3,5-diclorofenol está dentro de los límites aceptados de 5 a 30 mg/l. |
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Reactivos
1.6.1.1. Soluciones de la sustancia de prueba
Las soluciones de la sustancia objeto del ensaya se preparan en el momento de comenzar el estudio, usando una solución madre. Para esta solución, es apropiada una concentración de 0,5 gr/l, si se sigue el procedimiento recomendado más abajo.
1.6.1.2. Solución de la sustancia de control
Puede prepararse por ejemplo una Solución de 3,5-diclorofenol disolviendo 0,5 g de 3,5-diclorofenol en 10 ml de NaOH, 1 M, diluyendo esta mezcla con agua destilada hasta un volumen de 30 ml aproximadamente, añadiendo a continuación y a la ver que se agita H2SO40,5 M hasta el punto de precipitación incipiente, para lo que se requieren 8 ml aproximadamente de H2SO40,5 M, y finalmente diluyendo la mezcla con agua destilada hasta obtener un litro de solución. El pH de esta solución deberá estar entre 7 y 8.
1.6.1.3. Aguas residuales sintéticas
El agua residual sintética para alimentación de los lodos se obtiene disolviendo las siguientes cantidades de sustancias en un litro de agua:
|
— |
16 g de peptona, |
|
— |
11 g de extracto de carne, |
|
— |
3 g de urea, |
|
— |
0,7 g de NaCl, |
|
— |
0,4 g de CaCl2 2H2O, |
|
— |
0,2 g de MgSo4.7H2O, |
|
— |
2,8 g de K2HPO4. |
Nota 1: Estas aguas residuales sintéticas están 100 veces más concentradas que las descritas en el Informe Técnico de la OCDE, «Método propuesto para la determinación de la biodegradabilidad de los tensoactivos utilizados en los detergentes sintéticos» del 11 de junio de 1976; además se les añade fosfato bipotásico.
Nota 2: Si el medio así preparado no va a utilizarse inmediatamente, deberá conservarse en la oscuridad y a una temperatura de 0 a 4 oC, durante un período que no exceda de una semana y en condiciones que no provoquen cambio alguno en su composición. También puede esterilizarse antes de su conservación, o bien añadir los extractos de peptona y carne poco antes de efectuar la prueba. Antes de utilizar la solución, deberá mezclarse a fondo y regular su pH.
1.6.2. Aparato de medida
El dibujo exacto no es crítico. Sin embargo, no debe quedar espacio en la cabeza y la muestra debe encajar herméticamente en el cuello del frasco de medida.
Se necesita un equipamiento normal de laboratorio y, especialmente, lo siguiente:
|
— |
aparato de medida, |
|
— |
dispositivo de aireación, |
|
— |
electrodo de pH y equipo de medida, |
|
— |
electrodo de O2. |
1.6.3. Preparación del inóculo
El inóculo microbiano para esta prueba proviene del lodo activado procedente de una planta de tratamiento de aguas residuales predominantemente domésticas.
Si es necesario, al volver al laboratorio, pueden eliminarse las partículas gruesas o grumos dejando sedimentar un poco, por ejemplo durante 15 minutos, y decantando la capa superior de sólidos más finos para su uso. También puede mezclarse el lodo con un mezclador durante algunos segundos.
Si se piensa que puede hallarse presente material inhibidor, deberá lavarse el lodo con agua corriente o con una solución isotómica. Después de centrifugar se decanta el sobrenadante (este procedimiento se repite tres veces).
Seguidamente, se pesa y se seca una pequeña cantidad de lodo lavado. A partir de estos resultados, se puede calcular la cantidad de lodo húmedo que debe ser suspendida en agua para obtener un lodo activado con un rango de concentración de sólidos en suspensión de 2 a 4 gr/l. Este nivel supone una concentración entre 0,8 y 1,6 gr/l en el medio de prueba, si se sigue el procedimiento recomendado más adelante.
Si no pudiera utilizarse el lodo el mismo día en que se ha recogido, se añadirán 50 ml de aguas residuales sintéticas a cada litro de lodo activado que haya sido preparado como se describe anteriormente; esta mezcla se airea durante la noche a una temperatura de 20 ± 2 oC, y se mantiene en estas condiciones durante el día, preparado para su utilización, Antes de utilizarlo, se comprueba el pH y, si es necesario, este se ajusta hasta un valor de entre 6,0 y 8,0. El nivel de sólidos en suspensión se determina como se describe en el párrafo anterior.
Si en días posteriores (cuatro como máximo), es necesario utilizar el mismo lote de lodo, se le añaden 50 ml de aguas residuales sintéticas por litro de lodo al final de cada día de trabajo.
1.6.4. Realización de la prueba
|
Duración/tiempo de contacto: |
30 minutos y/o 3 horas, tiempo durante el cual tiene lugar la aireación. |
|
Agua: |
Agua potable (si es necesario, declorada). |
|
Suministro de aire: |
Aire limpio, sin aceite. Caudal de aire de 0,5 a 1 litro/minuto. |
|
Aparato de medida: |
Matraz de fondo plano, como por ejemplo, un frasco DBO (véase la figura 1). |
|
Medidor de oxígeno: |
Electrodo de oxígeno adecuado, con un registrador. |
|
Solución nutritiva: |
Aguas residuales sintéticas (véase más arriba). |
|
Sustancia de prueba: |
La solución de prueba se prepara en el momento de comenzar la prueba. |
|
Sustancia de referencia: |
Por ejemplo, 3,5-diclorofenol (tres concentraciones como mínimo). |
|
Muestra de control: |
Muestra inoculada sin sustancia de prueba. |
|
Temperatura: |
20 ± 2 oC. |
A continuación se sugiere un procedimiento experimental, que puede seguirse para ambas sustancias, la de prueba y la de referencia, y para un período de contacto de 3 horas:
Se utilizan varios recipientes (por ejemplo, vasos de precipitado de 1 litro),
Se usarán como mínimo cinco concentraciones, distanciadas por un factor constante que, preferiblemente, no exceda de 3,2.
A tiempo «O», se mezclan 16 ml de cultivo de aguas residuales sintéticas con agua, hasta obtener 300 mi, a los que se añaden 200 ml de inóculo microbiano, y la mezcla total (500 ml) se vierte en el primer recipiente (primera muestra de control, Ct). Los recipientes de la prueba deberán airearse continuamente para asegurar que el O2 disuelto no se hace menor de 2,5 mg/l y que, justo antes de la medida de la tasa de respiración, la concentración de O2 deberá ser de unos 6,5 mg/l.
A los 15 minutos (15 minutos es un intervalo arbitrario, aunque conveniente), se repite el procedimiento anterior, con la diferencia de que, antes de añadir agua hasta llegar a 300 ml e inóculo microbiano hasta obtener un volumen de 500 ml, se añaden 100 ml de la solución madre de sustancia de prueba a los 16 ml de aguas residuales sintéticas. Seguidamente, esta mezcla se vierte en un segundo recipiente y se airea como se ha expuesto anteriormente. Este proceso se repite a intervalos de 15 minutos con diferentes volúmenes de solución madre de sustancia de prueba, para obtener una serie de recipientes que contengan concentraciones diferentes de la sustancia de prueba, Finalmente, se prepara una segunda muestra de control (C2).
Después de tres horas se mide el pH y se vierte bien mezclado el contenido del primer recipiente en el aparato de medida, en el que se mide la tasa de respiración durante un período de hasta 10 minutos.
Esta determinación se repite a intervalos de 15 minutos con el contenido de cada recipiente, de tal manera que el tiempo de contacto de cada recipiente sea de 3 horas.
La sustancia de referencia se prueba en cada lote de inóculo microbiano siguiendo el mismo procedimiento.
Cuando vayan a tomarse las medidas después de 30 minutos de contacto, se necesitará un régimen diferente (por ejemplo, más de un medidor de oxígeno).
Cuando sea necesario medir el consumo químico de oxígeno, se preparan más recipientes que contengan la sustancia de prueba, el medio nutriente de aguas residuales sintéticas y agua, pero no lodo activado.
El consumo de oxígeno se mide y se registra después de un tiempo de aireación de 30 minutos y/o de tres horas (tiempo de contacto).
2. DATOS Y EVALUACIÓN
La tasa de respiración se calcula a partir del trazado del registrador en mg O2/l.h, con valores aproximados de entre: 6,5 mg O2/l y 2,5 mg O2/l, o cuando la tasa de respiración sea baja, durante un período de 10 minutos. La parte de la curva de respiración sobre la que se mide la tasa de respiración debe ser lineal.
Cuando las tasas de respiración de las dos muestras de control difieran en más del 15 por ciento o el valor de CE50 (30 minutos y/o 3 horas) de la sustancia de referencia no quede dentro de la gama aceptada (de 5 a 30 mg/l para el 3,5-diclorofenol), la prueba no será válida y deberá repetirse.
El porcentaje de inhibición se calcula para cada concentración de prueba como se ha descrito anteriormente, y se representa gráficamente en función de la concentración en un papel logarítmico normal (o logaritmo-probabilidad); para obtener el valor de CE50.
Los límites de confianza del 95 % de los valores de CE50 se pueden determinar usando procedimientos establecidos.
3. INFORMES
3.1. INFORME DE LA PRUEBA
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
sustancia de prueba: datos de identificación química, |
|
— |
sistema de prueba: fuente, concentración y cualquier tratamiento previo del lodo activado, |
|
— |
condiciones de la prueba:
|
|
— |
resultados:
|
3.2. INTERPRETACIÓN DE LOS DATOS
El valor de CE50 se considerará solamente como indicación de los probables efectos tóxicos de la sustancia de prueba sobre los microorganismos de aguas residuales o de depuración de lodo activado, si se tiene en cuenta que las complejas interacciones que ocurren en el medio ambiente no pueden ser simuladas con precisión de una prueba de laboratorio. Además, las Sustancias de prueba que pueden ejercer un efecto inhibitorio sobre la oxidación amónica pueden producir también curvas de inhibición atípicas. En razón de esto, dichas curvas deberán interpretarse con cautela.
---
REFERENCIAS
|
(1) |
International Standard ISO 8192-1986. |
|
(2) |
Broccker, B., and Zahn, R., Water Research 11, 165 (1977). |
|
(3) |
Brown, D., Hitz, H. R., and Schaefer, L., Chemosphere 10, 245 (1981). |
|
(4) |
ETAD (Ecological and Toxicological Association of Dyestuffs Manufacturing Industries), Recommended Method No. 103, also describid by: |
|
(5) |
Robra, B., Vasser/Abwasser 117, 80 (1976). |
|
(6) |
Schefer, W., Textilveredlung 6, 247 (1977). |
|
(7) |
OCDE, París, 1981, Directriz para pruebas 209, Decisión del Consejo C(81) 30 final. |
C.12. BIODEGRADACIÓN
PRUEBA LASC MODIFICADA
1. MÉTODO
1.1. INTRODUCCIÓN
El objetivo del método es la evaluación del potencial de biodegradabilídad última o total de sustancias orgánicas hidrosolubles y no-volátiles, cuando se exponen a concentraciones relativamente altas de microorganismos durante un largo período de tiempo. Se mantiene durante este período la viabilidad de los microorganismos añadiendo diariamente una alimentación de aguas residuales decantadas. Para las necesidades del fin de semana, pueden almacenarse las aguas residuales a 4 oC. Pueden emplearse como alternativa las aguas residuales sintéticas de la prueba de confirmación OCDE.
Puede producirse una absorción fisicoquímica sobre los sólidos en suspensión, y este hecho deberá tomarse en cuenta al interpretar los resultados (véase el punto 3.2).
Debido al largo período de retención de la fase líquida (36 horas) y a la adición intermitente de nutrientes, la prueba no simula las condiciones reinantes en una planta depuradora de aguas residuales. Los resultados obtenidos con diversas sustancias de prueba indican que la prueba tiene un elevado potencial de biodegradación.
Las condiciones que la prueba establece son muy favorables a la selección y/o adaptación de microorganismos capaces de degradar el compuesto de prueba (el procedimiento puede utilizarse también para obtener inóculos aclimatados destinados a emplearse en otras pruebas).
En este método se emplea la medida de la concentración del carbono orgánico disuelto para evaluar la biodegradabilidad total de las sustancias de prueba. Resulta preferible determinar el COD por acidificación y purgado más que por la diferencia de Ctotal-Ciriorginico..
El empleo simultáneo de un método analítico específico puede permitir también evaluar la degradación primaria de la sustancia (desaparición de la estructura química madre).
El método solo es aplicable a las sustancias orgánicas de prueba que, a la concentración empleada en la prueba,
|
— |
sean solubles en agua, (como mínima, 20 mg de carbono orgánico dísuelto/l), |
|
— |
posean una presión de vapor despreciable, |
|
— |
no posean efecto inhibidor sobre las bacterias, |
|
— |
no se absorban significativamente dentro del sistema de prueba, |
|
— |
no escapen de la solución de prueba por formación de espumas. |
Deberá determinarse el contenido de carbono orgánico del material de prueba.
Para interpretar los resultados obtenidos, será útil la información sobre las proporciones relativas de los principales componentes de la sustancia de prueba, particularmente en los casos para los que los resultados sean bajos o marginales.
Para la interpretación de resultados bajos y para la selección de una concentración de prueba idónea podrá ser útil la información sobre la toxicidad de la sustancia de prueba para los microorganismos.
1.2. DEFINICIONES DE U NIDADES
|
CT |
= |
concentración del compuesto de prueba como carbono orgánico presente o añadido a las aguas residuales sedimentadas al comienzo del período de aireación (mg/l). |
|
C2 |
= |
concentración del carbono orgánico disuelto, encontrada en el líquido sobrenadante de la prueba al término del período de aireación (mg/l). |
|
Cc |
= |
concentración del carbono orgánico disuelto, encontrada en el líquido sobrenadante del control al término del período de aireación (mg/l). |
En este método, la biodegradación se define como la desaparición del carbono orgánico. La biodegradación puede expresarse como:
|
1. |
La eliminación porcentual D2d de la cantidad de sustancia añadida diariamente;
|
|
2. |
La eliminación porcentual Did de la cantidad de sustancia presente al inicio de cada día:
Donde los índices i e (i + 1) se refieren al día de la medición. Se recomienda la ecuación [2 a)] si el COD efluente varía de día a día, mientras que la ecuación [2 b)] puede emplearse cuando el COD efluente permanezca relativamente constante de un día para otro. |
1.3. SUSTANCIAS DE REFERENCIA
En algunos casos, al investigar una sustancia nueva, serán útiles las sustancias de referencia; sin embargo, no se recomienda en el método ninguna sustancia de referencia específica.
Se proporcionan los datos de diversos compuestos evaluados en pruebas efectuadas en varios laboratorios (véase el anexo 1). En principio esto puede ser llevado a cabo periódicamente como calibración del método y para comparar los resultados obtenidos cuando se emplea un método distinto.
1.4. PRINCIPIO DEL MÉTODO DE LA PRUEBA
Se coloca en una unidad de lodo activado semicontinuo (LASC) lodo activado procedente de una planta depuradora de aguas residuales. Se añaden el compuesto de prueba y aguas residuales domésticas decantadas, aireándose la mezcla durante 23 horas. Se interrumpe entonces la aireación, permitiéndose que el lodo se deposite por sedimentación y retirándose el líquido sobrenadante.
Se mezcla a continuación el lodo que permanece en la cámara de aireación con una cantidad igual del compuesto de prueba y de aguas residuales, repitiéndose el ciclo.
La biodegradación se determina por el contenido en carbono orgánico disuelto del líquido sobrenadante. Se compara dicho valor con el obtenido en un tubo de control en el que se han introducido solo aguas residuales decantadas.
Cuando se utiliza un método analítico específico, pueden medirse los cambios en la concentración de la molécula madre debidos a la biodegradación (biodegradabilidad primaria).
1.5. CRITERIOS DE CALIDAD
No ha sido bien establecida aún la reproducibilidad de este método basado en la eliminación del carbono orgánico disuelto (cuando se considera la biodegradación primaria, se obtienen datos muy precisos para las sustancias degradadas en gran medida),
La sensibilidad del método viene determinada en gran parte por la variabilidad del control y, en menor grado, por la precisión de la determinación del carbono orgánico disuelto y por el contenido del compuesto de prueba en el líquido al comienzo de cada ciclo.
1.6. DESCRIPCIÓN DEL PROCEDIMIENTO DE LA PRUEBA
1.6.1. Preparativos
Se reúne un número suficiente de unidades limpias de aireación (también puede usarse alternativamente la unidad original de prueba LASC de 1,5 l) y de tubos de entrada de aire (figura 1) para cada sustancia de prueba y para los controles. El aire comprimido suministrada a las unidades de prueba, que se purifica pasándolo por un filtro de lana de algodón, debe hallarse exento de carbono orgánico y presaturado con agua, a fin de reducir las pérdidas por evaporación.
Se toma una muestra de líquido mixto, que contenga de 1 a 4 g de sólidos en suspensión por litro de una central depuradora de lodo activado en la que se depuren sobre todo aguas residuales domésticas. Para cada unidad de aireación se requieren aproximadamente 150 ml de líquido mixto.
Se preparan en agua destilada soluciones madre de la sustancia de prueba; la concentración exigida normalmente es de 400 mg/l como carbono orgánico, lo que supone una concentración del compuesto de prueba de 20 mg/l de carbono al inicio de cada ciclo de aireación, si no tiene lugar una biodegradación.
Pueden emplearse concentraciones más altas si la toxicidad frente a los microorganismos lo permite.
Se mide el contenido en carbono orgánico de las soluciones madre.
1.6.2. Condiciones de la prueba
La prueba deberá llevarse a cabo a 20 a 25 oC.
Se utiliza una concentración elevada de microorganismos aerobios (de 1 a 4 g/l de sólidos en suspensión), y el período efectivo de retención es de 36 horas. La materia carbonada existente en las aguas residuales que sirven de alimentación resulta oxidada en gran medida, normalmente al cabo de 8 horas tras el inicio de cada ciclo de aireación. A continuación, el lodo respira endógenamente durante el resto del período de aireación, siendo entonces el compuesto de prueba el único sustrato disponible, a menos que este resulte también rápidamente metabolizado. Estos factores, junto con el de la reinoculación diaria de la solución de prueba cuando se utilizan como medio aguas residuales domesticas, ofrecen condiciones muy favorables para que se produzcan la aclimatación y un alto grado de biodegradación.
1.6.3. Ejecución de la prueba
Se toma una muestra de líquido mixto de una estación de depuración de lodo activado predominantemente de origen doméstico, ó bien de un laboratorio, y se mantiene en condiciones aerobias hasta emplearla en el laboratorio. Se llena cada unidad de aireación, y la unidad de control, con 150 ml (si se utiliza la unidad original de' prueba LASC, deben multiplicarse los volúmenes dados por 10) de líquido mixto, y se da inicio a la aireación. Transcurridas 23 horas, se interrumpe la aireación y se permite que el lodo sedimente durante 45 minutos. Se abre sucesivamente la espita de cada recipiente, y se retiran de cada uno 100 ml del líquido sobrenadante. Se toma, inmediatamente antes de su uso, una muestra de aguas residuales domésticas decantadas y se añaden 100 ml al lodo que permanece en cada unidad de aireación. Se vuelve a comenzar la aireación. En esta fase no se añaden sustancias de prueba, y las unidades se alimentan diariamente con aguas residuales domésticas solo hasta que se obtenga un líquido sobrenadante transparente después de la decantación. El tiempo necesario para ello es por lo general de 2 semanas, transcurridas las cuales el carbono orgánico disuelto contenido en el líquido sobrenadante al término de cada ciclo de aireación se acerca a un valor constante,
Al término de este período, se mezclan Jos diferentes lodos decantados, y se añaden a cada unidad 50 mL del lodo compuesto resultante.
Se añaden 95 ml de aguas residuales decantadas y 5 ml de agua a las unidades control; por su parte, a las unidades de ensayo se añaden 95 ml de aguas residuales decantadas y 5 ml de la solución madre (400 mg/l) del compuesto en estudio. Se recomienza la aireación y se mantiene durante 23 horas. Se deja entonces que el lodo decante durante 45 minutos, recogiéndose a continuación el líquido sobrenadante y analizándose el mismo, a fin de establecer su contenido en carbono orgánico disuelto.
Se repite diariamente durante toda la duración de la prueba el procedimiento mencionado de llenado y recogida.
Antes de la sedimentación, puede resultar necesario limpiar las paredes de las unidades para impedir que se acumulen sólidos por encima del nivel de líquido. Se utiliza para cada unidad un raspador o un cepillo individual, a fin de impedir que se produzca una contaminación cruzada.
Idealmente, eí carbono orgánico disuelto en los líquidos sobrenadantes se determinará diariamente, aunque puede permitirse una frecuencia menor de análisis. Antes de efectuar el análisis, se filtran los líquidos a través de filtros de membrana de 0,45 μm lavados o se centrifugan los mismos. Los filtros de membrana son adecuados si es seguro que ni liberan carbono ni absorben sustancia en la fase de filtración. La temperatura de la muestra no debe superar los 40 oC mientras se halle dentro de la centrífuga.
La duración de la prueba para compuestos con poca o ninguna degradación es indeterminada, pero la experiencia sugiere que generalmente debería alcanzar al menos 12 semanas, sin superar las 26.
2. DATOS Y EVALUACIÓN
Se representan en un diagrama frente al tiempo, los valores del carbono orgánico disuelto en los líquidos sobrenadantes de las unidades de prueba y de las unidades de control.
Según se va produciendo la biodegradación, el nivel encontrado en la muestra se aproximará al nivel del control. Una vez que la diferencia entre los dos niveles resulta constante para tres medidas consecutivas, se efectúa el número de medidas adicionales que sea suficiente para permitir el tratamiento estadístico de los datos, y se calcula con ellos la biodegradación porcentual del compuesto de prueba (Dad o Dsid, véase el punto 1.2).
3. INFORME
3.1. INFORME DE LA PRUEBA
El informe sobre el ensayo incluirá, si fuera posible:
|
— |
toda la información sobre el tipo de aguas residuales, el tipo de unidad empleado y los resultados experimentales relativos a la sustancia probada, la sustancia de referencia (si se usa), y el control, |
|
— |
la temperatura, |
|
— |
la curva de eliminación, con descripción y método de cálculo (véase 1,2), |
|
— |
la fecha y el emplazamiento donde se tomó la muestra del lodo activado y de las aguas residuales, el estado de adaptación, concentración, etc., |
|
— |
razones científicas para efectuar cualquier modificación del procedimiento de la prueba, |
|
— |
firma y fecha. |
3.2. INTERPRETACIÓN DE LOS RESULTADOS
Dado que la sustancia que deberá probarse con este método no será fácilmente biodegradable, toda eliminación del COD debida exclusivamente a la biodegradación será normalmente gradual, extendiéndose durante días o semanas, excepto en los casos en los que la aclimatación sea brusca, como indica una desaparición abrupta que tenga lugar tras algunas semanas.
Sin embargo, la absorción fisicoquímica puede jugar en ocasiones un papel importante; esto se apreciará cuando se produzca desde el principio una eliminación completa o parcial del COD añadido. Lo que sucede a continuación depende de factores tales como los grados de absorción y la concentración de sólidos en suspensión en la descarga del efluente. Generalmente, la diferencia entre las concentraciones del COD en los líquidos de control y en los líquidos sobrenadantes de la prueba se incrementa gradualmente a partir de un valor inicial bajo, permaneciendo a continuación con el nuevo valor durante el resto del experimento a menos que tenga lugar la aclimatación.
Si debe llevarse a cabo una distinción entre la biodegradación (o la biodegradación parcial) y la absorción, son necesarias otras pruebas. Estas pueden efectuarse de diversas maneras, pero la más convincente consiste en utilizar el líquido sobrenadante, o el lodo, como inóculo para una prueba de grupo base (preferiblemente una prueba respirométrica).
Las sustancias de prueba que produzcan una eliminación alta y no adsortiva del COD en esta prueba deberán considerarse como potencialmente biodegradables. La eliminación parcial y no adsortiva indica que el producto químico experimenta al menos algo de biodegradación.
Las eliminaciones bajas o nulas del COD pueden ser debidas a que la sustancia de prueba tiene efecto inhibidor sobre los microorganismos, y esto puede quedar de manifiesto también por lisis y pérdida de lodo, produciéndose sobrenadantes turbios. En este caso, deberá repetirse la prueba empleando una concentración menor de la sustancia de prueba.
El empleo de un método analítico específico, o de una sustancia de prueba marcada con 14C puede proporcionar una mayor sensibilidad. En el caso del compuesto de prueba marcado con I4C, la recuperación de 14CO2 confirmará que ha tenido lugar la biodegradación.
Cuando los resultados se expresen también en términos de biodegradación primaria, deberá explicarse también, si ello es posible, el cambio en la estructura química que produce la pérdida de respuesta de la sustancia inicial de prueba.
Deberá indicarse la validación del método analítico junto con la respuesta encontrada con el medio del control.
4. REFERENCIAS
|
(1) |
OCDE, París, 1981, Directriz para pruebas 302 A, Decisión del Consejo C(81) 30 final. |
Apéndice 1
Prueba LASC: Ejemplo de resultados
|
Sustancia |
CT (mg/l) |
CT-Ce (mg/l) |
Porcentaje de biodegradación (Dod) |
Duración del ensayo (días) |
|
4-acetil aminobenceno sulfonato |
17,2 |
2,0 |
85,0 |
40 |
|
Tetra propilen benceno sultanato |
17,3 |
8,4 |
51,4 |
40 |
|
4-nitrofenil |
16,9 |
0,8 |
95,3 |
40 |
|
Dieril glicol |
16,5 |
0,2 |
98,8 |
40 |
|
Anilina |
16,9 |
1,7 |
95,9 |
40 |
|
Tetra ciclopentano carboxilato |
17,9 |
3,2 |
81,1 |
120 |
Apéndice 2
Ejemplo del aparato de ensayo
Figure 1
C.13 BIOCONCENTRACIÓN: ENSAYO DINÁMICO CON PECES
1. MÉTODO
El presente método de bioconcentración reproduce las directrices de la OCDE TG 305 (1996).
1.1. INTRODUCCIÓN
El presente método describe un procedimiento para caracterizar el potencial de bioconcentración de algunas sustancias en peces en condiciones dinámicas. Son preferibles los regímenes de ensayos dinámicos, pero los regímenes semiestátícos son también aceptables en la medida en que se satisfagan los criterios de validez.
El método da toda la información necesaria para la ejecución del ensayo pero deja libertad suficiente para la adaptación del diseño experimental a las condiciones específicas de cada laboratorio y para variar las características de las sustancias de ensayo. Conviene especialmente a los productos orgánicos estables cuyos valores de log Pow están entre 1,5 y 6,0 (1) pero también es aplicable a las sustancias superlipofílicas (log Pow > 6,0). Para estas últimas, el valor estimado del factor de bioconcentración (FBC), a veces denominado KB, será probablemente superior al valor del factor de bioconcentración en estado de equilibrio (FBCSS) que se puede esperar en un experimento de laboratorio. Pueden calcularse valores estimados del factor de bioconcentración de los productos orgánicos cuyos valores de log Pow llegan hasta alrededor de 9,0 con ayuda de la ecuación de Binteín et al. (2). El potencial de bioconcentración se caracteriza por algunos parámetros, como la constante de la velocidad de absorción (k1), la constante de la velocidad de depuración (k2) y el FBCSS.
Puede facilitarse el análisis de las muestras de agua y de peces con ayuda de sustancias de ensayo radiomarcadas, que pueden también servir para definir si se deben identificar y cuantificar las sustancias degradadas. Si se miden los residuos radiactivos totales (por ejemplo, por combustión o disolución de los tejidos), el FBC se basará en el compuesto de origen, todos los metabolitos retenidos y el carbono asimilado. El FBC basado en los residuos radiactivos totales puede, entonces, no ser directamente comparable a un FBC derivado de un análisis químico específico exclusivamente del compuesto de origen.
Se podrán aplicar procedimientos de limpieza en los estudios radiomarcados para determinar el FBC sobre la base del compuesto de origen; los principales metabolitos se determinarán sí se considera necesario. Es posible también combinar un estudio del metabolismo de los peces con un estudio de bioconcentración por análisis e identificación de los residuos en los tejidos orgánicos y órganos.
1.2. DEFINICIONES Y UNIDADES
Bioconcentración/bioacumulación: es el aumento de la concentración de la sustancia de ensayo en el interior o en la superficie de un organismo (en tejidos específicos de este) en relación con la concentración de esta sustancia en el medio ambiente.
Factor de bioconcentración (FBC o KB): en cualquier momento durante la fase de absorción de este ensayo de acumulación es la concentración de la sustancia de ensayo en el pez, o en tejidos determinados de este, [Cp expresada en μg/g (ppm)] dividida por la concentración de la sustancia en el medio ambiente [Cw, expresada en μg/ml (ppm)].
Factor de bioconcentración en estado de equilibrio (FBCSS o KB): no cambia notablemente durante un plazo de tiempo prolongado, al ser constante la concentración de la sustancia de ensayo en el medio ambiente durante este mismo período.
Meseta o estado de equilibrio: en la representación gráfica de la concentración de sustancia de ensayo en el pez (Cf) en función del tiempo, se alcanza cuando la curva se hace paralela al eje del tiempo y tres análisis sucesivos de C realizados con muestras tomadas a intervalos de, al menos, dos días presentan una diferencia máxima del ± 20 % entre sí, y no hay diferencias significativas entre los tres períodos de muestreo. Cuando se analizan muestras puestas conjuntamente, es necesario proceder al menos a cuatro análisis sucesivos. Si las sustancias de ensayo son absorbidas con lentitud, se optará preferiblemente por intervalos semanales.
Factores de bioconcentración: calculados directamente a partir de las constantes cinéticas (k1/k2) se llaman factores de concentración cinéticos, FBCk.
Coeficiente de reparto octanol-agua (Pow): es la relación entre la solubilidad de una sustancia en n-octanol y en agua, en equilibrio (método A.8); también se denomina Kow. El logaritmo de Pow indica el potencial de bioconcentración de una sustancia por los organismos acuáticos.
Fase de exposición o de absorción: es el tiempo durante el cual se exponen los peces a la sustancia de ensayo.
Constante de velocidad de absorción (k,): es el valor numérico que define la velocidad de aumento de la concentración de la sustancia de ensayo en los peces del ensayo (o en tejidos específicos de estos) cuando se expone a esta sustancia (k1 se expresa en días-1).
Fase de post-exposición o de depuración (pérdida): es el tiempo, tras la transferencia de los peces de un medio que contiene la sustancia de ensayo a un medio libre de esta, durante el cual se estudia la depuración (o pérdida neta) de la sustancia a partir de los peces del ensayo (o de los tejidos específicos).
Constante de velocidad de depuración (pérdida) (k2): es el valor numérico que define la velocidad de disminución de la concentración de la sustancia de ensayo en los peces del ensayo (o en tejidos específicos) tras su transferencia de un medio que contiene la sustancia de ensayo a un medio que está libre de ella (k2 se expresa en días-1).
1.3. PRINCIPIO DEL MÉTODO
El ensayo se divide en dos fases: la de exposición (absorción) y la de post-exposición (depuración). Durante la fase de absorción, se exponen grupos distintos de peces de una sola especie a al menos dos concentraciones de la sustancia de ensayo. Luego se transfieren a un medio libre de esta sustancia, para la fase de depuración. Esta última es siempre necesaria, excepto cuando la absorción de la sustancia durante la fase de absorción es poco importante (por ejemplo, si el FBC es inferior a 10). La concentración de la sustancia de ensayo en los peces (o tejidos específicos) se mide durante las dos fases del ensayo. Además de las dos concentraciones de ensayo, se mantiene un grupo testigo de peces en condiciones idénticas, salvo que no se exponen a la sustancia de ensayo. Esto sirve para relacionar los posibles efectos nocivos observados en el ensayo de bioconcentración con un grupo testigo correspondiente, y deducir las concentraciones de fondo de la sustancia de ensayo.
La fase de absorción dura 28 días salvo si se demuestra que el equilibrio se alcanza antes. Puede preverse la duración de la fase de absorción y el tiempo necesario para la obtención del estado de equilibrio gracias a la ecuación del anexo 3. El período de depuración comienza entonces con la transferencia de los peces a otro recipiente limpio que contiene el mismo medio, pero esta vez sin la sustancia de ensayo. Cuando sea posible, se calculará el factor de bioconcentración preferiblemente como relación entre (FBCSS) de la concentración en los peces (Cf) y en el agua (Cw) en el estado de equilibrio aparente y también como factor de bioconcentración cinético, FBCK, que es la relación entre las constantes de velocidad de absorción (k1) y de depuración (k2), suponiendo una cinética de primer orden. Si es evidente que no se sigue una cinética de primer orden, deberán aplicarse modelos más complejos (véase el anexo 5).
Si no se llega a un estado de equilibrio en el plazo de 28 días, la fase de absorción se prolongará hasta alcanzar este estado de equilibrio, o durante 60 días si esto supone un plazo menor; luego comenzará la fase de depuración.
La constante de la velocidad de absorción, la constante de la velocidad de depuración (pérdida) (o las constantes, cuando entren en juego modelos más complejos), el factor de bioconcentración y, si es posible, los intervalos de confianza de cada uno de estos parámetros, se calculan a partir del modelo que describa mejor las concentraciones medidas de la sustancia de ensayo en los peces y en el agua.
El FBC se expresa en función del peso total húmedo de los peces. Sin embargo, para algunos estudios pueden utilizarse tejidos u órganos específicos (por ejemplo, músculos, hígado) si los peces son suficientemente grandes o si es posible separar las partes comestibles (filetes) y no comestibles (vísceras). Como hay una clara relación entre el potencial de bioconcentración y la lipofilia de numerosas sustancias orgánicas, también hay una relación correspondiente entre el contenido en lípidos de los peces del ensayo y las bioconcentraciones observadas de estas sustancias. Así pues, para reducir esta fuente de variabilidad en los resultados de los ensayos relativos a las sustancias con fuerte lipofilia (es decir, con log Pow > 3), la bioconcentración debe expresarse en función del contenido lipídico, ademas del peso corporal total.
El contenido lipídico se establecerá, si es posible, con los mismos materiales biológicos que se utilicen para determinar la concentración de la sustancia de ensayo.
1.4. INFORMACIÓN SOBRE LA SUSTANCIA DE ENSAYO
Antes de emprender el ensayo de bioconcentración, deberán reunirse los datos siguientes respecto a la sustancia de ensayo:
|
a) |
solubilidad en el agua; |
|
b) |
coeficiente de reparto octanol-agua Pow (denominado también K ow, determinado por un método de CLAR en A.8); |
|
c) |
hidrólisis; |
|
d) |
características de fototransformación en el agua bajo irradiación solar natural o artificial y en las condiciones de irradiación del ensayo de bioconcentración (3); |
|
e) |
tensión superficial (para las sustancias cuyo log Pow no pueda determinarse); |
|
f) |
presión de vapor; |
|
g) |
biodegradabilidad «fácil» (en su caso). |
Será necesario también conocer la toxicidad relativa a la especie de peces utilizada en el ensayo, preferiblemente la CL5O asintótica (es decir, independiente del tiempo). Es indispensable disponer de un método analítico correcto, de una exactitud, de una precisión y de una sensibilidad conocidas para la cuantificación de la sustancia de ensayo en las soluciones de ensayo y en el material biológico, así como de datos precisos sobre la preparación y la conservación de las muestras. Es necesario también conocer el límite de detección analítica de la sustancia de ensayo tanto en el agua como en los tejidos de los peces. Si se utiliza una sustancia de ensayo marcada con 14C, deberá conocerse el porcentaje de radiactividad asociada a las impurezas.
1.5. CONDICIONES DE VALIDEZ DEL ENSAYO
Para que un ensayo sea válido, deberán darse las condiciones siguientes:
|
— |
la variación de temperatura será inferior a ± 2 oC, |
|
— |
la concentración de oxígeno disuelto no caerá por debajo del 60 % del nivel de saturación, |
|
— |
la concentración de la sustancia de ensayo en los recipientos o armarios se mantendrá en la banda del ± 20 % de la media de los valores medidos durante la fase de absorción, |
|
— |
la mortalidad y otros efectos adversos o enfermedades tanto entre los peces testigo como entre los tratados serán inferiores al 10 % al final del ensayo; cuando el ensayo se prolongue a lo largo de varias semanas o meses, la mortalidad y los otros efectos adversos en los dos conjuntos de peces deberán ser inferiores al 5 % al mes y no exceder del 30 % en total. |
1.6. COMPUESTOS DE REFERENCIA
La utilización de compuestos de referencia con un potencial de bioconcentración conocido podrá ayudar al control del procedimiento experimental, en caso necesario. No se puede aún, sin embargo, recomendar ninguna sustancia específica.
1.7 DESCRIPCIÓN DEL MÉTODO
1.7.1. Equipo
Se tendrá cuidado de evitar los materiales que puedan presentar fenómenos de disolución, sorción o lixiviación o tener algún efecto adverso sobre los peces, y esto en todas las partes del equipo. Se utilizarán tanques normales rectangulares o cilíndricos, de materiales químicamente inertes y de una capacidad adaptada a la tasa de carga. Se reducirá al mínimo el uso de tubos de plástico flexible. Serán preferibles los tubos de teflon (R), acero inoxidable o vidrio. La experiencia pone de manifiesto que para las sustancias que presentan fuertes coeficientes de adsorción, como los piretroides sintéticos por ejemplo, pudiera ser necesario el vidrio silanizado. Será necesario, en estos casos, deshacerse de los equipos una vez usados.
1.7.2. Agua
Se utiliza generalmente para el ensayo en agua natural procedente de una fuente no contaminada y de calidad uniforme. El agua de dilución debe ser tal que permita la supervivencia de la especie de la especie de peces elegida, durante los períodos de aclimatación y de ensayo, sin que aparezcan ningún comportamiento ni aspecto anormales. Idealmente, se debería demostrar que las especies sometidas al ensayo pueden sobrevivir, crecer y reproducirse en el agua de dilución (por ejemplo, mediante cría en laboratorio o un estudio de toxicidad sobre un ciclo biológico). El agua se caracterizará al menos por su pH, dureza, materia sólida total, carbono orgánico total y también, preferentemente, su contenido en amoniaco y en nitratos, su alcalinidad y, para las especies marinas, su salinidad. Se conocen perfectamente los parámetros importantes para el bienestar óptimo de los peces, pero el anexo 1 indica las concentraciones máximas recomendadas de una serie de parámetros relativos al agua de ensayo, dulce y salada.
Durante toda la duración de un ensayo, el agua tendrá calidad constante. El pH se mantendrá entre 6,0 y 8,5, pero durante un ensayo dado permanecerá dentro de una gama de ±0,5 unidades de pH. Para garantizar que el agua de dilución no influye indebidamente en los resultados del estudio (por ejemplo, por complejación de la sustancia de ensayo) ni afecta negativamente al comportamiento de las poblaciones de peces, se tomarán regularmente muestras para análisis. Cuando se sepa que un agua de dilución es de calidad relativamente constante, convendrá proceder, por ejemplo cada tres meses, a la determinación de los metales pesados (por ejemplo, Cu, Pb, Zn, Hg, Cd, Ni), aniones y cationes principales (por ejemplo, Ca, Mg, Na, K, Cl, SO4), plaguicidas (por ejemplo, organofosforados totales y organoclorados totales), carbono orgánico total y sólidos en suspensión. Si se demuestra que la calidad del agua es constante al menos durante un año, estos análisis podrán ser menos frecuentes y sus intervalos más espaciados (por ejemplo, cada seis meses).
El contenido en partículas naturales así como el carbono orgánico total (COT) del agua de dilución será lo más bajo posible para evitar la adsorción de la sustancia de ensayo en la materia orgánica, lo que podría reducir su biodisponibilidad (4). El valor máximo admisible es de 5 mg/l para las partículas (materia seca que no atraviesa un filtro de 0,45 μm) y 2 mg/l para el carbono orgánico total (véase el anexo 1). En caso necesario, el agua se filtrará antes de usarse. La aportación de los peces (excretas) y los residuos alimentarios al contenido del agua en carbono orgánico deberá ser lo más baja posible. A lo largo del ensayo, la concentración de carbono orgánico en el recipiente del ensayo no deberá sobrepasar en más de 10 mg/l (± 20 %) la del carbono orgánico procedente de la sustancia de ensayo y, en su caso del agente de disolución.
1.7.3. Soluciones de ensayo
Se prepara una solución madre de la sustancia de ensayo, a una concentración adecuada. La solución madre será preparada preferiblemente por simple mezcla o agitación de la sustancia de ensayo en el agua de dilución. No se recomienda la utilización de disolventes o de dispersantes (agentes de disolución); se puede recurrir a ellos, sin embargo, en algunos casos para obtener una solución madre de la concentración necesaria. Los disolventes que pueden utilizarse son el etanol, el metanol, el éter monometílico de etilenglicol, el éter dimetílico de etilenglicol, la dimetilformamida y el trietilenglicol. Los dispersantes utilizables son Cremaphor RH40, Tween 80, metilcelulosa al 0,01 % y HCOI-40. Se tomarán precauciones en caso de utilización de agentes fácilmente biodegradables, ya que estos pueden causar problemas de crecimiento bacteriano en los ensayos dinámicos. La sustancia de ensayo puede ir radiomarcada y deberá ser del nivel más alto de pureza (por ejemplo, preferiblemente > 98 %).
Para los ensayos dinámicos, un equipo aportará y diluirá continuamente la solución madre de la sustancia de ensayo (por ejemplo, bomba dosificadora, diluyente proporcional, sistema saturador) para conseguir las concentraciones de ensayo en los recipientes. Será preferible prever al menos cinco volúmenes de sustitución al día para cada recipiente de ensayo. Se preferirá el método dinámico, pero en caso de imposibilidad (por ejemplo, cuando los organismos del ensayo sufran efectos adversos) podrá aplicarse una técnica semiestática sí siguen cumpliéndose los criterios de validez. Los regímenes de flujo de las soluciones madre y del agua de dilución se controlarán 48 horas antes del ensayo y luego al menos diariamente durante este. Este control incluirá la determinación del flujo en cada recipiente de ensayo y garantizará que dicho flujo no varía en más del 20 % dentro de cada recipiente ni entre recipientes distintos.
1.7.4. Selección de las especies
Entre los criterios importantes de selección de las especies figuran el que sean de fácil disponibilidad, que puedan obtenerse de los tamaños adecuados y que puedan mantenerse satisfactoriamente en el laboratorio. Otros criterios de selección de las especies de peces son su importancia recreativa, comercial o ecológica, así como una sensibilidad comparable, haber producido buenos resultados anteriormente, etc.
En el anexo 2 figura una lista de especies recomendadas para los ensayos. Pueden utilizarse otras especies pero es posible entonces tener que adaptar el procedimiento de ensayo para obtener condiciones experimentales convenientes. En tal caso se expondrá la motivación de la elección de las especies y el método experimental elegido.
1.7.5. Mantenimiento de los peces
Es necesario aclimatar la población de peces durante dos semanas al menos en agua a la temperatura del ensayo y aportar una cantidad de comida suficiente y del mismo tipo que se vaya a utilizar durante el ensayo.
Después de un período de adaptación de 48 horas, se registra la mortalidad y se aplican los criterios siguientes:
|
— |
mortalidad superior al 10 % de la población en 7 días: se rechaza el lote entero, |
|
— |
mortalidad entre el 5 y el 10 % de la población en 7 días: se prolonga la aclimatación durante 7 días más, |
|
— |
mortalidad inferior al 5 % de la población en 7 días: se acepta el lote; si la mortalidad sobrepasa el 5 % durante los siete días siguientes, se rechaza el lote entero. |
Hay que asegurarse de que los peces utilizados para los ensayos no presentan enfermedades ni anomalías observables. Se descartan todos los peces enfermos. Los peces no deben recibir tratamiento contra ninguna enfermedad durante las dos semanas que preceden al ensayo ni durante este.
1.8. REALIZACIÓN DEL ENSAYO
1.8.1. Ensayo preliminar
Puede ser útil proceder a una experimentación preliminar para optimizar las condiciones de realización del ensayo definitivo, como, por ejemplo, la selección de las concentraciones de la sustancia de ensayo o la duración de las fases de absorción y de depuración.
1.8.2. Condiciones de exposición
1.8.2.1. Duración de la fase de absorción
Se puede prever la duración de la fase de absorción basándose en la experiencia práctica (por ejemplo, a partir de un estudio previo o de un producto químico que tenga propiedades similares de acumulación) o a partir de algunas relaciones empíricas fundadas en el conocimiento de la solubilidad en el agua o de coeficiente de reparto octanol/agua de la sustancia de ensayo (véase el anexo 3).
La fase de absorción durará 28 días salvo si puede demostrarse que se alcanza antes el equilibrio. Si no se llega al estado de equilibrio en el plazo de 28 días, se prolongará la fase de absorción y se procederá a otras mediciones hasta conseguir el estado de equilibrio, o durante 60 días si esto supone un plazo menor.
1.8.2.2. Duración de la fase de depuración
La mitad de la duración de la fase de absorción es generalmente suficiente para que se produzca una reducción conveniente (por ejemplo, 95 %) de la carga corporal de la sustancia de ensayo (véanse las explicaciones del cálculo en el anexo 3). Si el tiempo necesario para llegar a una pérdida del 95 % es excesivamente largo, sobrepasando por ejemplo dos veces la duración normal de la fase de absorción (es decir, más de 56 días), se podrá utilizar un período más corto (hasta que la concentración de la sustancia de ensayo sea inferior al 10 % de la concentración en el estado de equilibrio). Sin embargo, para las sustancias con prepiedades de absorción y depuración más complejas que el modelo de peces con un único compartimento, que da una cinética de primer orden, se aplicarán fases de depuración más largas para determinar las constantes de la velocidad de pérdida. Sin embargo, este plazo de tiempo puede regirse por el período durante el cual la concentración de la sustancia de ensayo en los peces permanece por encima del límite de detección analítico.
1.8.2.3. Número de peces del ensayo
El número de peces por concentración del ensayo debe elegirse de modo que, como mínimo, se disponga de cuatro peces por muestra en cada muestreo. Si se desea una estadística más fina, deberá aumentar el número de peces por muestra.
Si se utilizan peces adultos, hay que indicar si son machos o hembras o si se utilizan los dos sexos en el experimento. En este último caso, antes de empezar la exposición, será necesario comprobar que no son significativas las diferencias de contenido lipídico entre sexos; podrá ser necesario reunir todos los machos y todas las hembras.
En todos los ensayos se elegirán peces de peso similar, de modo que el peso del más pequeño no sea inferior a dos tercios del peso del mayor. Pertenecerán a la misma clase de edad y procederán de la misma fuente. Dado que, al parecer, el peso y la edad de un pez tienen a veces efectos notables sobre los valores de FBC (1), estos datos se registrarán con precisión. Se recomienda pesar una submuestra de la población de peces antes del ensayo, para calcular el peso medio.
1.8.2.4. Carga
Se utiliza una relación elevada agua/peces con el fin de minimizar la reducción de Cw debida a la introducción de los peces al principio del ensayo, y también para evitar el descenso de la concentración de oxígeno disuelto. Es importante que la tasa de carga sea adaptada a la especie utilizada para el ensayo. De todos modos, se recomienda una tasa de carga de 0,1-1,0 g de peces (peso húmedo) por litro de agua al día. Se pueden realizar cargas de tasa elevada si se demuestra que la concentración requerida de sustancia de ensayo puede mantenerse en la banda del ± 20 %, y que la concentración de oxígeno disuelto no cae por debajo del 60 % del nivel de saturación.
Se tendrá en cuenta el hábitat normal de la especie de peces al elegir la tasa de carga. Los peces bentónicos, por ejemplo, pueden necesitar un acuario que tenga más superficie de fondo que las especies pelágicas, para un mismo volumen de agua.
1.8.2.5. Alimentación
Durante los períodos de aclimatación y de ensayo, se alimentará a los peces con una dieta conveniente que tenga un contenido conocido en lípidos y proteínas totales, en cantidades suficientes para mantenerlos en buena salud y conservar el peso corporal. Se alimentará a los peces diariamente durante los períodos de aclimatación y de ensayo en la proporción de alrededor del 1 o 2 % del peso corporal cada día; se mantiene así en la mayoría de las especies una concentración lipídica de un nivel relativamente constante durante el ensayo. La cantidad de comida deberá volver a calcularse una vez por semana, por ejemplo, con el fin de mantener constantes el peso corporal y el contenido lipídico. Para ese cálculo, el peso de los peces de cada recinto de ensayo se evaluará a partir del peso de los peces muestreados últimamente en ese recinto. No hay que pesar los peces que permanecen en el recinto.
La comida no consumida y los excrementos son evacuados de los recipientes de ensayo mediante sifón cada día poco después de la alimentación (30 minutos a una hora). Los recipientes se tendrán lo más limpios posible a lo largo del ensayo de modo que la concentración de materia orgánica permanezca al nivel más bajo posible, puesto que la presencia de carbono orgánico puede limitar la biodisponibilidad de la sustancia de ensayo (1).
Como muchos alimentos son derivados de harinas de pescado, se analizarán para determinar su contenido en sustancia de ensayo. Es conveniente también analizar el contenido de estos alimentos en plaguicidas y metales pesados.
1.8.2.6. Iluminación y temperatura
El período de iluminación es generalmente de 12 a 16 horas y la temperatura (± 2 oC) corresponderá a la especie utilizada (véase el anexo 2). Se definirán claramente el tipo y las características de la iluminación. Será necesario prestar atención a la posible fototransformación de la sustancia de ensayo en las condiciones de irradicación del estudio. Se utilizará un alumbrado adecuado que no exponga a los peces a fotoproductos no naturales. En algunos casos, puede ser conveniente utilizar un filtro para eliminar los rayos UV por debajo de 290 nm.
1.8.2.7. Concentraciones del ensayo
Se exponen los peces en condiciones dinámicas a dos concentraciones al menos de la sustancia de ensayo en agua. Normalmente, la concentración mas alta de la sustancia de ensayo será del 1 % de su CL50 aguda asintótica, y al menos diez veces superior a su límite de detección en el agua por el método de análisis elegido.
La concentración mas alta del ensayo puede establecerse también dividiendo la CL50 a 96 horas por una relación adecuada aguada/crónica (las relaciones adecuadas de algunos productos químicos pueden ir de 3 a 100). Si es posible, la otra u otras concentraciones deben elegirse de modo que la diferencia con la antes citada alcance un factor de diez. En caso de imposibilidad debido al criterio del 1 % de la CL50 y del límite analítico, podrá utilizarse un factor inferior a diez o se pensará en el uso de una sustancia radiomarcada con 14C. Ninguna concentración podrá superar la solubilidad en agua de la sustancia de ensayo.
Cuando se utilice un agente de disolución, su concentración no deberá sobrepasar 0,1 ml/l y deberá ser idéntica en todos los recipientes del ensayo. Debe conocerse su contribución, junto con la de la sustancia de ensayo, al contenido global de carbono orgánico en el agua del ensayo. Sin embargo, se hará todo lo posible para evitar recurrir a este tipo de materiales.
1.8.2.8. Controles
Se utilizará un control (testigo) del agua de dilución o, en su caso, un control con el agente de disolución, además de la serie de ensayo, en la medida en que se haya establecido que el agente no tiene ningún efecto sobre los peces. En caso contrario, se establecerán ambos controles.
1.8.3. Frecuencia de las medidas de la calidad del agua
Durante el ensayo se medirán en todos los recipientes el oxígeno disuelto, el COT, el pH y la temperatura. La dureza total, y la salinidad eventualmente, se medirán en los controles y en un recipiente que contenga la concentración más alta. Por lo menos, el oxígeno disuelto y eventualmente la salinidad se medirán tres veces —al principio, hacia el medio y al final de período de absorción— y una vez por semana durante el período de depuración. El COT se medirá al principio del ensayo (24 y 48 horas antes del comienzo de la fase de absorción), antes de la adición de los peces y al menos una vez por semana durante los períodos de absorción y de depuración. La temperatura se medirá diariamente, el pH al principio y al final de cada período y la dureza una vez en cada ensayo. La temperatura se medirá preferiblemente de forma continua en un recipiente al menos.
1.8.4. Muestreo y análisis de los peces y del agua
1.8.4.1. Calendario de muestreo de los peces y del agua
Se tomará agua de los recipientes de ensayo para determinar la concentración de la sustancia de ensayo antes de la introducción de los peces y durante las fases de absorción y de depuración. Por lo menos, el agua se tomará al mismo tiempo que los peces y antes de darles de comer. Durante la fase de absorción, se determinarán las concentraciones de sustancia de ensayo para comprobar que satisfacen los criterios de validez.
Se muestrearán los peces en cinco ocasiones al menos durante la fase de absorción y en cuatro ocasiones al menos durante la fase de depuración. Como a veces es difícil calcular una estimación razonablemente precisa del FBC a partir de este número de muestras, en particular cuando hay una cinética de depuración distinta de la simple de primer orden, puede ser recomendable muestrear con mayor frecuencia en los dos períodos (véase el anexo 4). Las muestras suplementarias se conservarán y analizarán solamente si los resultados de la primera serie de análisis se revelan insuficientes para el cálculo del FBC con la precisión requerida.
En el anexo 4 se encuentra un ejemplo de calendario de muestreo aceptable. Pueden establecerse otros calendarios fácilmente sobre la base de otros valores supuestos del Pow para calcular el tiempo de exposición que corresponde a una absorción del 95 %.
Se prosigue el muestreo durante la fase de absorción hasta el establecimiento de un estado de equilibrio o durante 28 días, si esto supone un plazo menor. Si no se llega a un estado de equilibrio en 28 días, el muestreo continúa hasta la obtención de un estado de equilibrio o durante 60 días, si esto supone un plazo menor, Antes de empezar la fase de depuración, se trasladan los peces a los tanques limpios.
1.8.4.2. Muestreo y preparación de las muestras
Se toman las muestras de agua destinadas a los análisis, por ejemplo mediante sifonado a través de un tubo inerte desde un punto central del recinto de ensayo. Puesto que parece que no siempre se puede separar ni por centrifugación ni por filtración la fracción no biodisponible de la sustancia de ensayo de la que es biodisponible (en particular en el caso de los productos químicos superlipofílicos, es decir, que tienen un log Pow > 5) (1) (5), las muestras pueden no someterse a estos tratamientos.
En su lugar, será necesario procurar que los tanques se mantengan lo más limpios posible y que el contenido en carbono orgánico total se mida a lo largo de las fases de absorción y de depuración.
Se toma un número conveniente de peces (normalmente 4 como mínimo) de cada recipiente de ensayo en cada momento de muestreo. Los peces tomados se lavan rápidamente con agua, se secan con material absorbente, y se sacrifican instantáneamente de la manera más adecuada e incruenta posible, y luego se pesan.
Es preferible analizar los peces y el agua inmediatamente después del muestreo para evitar toda degradación u otras pérdidas y para calcular aproximadamente las tasas de absorción y de depuración mientras el ensayo continúa. El análisis inmediato evita también retrasos en la determinación del momento en que se alcanza una meseta.
A falta de un análisis inmediato, las muestras se conservan según un método conveniente. Antes del principio del estudio, se reunirá información sobre el método de conservación adecuado de la sustancia de ensayo correspondiente como, por ejemplo, congelación, mantenimiento a 4 oC, duración de la conservación, extracción, etc.
1.8.4.3. Calidad del método analítico
Como la totalidad del procedimiento se regula principalmente por la exactitud, la precisión y la sensibilidad del método analítico utilizado para la sustancia de ensayo, es necesario controlar experi-mentalmente que la precisión y la reproducibilidad del análisis químico, así como la recuperación de la sustancia de ensayo tanto en el agua como en los peces son satisfactorias en el método particular. Ha de comprobarse también que la sustancia de ensayo no es detectable en el agua de dilución utilizada.
En caso necesario, los valores de C y C, derivados de los ensayos se corregirán a la luz de los valores de recuperación y de fondo de los controles. Las muestras de peces y de agua se manejan permanentemente para minimizar las contaminaciones y las pérdidas (derivadas, por ejemplo de la adsorción por el equipo de muestreo).
1.8.4.4. Análisis de los peces muestreados
Si se utilizan para el ensayo materiales radiomarcados, se puede analizar el marcado radiactivo total (es decir, compuestos de origen y metabolitos) o se pueden purificar las muestras de modo que sea posible analizar separadamente los compuestos de origen. Por otro lado, los principales metabolitos pueden caracterizarse en el estado de equilibrio al final de la fase de absorción, si esto supone un plazo menor. Si el FBC en términos de residuos radiomarcados totales es ≥ 1 000 %, puede ser bueno (y, para algunas categorías de productos químicos como los plaguicidas, muy recomendable), identificar y cuantificar los productos de degradación que representen ≥ 10 % de los residuos totales en los tejidos de los peces en el estado de equilibrio. Si estos productos que representan > 10 % de los residuos totales radiomarcados en los tejidos de peces se identifican y cuantifican, se recomienda entonces identificarlos y cuantificarlos también en el agua del ensayo.
La concentración de la sustancia de ensayo debería generalmente determinarse en cada pez pesado aparte. Si eso no es posible, se podrán poner conjuntamente las muestras en cada muestreo, pero esta mezcla limita los procedimientos estadísticos que pueden aplicarse a los datos. Si se desean un procedimiento y una potencia estadísticos, convendrá entonces utilizar en el ensayo un número adecuado de peces para conciliar el procedimiento de mezcla con la precisión estadística deseada (6) (7).
El FBC se expresará en función del peso húmedo total y también, en caso de sustancias muy lipofílicas, en función del contenido lipídico. El contenido lipídico de los peces se determina si es posible en cada muestreo. Se utilizarán métodos adaptados para determinar el contenido en lípidos (referencias 8 y 2 del anexo 3). Se puede recomendar la técnica de extracción por cloroformo/metanol como método normal (9). Los diversos métodos no dan valores idénticos (10), por lo que es importante precisar bien el método utilizado. El análisis de los lípidos se hará, si es posible, con el mismo extracto que el consagrado al análisis de la sustancia de ensayo, puesto que los lípidos deben a menudo retirarse del extracto antes de que este se pueda analizar por cromatografía. El contenido lipídico de los peces (en rng/kg de peso húmedo) al final del experimento no debería separarse de la del principio en más del ± 25 %. El porcentaje en solidos de los tejidos también se indicará para permitir la conversión de la concentración lipídica de peso húmedo a la de peso seco.
2. DATOS
2.1. TRATAMIENTO DE LOS RESULTADOS
La curva de absorción de la sustancia de ensayo se obtiene representando su concentración en los peces (o tejidos específicos) durante la fase de absorción en función del tiempo, con una escala aritmética. Si la curva alcanza una meseta, es decir, adopta aproximadamente una disposición asintótica al eje del tiempo, se calculará del siguiente modo el FBCSS en el estado de equilibrio:
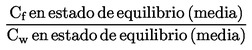
Cuando no se llega a ningún estado de equilibrio, es posible calcular un FBCSS de una precisión suficiente para la evaluación de los riesgos a partir de un «estado de equilibrio» al 80 % (1,6/k,) o 95 % (3,0/k2) del equilibrio.
También se determina el factor de concentración (FBCJ como la relación de k1/k2 las dos constantes cinéticas de primer orden. La constante de la velocidad de depuración (k2) se determina generalmente a partir de la curva de depuración (es decir, representación de la disminución de la concentración de la sustancia de ensayo en los peces en función del tiempo). La constante de la velocidad de absorción (k1) se calcula entonces en función de k2 y de un valor de Cf derivado de la curva de absorción (véase también el anexo 5). El método de elección para obtener FBCk y las constantes de velocidad k, y k2 es recurrir a métodos informáticos de estimación de parámetros no lineales (11). Alternativamente, se pueden utilizar métodos gráficos para calcular k1 y k2. Si la curva de depuración es evidentemente de orden distinto del primero, entonces convendrá utilizar modelos más complejos (véanse las referencias en el anexo 3) y buscar el consejo de un bioestadístico.
2.2. INTERPRETACIÓN DE LOS RESULTADOS
Los resultados se interpretarán con precaución cuando las concentraciones medidas de las soluciones de ensayo se encuentren a niveles cercanos al límite de detección del método de análisis.
La claridad de definición de las curvas de absorción y de pérdida indican la buena calidad de los datos de bioconcentración. La variación de las constantes de absorción/depuración entre las dos concentraciones de ensayo debería ser inferior al 20 %. Las diferencias notables observadas en las velocidades de absorción/depuración entre las dos concentraciones de ensayo se registrarán y explicarán si es posible. En general el límite de confianza del FBC se acerca al ± 20 % en los estudios bien diseñados.
3. INFORME
El informe del ensayo debe incluir la información siguiente:
3.1. SUSTANCIA DE ENSAYO
|
— |
naturaleza física y, en su caso, propiedades físico-químicas, |
|
— |
datos de identificación química (incluido el contenido en carbono orgánico, si procede), |
|
— |
en caso de marcado radiactivo, posición precisa de los átomos marcados y porcentaje de radiactividad asociada a las impurezas. |
3.2. ESPECIES UTILIZADAS
|
— |
Denominación científica, estirpe, fuente, eventuales tratamientos previos, aclimatación, edad, rango de tamaños, etc. |
3.3. CONDICIONES DEL ENSAYO
|
— |
procedimiento seguido (por ejemplo, dinámico o semiestático), |
|
— |
tipo y características del alumbrado utilizado y períodos de iluminación, |
|
— |
diseño del ensayo (por ejemplo, número y tamaño de los recipientes de ensayo, régimen de sustitución de los volúmenes de agua, numero de grupos y de peces por grupo, número de concentraciones de ensayo, duración de las fases de absorción y depuración, frecuencia de los muestreos de peces y de agua), |
|
— |
método de preparación de las soluciones madre y frecuencia de renovación (se indicarán el agente de disolución, su concentración y su contribución al contenido en carbono orgánico del agua del ensayo, en su caso), |
|
— |
las concentraciones de ensayo nominales, las medias de los valores medidos y sus desviaciones típicas en los recipientes de ensayo y su método de obtención, |
|
— |
fuente del agua de dilución, descripción de los eventuales tratamientos previos, resultados de las posibles demostraciones de la aptitud de los peces utilizados para vivir en esta agua y características de esta: pH, dureza, temperatura, concentración en oxígeno disuelto, niveles de cloro residual (si se mide), carbono orgánico total, sólidos en suspensión, salinidad del medio de ensayo (en su caso), y todas las demás medidas realizadas, |
|
— |
calidad del agua en los recipientes de ensayo, pH, dureza, COT, temperatura y concentración en oxígeno disuelto, |
|
— |
información precisa sobre la alimentación (por ejemplo tipo de comida, fuente, composición —al menos contenido en lípidos y proteínas si es posible— cantidad dada y frecuencia), |
|
— |
información sobre el tratamiento de las muestras de peces y de agua, incluidos datos de preparación, almacenamiento, extracción y procedimientos (y precisión) del análisis de la sustancia de ensayo y del contenido en lípidos (si se mide). |
3.4. RESULTADOS
|
— |
resultados de los eventuales estudios preliminares efectuados, |
|
— |
mortalidad de los peces de control y de los peces de cada recinto de ensayo y todos los comportamientos anormales observados, |
|
— |
contenido lipídico de los peces (si se determina con motivo del ensayo), |
|
— |
curvas (con todos los datos medidos) que muestren la absorción y la depuración de las sustancias de ensayo en los peces, el tiempo de acceso al estado de equilibrio, |
|
— |
Cf y Cw (con desviación típica y gama, en su caso) en el momento de cada muestreo [Cf expresado en μg/g de peso húmedo (ppm) de todo el animal o de algunos de sus tejidos, por ejemplo lípidos, y Cw en (μg/ml (ppm)] valores de Cw de la serie de control (indicar también la concentración de fondo), |
|
— |
factor de bioconcentración en el estado de equilibrio (FBCSS) o factor de concentración cinético (FBCk) y, en su caso, límites de confianza del 95 % de las constantes de la velocidad de absorción y de depuración (pérdida) todo expresado respecto al cuerpo entero y al contenido total en lípidos, si se mide, del animal (o de sus tejidos especificados), límites de confianza y desviación típica (si se dispone de ella), métodos de cálculo/análisis de los datos para cada concentración de sustancia de ensayo utilizada, |
|
— |
cuando se utilicen sustancias radiomarcadas, podrá exponerse la acumulación de todos los metabolitos detectados sí es necesario, |
|
— |
toda observación inusual relativa al ensayo, toda divergencia respecto a estos procedimientos y toda la información pertinente. |
Minimizar los resultados «no detectado al límite de detección» mediante la aplicación de un método de ensayo preliminar y el diseño experimental, puesto que estos resultados son inutilizables para los cálculos de las constantes de velocidad.
4. REFERENCIAS
|
(1) |
Connell D.W. (1988). «Bioaccumulation behaviour of persistent chemicals with aquatic organisms». Rev. Environ. Contam. Toxicol. 102, pp. 117-156. |
|
(2) |
Bintein S., Devillers, J. and Karcher W. (1993). Nonlinear dependence of fish bioconcentration on n-octano/water partition coefficient. SAR and OSAR in Environmental Research. 1, 29-390. |
|
(3) |
OECD, París (1996). Direct Phototransformation of chemicals in water. Environmental Health and Safety guidance document series on Testing and Assessment of Chemicals No 3. |
|
(4) |
Kristensen P. (1991). Bioconcentration in fish: Comparison of bioconcentration factors derived from OECD and ASTM testing methods: influence of particulate organic matter to the bioavailability of chemicals. Water Quality Institute, Denmark. |
|
(5) |
US EPA 822-R-94-002 (1994) Great Lake Water Ouality Initiative Technical Support Doc. for the Procedure to Determine Bioaccumulation Factors. July 1994. |
|
(6) |
US FDA (Food and Drug Administration) Revision. Plaguicide analytical manual, 1, 5600 Fisher's Lane Rockville, MD 20852, July 1975. |
|
(7) |
US EPA (1974). Section 5, A(l). «Analysis of Human or Animal Adipose Tissue», in Analysis of Plaguicide Residues in Human and Evironmental Samples, Thompson J.F. (ed.) Research Triangle Park, N.C. 27711. |
|
(8) |
Compaan H. (1980) en «The determination of the possible effects of chemicals and wastes on the aquatic invironment: degradation, toxicity, bioaccumulation», Ch. 2.3, Part II. Government Publi- shing Office, The Hague, The Netherlands. |
|
(9) |
Gardner et al, (1995). Limn. & Oceanogr. 30, 1099-1105. |
|
(10) |
Randall R.C, Lee H., Ozretich R.J., Lake J.L. and Pruell R.J. (1991). Evaluation of selected lipid methods for normalising pollutant bioaccumulation. Envir. Toxicol. Chem. 10, pp. 1431-1436. |
|
(11) |
CEC. Bioconcentration of chemical substances in fish: the flow-through metbod-Ring test programme, 1984-1985. Final report March 1987. Authors: P. Kristensen and N. Nyholm. |
|
(12) |
ASTME E-1022-84 (Reapproved 1988) Standard Practice for conducting Bioconcentration Tests with Fisches and Saltwater Bivalve Molluscs. |
Anexo 1
Características químicas aceptables del agua de dilución
|
|
Sustancia |
Límite de concentración |
|
1 |
Materia en suspensión |
5 mg/l |
|
2 |
Carbono orgánico total |
2mg/l |
|
3 |
Amoníaco no ionizado |
1 μg/l |
|
4 |
Cloro residual |
10 ng/l |
|
5 |
Plaguicidas organofosforados totales |
50 μg/l |
|
6 |
Plaguicidas organoclorados totales más policlorobifenilos |
50 μg/l |
|
7 |
Cloro orgánico total |
25 ng/l |
|
8 |
Aluminio |
1 ng/l |
|
9 |
Arsénico |
1 μg/l |
|
10 |
Cromo |
1 μg/l |
|
11 |
Cobalto |
1 μg/l |
|
12 |
Cobre |
1 μg/l |
|
13 |
Hierro |
1 μg/l |
|
14 |
Plomo |
1 μg/l |
|
15 |
Níquel |
1 μg/l |
|
16 |
Cinc |
1 μg/l |
|
17 |
Cadmio |
100 ng/l |
|
18 |
Mercurio |
100 ng/l |
|
19 |
Plata |
100 μg/l |
Anexo 2
Especies de peces recomendadas para los ensayos
|
|
Especies recomendadas |
Gamas de temperaturas recomendadas para los ensayos (en oC) |
Longitud total recomendada de los animales utilizados (en cm) |
|
1 |
Danio rerio (16) (Teleostei, Cyprinidae) (Hamilton-Buchanan) pez cebra |
20-25 |
3,0±0,5 |
|
2 |
Pimephales promelas (Teleostei, Cyprinidae) (Rafinesque) pez cabeza gorda |
20-25 |
5,0±2,0 |
|
3 |
Cyprinus carpio (Teleostei, Cyprinidae) (Linnaeus) carpa común |
20-25 . |
5,0±3,0 |
|
4 |
Oryzias latipes (Teleostei, Poeciliidae) (Temminck y Schlegel) medaka |
20-25 |
4,0±1,0 |
|
5 |
Poecilia reticulata (Teleostei, Poeciliidae) (Peters) guppy |
20-25 |
3,0±1,0 |
|
6 |
Lepomis macrochirus (Teleostei, Cenirarchidae) (Rafinesque) agallas azules |
20-25 |
5,0±2,0 |
|
7 |
Oncorhynchus mykiss (Teleostei Salmortidae) (Walbaum) trucha arco iris |
13-17 |
8,0±4,0 |
|
8 |
Gasterosteus aculeatus (Teleostei, Gasterosteidae) (Linnaeus) espinoso |
18-20 |
3,0±1,0 |
Se han utilizado distintas especies marinas y de estuarios en algunos países como, por ejemplo:
|
|
Leiostomus xanthurus |
|
|
Cyprinodon variegatus, |
|
|
Menidia beryllina |
|
|
Cymatogaster aggregata, |
|
|
Parophrys vetulus |
|
|
Leptocottus armatus, |
|
|
Gasterosteus aculeatus |
|
|
Dicentracus labrax |
|
|
Alburnus alburnus. |
Aprovisionamiento
Los peces de agua dulce enumerados en el cuadro anterior son fáciles de criar o están fácilmente disponibles a lo largo del año, mientras que la disponibilidad de las especies marinas o de estuario se limita en parte a los países correspondientes. Pueden reproducirse y desarrollarse en explotaciones piscícolas o en laboratorio, en condiciones donde las enfermedades y los parásitos están bajo control; los animales utilizados serán, pues, sanos y de origen conocido. Estos peces se encuentran en muchas partes del mundo.
Anexo 3
Previsión de la duración de las fases de absorción y de depuración
Previsión de la duración de la fase de absorción
Antes de realizar el ensayo, podrá obtenerse una estimación de k2 y, en consecuencia, un porcentaje del tiempo necesario para llegar al estado de equilibrio, a partir de relaciones empíricas entre k3 y el coeficiente de reparto n-octanol/agua (Pow) o k2 y la solubilidad en el agua (s).
Se puede estimar k2 (días-1), por ejemplo a partir de la relación empírica siguiente (1):
|
log10k2 = -0,414 log10(Pow) + 1,47 (r2 = 0,95) |
(ecuación 1) |
Para las otras relaciones, véase la referencia (2).
Si el coeficiente de reparto (Pow) es desconocido, se procederá a una estimación (3) a partir de la solubilidad de la sustancia en el agua según:
|
log10(Pow) = 0,862 log10(s) + 0,710 (r2 = 0,994) |
(ecuación 2) |
donde s = solubilidad (moles/l): (n = 36).
Estas relaciones solo se aplican a los productos químicos cuyos valores de log Pow se sitúan entre 2 y 6,5 (4).
El tiempo necesario para alcanzar un determinado porcentaje del estado de equilibrio puede obtenerse, aplicando la estimación de k2, de la ecuación cinética general que describe la absorción y la depuración (cinética de primer orden):

o, si C . es constante:
|
|
(ecuación 3) |
 or Cf/Cw = k1/k2 = BCF
or Cf/Cw = k1/k2 = BCF
La relación k1/k2 . Cw se acerca entonces a la concentración en el pez en el «estado de equilibrio» (Cfs).
La ecuación 3 puede así transcribirse:
|
(ecuación 4) |
El tiempo necesario para alcanzar un determinado porcentaje del estado de equilibrio puede preverse aplicando la ecuación 4, cuando k2 se estima con ayuda de las ecuaciones 1 o 2.
A título indicativo, la duración óptima estadísticamente de la fase de absorción que permite obtener datos estadísticamente aceptables (FBCk) es el período necesario para que la curva del logaritmo de la concentración de la sustancia de ensayo en el pez representado frente al tiempo lineal, llegue a su punto medio, o 1,6/k, o un 80 % del estado de equilibrio, pero sin sobrepasar 3,0/k2 o un 95 % del estado de equilibrio (7).
El tiempo para llegar al 80 % del estado de equilibrio es (ecuación 4):
|
(ecuación 5) |
Del mismo modo, para un 95 % del estado de equilibrio:
|
|
[equation 6] |
Por ejemplo, la duración de la fase de absorción (abs.) de una sustancia de ensayo que tenga un log Pow = 4 sería (con las ecuaciones 1, 5 y 6):
|
log10k2 = -0,414.(4) + 1,47 |
k2 = 0,652 días-1 |
abs. (80 %) = 1,6/0,652, es decir, 2,45 días (59 horas)
o abs. (95 %) = 3,0/0,652, es decir, 4,60 días (110 horas)
Del mismo modo, para una sustancia de ensayo con s = 10-5 mol/l (log(s) = -5,0), la duración de la absorción sería (ecuaciones 1, 2, 5 y 6):
log10 (Pow) = -0,862. (-5,0) + 0,710 = 5,02
log10k2 = -0,414. (5,02) + 1,47
k2 = 0,246 días-1
abs. (80 %) = 1,6/0,246, es decir, 6,5 días (156 horas)
o abs. (95 %) = 3,0/0,246, es decir, 12,2 días (293 horas)
Se utiliza, como alternativa, la expresión:
teq = 6,54 x 10-3 Pow + 55,31 (horas)
para calcular el tiempo de llegada al estado de equilibrio efectivo (4).
Previsión de la duración de la fase de depuración
Se podrá también prever el tiempo necesario para que (a carga corporal se reduzca a un determinado porcentaje de la concentración inicial gracias a la ecuación general que describe la absorción y la depuración (cinética de primer orden) (1) (8).
Para la fase de depuración, Cw se supone nulo. La ecuación puede reducirse a:
 o
o 
donde Cf.o es la concentración al principio del período de depuración. Se llegará a una depuración del 50 % en el tiempo (t50):
 o
o 
Del mismo modo, el 95 % de depuración se alcanzará en el tiempo:

Si se utiliza el 80 % de la absorción para el primer período (l,6/k2) y el 95 % de perdida en la fase de depuración (3,0/k2), entonces la fase de depuración será aproximadamente el doble de la depuración de la fase de absorción.
Es importante, sin embargo, tener en cuenta que estas estimaciones se basan en la hipótesis de que los esquemas de absorción y de depuración siguen una cinética de primer orden. Si es evidente que no se cumple esta condición, deberán emplearse modelos más complejos [por ejemplo, la referencia (1)].
REFERENCIAS
|
(1) |
Spacie A. and Hamelink J.L. (1982) Alternative models for describing the bioconcentration of organics in fisb. Environ. Toxicol. and Ch. 1, pp. 309-320. |
|
(2) |
Kristensen P. (1991) Bioconcentration is fisb: comparison of BCF's derived from OECD and ASTM testing methods; influence of particulate matter to the bioavailability of chemicals. Danish Water Quality Institute. |
|
(3) |
Chiou C.T. and Schmedding D.W. (1982). Partitionmg of organic compounds in octanol-water systems. Environ: Sci. Technol. 16 (1), pp. 4-10. |
|
(4) |
Hawker D.W. and Connell D.W. (1988). Influence of partition coefficient of lipophilic compounds on bioconcentration kinewtics with fish. Wat. Res. 22 (6), pp. 701-707. |
|
(5) |
Branson D.R., Blau G.E., Alexander H.C. and Neely W.B. (1975). Transactions of the American Fisheries Society, 104 (4), pp. 785-792. |
|
(6) |
Ernst W. (1985). «Accumulation in Acuatic Organisms». In: Appraisal of tests to predict the environe-mental behaviour of chemicah. Ed. by Sheehman P., Korte F., Klein W. and Bourdeau P.H. Part 4.4, pp. 243-255. SCOPE, 1985, John Wiley & Sons Ltd, N.Y. |
|
(7) |
Reilly P.M., Bajramovic R., Blau G.E., Branson D.R. and Sauerhoff M.W. (1977). Guidelines for the optimal design of experiments lo estimate parameters in first order kenetic models, Can. J. Chem. Eng. 55, pp. 614-622. |
|
(8) |
Kónemann H. and Van Leeuwen K. (1980). Toxicokinetics in fish: Accumulation and elimination of six Chlorobenzenes by Guppies. Chemosphere, 9, pp. 3-19, |
Anexo 4
Ejemplos teóricos de calendarios de muestreo para los ensayos de bioconcentración de sustancias con log Pow = 4
|
Muestreos de peces |
Calendario de muestreo |
Número de muestra |
Número de peces por muestra |
|
|
Frecuencia mínima requerida (días) |
Muestreos suplementarios |
|||
|
Fase de absorción |
- 1 0 |
|
2 (17) 2 |
añadir 45-80 peces |
|
V |
0,3 |
0,4 |
2 (2) |
4 (4) |
|
|
0,6 |
0,9 |
2 (2) |
4 (4) |
|
3 a |
1,2 |
1,7 |
2 (2) |
4 (4) |
|
4a |
2,4 |
3,3 |
2 (2) |
4 (4) |
|
5a |
4,7 |
|
2 |
6 |
|
Fase de depuración |
|
|
|
Trasladar los peces a agua libre del producto de ensayo |
|
6a |
5,0 |
5,3 |
|
4 (4) |
|
7a |
5,9 |
7,0 |
|
4 (4) |
|
8a |
9,3 |
11,2 |
|
4 (4) |
|
9a |
14,0 |
17,5 |
|
6 (4) |
|
Los valores entre paréntesis son los números de muestras (agua, peces) que se toman si se procede a un muestreo suplementario.
|
Anexo 5
Discriminación de los modelos
Se supone que la mayoría de los datos de bioconcentración se describen «razonablemente» bien con un modelo simple de dos compartimentos/dos parámetros, según indica la curva rectilínea que une aproximadamente los puntos de las concentraciones medidas en los peces durante la fase de depuración, cuando se representan sobre papel semilogarítmico [cuando estos puntos no pueden unirse con una recta, conviene recurrir a modelos más complejos; véase, por ejemplo, Spacie and Hamelink, referencia (1) en el anexo 3].
Método gráfico de determinación de la constante de la velocidad de depuración (pérdida) k2
Llevar sobre papel semilogarítmico la concentración de la sustancia de ensayo encontrada en cada muestra de peces, frente al tiempo de muestreo. La pendiente de esta recta es k2.

Atención: las divergencias en relación con esta línea recta pueden indicar un régimen de depuración más complejo que una cinética de primer orden. Puede aplicarse un método gráfico para solucionar los tipos de depuración que se desvían de la cinética de primer orden.
Método gráfico de determinación de la constante de la velocidad de absorción k1
Dada K2, se calcula k1 del modo siguiente:
|
|
(ecuación 1) |
Se lee el valor de Ct en el punto medio de la curva de absorción suavizada obtenida con los datos cuando se representa la concentración logarítmica frente al tiempo (en escala aritmética).
Método de cálculo informático de las constantes de las velocidades de absorción y de depuración (pérdida)
Para obtener el factor de bioconcentración y las constantes de velocidad k1 y k 2 se utilizarán preferiblemente métodos informáticos de estimación de parámetros no lineales. Estos programas establecen los valores de k1 y de k2 en función de un conjunto de datos secuenciales de concentración en el tiempo y del modelo:
|
(ecuación 2) |
|
(ecuación 2) |
donde tc = tiempo al final de la fase de absorción.
Este proceso desemboca en una estimación de la desviación típica para k1 y k2
Puesto que k2 puede estimarse en la mayoría de los casos a partir de la curva de depuración con una precisión relativamente grande, y puesto que existe una fuerte correlación entre los dos parámetros kl y k2 si se estiman simultáneamente, puede ser deseable calcular en primer lugar k2 a partir de los datos de depuración solamente, y luego calcular k1 a partir de los datos de absorción con ayuda de una regresión lo lineal.
C.14. ENSAYO DE CRECIMIENTO EN PECES JUVENILES
1. MÉTODO
El presente método de ensayo de toxicidad para el crecimiento reproduce las directrices del documento OCDE TG 215 (2000).
1.1. INTRODUCCIÓN
El presente ensayo tiene por objeto determinar los efectos de la exposición prolongada a sustancias químicas sobre el crecimiento de peces juveniles y se funda en un método elaborado y sometido a un estudio interlaboratorios (1) (3) en la Unión Europea para valorar los efectos de las sustancias químicas en el crecimiento de juveniles de trucha arco iris (Oncorhynchus mykiss) en condiciones dinámicas. Pueden emplearse otras especies de peces bien estudiadas. Por ejemplo, se dispone de una cierta experiencia en ensayos de crecimiento con el pez cebra (Danio rerio)(2) (4) (5) y el medaka (Oryzias (atipes) (6) (7) (8).
Véase también la parte C de la introducción general.
1.2. DEFINICIONES
Concentración mínima con efecto observado (LOEC): concentración más baja de sustancia de ensayo con la que se observa un efecto significativo (para p < 0,05) en comparación con el control. Además, todas las concentraciones de ensayo superiores a la LOEC han de provocar un efecto nocivo superior o igual al que se observa con dicha concentración.
Concentración (máxima) sin efecto observado (NOEC): concentración de ensayo inmediatamente inferior a la LOEC.
ECX: en el presente método de ensayo, concentración de sustancia de ensayo que produce una variación del x % en la tasa de crecimiento de los peces respecto a los controles.
Tasa de carga: peso húmedo de peces por unidad de volumen de agua.
Densidad de población: número de peces por unidad de volumen de agua.
Tasa de crecimiento específico de cada pez: tasa de crecimiento de un pez respecto a su peso inicial.
Tasa de crecimiento específico medio por recipiente: tasa de crecimiento medio de la población de un recipiente a una concentración determinada.
Tasa de crecimiento pseudoespecífico: tasa de crecimiento individual respecto al peso inicial medio de la población del recipiente.
1.3. PRINCIPIO DEL MÉTODO DE ENSAYO
Después de haberlos pesado, se colocan peces juveniles en fase de crecimiento exponencial en recipientes de ensayo y se exponen a una gama de concentraciones subletales de la sustancia de ensayo disuelta en agua, preferiblemente en condiciones dinámicas (flujo continuo) o, de no ser posible, en condiciones semiestáticas adecuadas (renovación discontinua). La duración del ensayo es de 28 días. Se proporciona alimento a los peces todos los días. La ración alimentaria se determina en función del peso inicial de los peces y puede volverse a calcular transcurridos 14 días. Al final del ensayo, se vuelven a pesar los peces. Se analizan los efectos sobre la tasa de crecimiento mediante un modelo de regresión con el fin de estimar la concentración que produciría una variación del x % de dicha tasa, es decir, ECx (EC10, EC20 o EC30, por ejemplo). También pueden compararse los datos con los valores de los controles para determinar la concentración mínima con efecto observado (LOEC) y. de ahí, la concentración (máxima) sin efecto observado (NOEC).
1.4. INFORMACIÓN RELATIVA A LA SUSTANCIA DE ENSAYO
Debe disponerse de los resultados de un ensayo de toxicidad aguda (véase el método C.1) efectuado preferiblemente con la especie objeto del presente ensayo y conocerse la solubilidad en el agua y la presión de vapor de la sustancia de ensayo. Para calcular la cantidad de sustancia en las soluciones de ensayo debe aplicarse un método analítico fiable, cuyo limite de detección y precisión sean conocidos y figuren en el informe.
La información útil para establecer las condiciones de ensayo incluye la fórmula estructural de la sustancia de ensayo, su pureza, estabilidad en el agua y a la luz, pKa, Pow y los resultados de un ensayo de biodegradabilidad fácil (véase el método C.4).
1.5. VALIDEZ DEL ENSAYO
Para que el ensayo sea válido deben darse las condiciones siguientes:
|
— |
la mortalidad en los controles no ha de superar el 10 % al final del ensayo, |
|
— |
el peso medio de los peces de los controles debe haber aumentado lo suficiente para poder detectar la variación mínima de la tasa de crecimiento que se considere significativa. Un ensayo interlaboratorios (3) ha puesto de manifiesto que, en el caso de la trucha arco iris, al cabo de 28 días el peso medio de los peces de los controles ha de haberse incrementado al menos en la mitad (50 %) de su peso medio inicial. Por ejemplo, si el peso inicial es de 1 g/pez (= 100 %), el peso final tras 28 días ha de ser ≥1,5 g/pez (≥ 150 %), |
|
— |
la concentración de oxígeno disuelto ha de ser, al menos, del 60 % del valor de saturación en el aire a lo largo de todo el ensayo, |
|
— |
en ningún momento del ensayo la temperatura del agua debe variar en más de ± 1 oC entre los recipientes de ensayo y debe mantenerse en un intervalo de 2 oC dentro de las gamas establecidas para la especie sometida a ensayo (apéndice 1). |
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Equipo
Se empleará el equipo común de laboratorio y, en particular:
|
a) |
medidores de oxígeno y de pH; |
|
b) |
equipo para determinar la dureza y alcalinidad del agua: |
|
c) |
dispositivo adecuado de regulación de la temperatura, preferiblemente continua: |
|
d) |
recipientes de material químicamente inerte y de capacidad adecuada a la carga y la densidad de población recomendadas (véase el punto 1.8.5 y el apéndice 1); |
|
e) |
balanza suficientemente precisa (precisión de ±0,5 %). |
1.6.2. Agua
Puede utilizarse para el ensayo toda agua en la que la especie sometida a ensayo muestre una tasa de crecimiento y supervivencia a largo plazo adecuadas. La calidad ha de ser constante a todo lo largo del ensayo. El pH debe hallarse entre 6,5 y 8,5, si bien a lo largo de un mismo ensayo debe permanecer en un intervalo de ±0,5 pH unidades. Se recomienda una dureza superior a 140 mg/l (CaCO3). Deben tomarse periódicamente muestras para análisis con el fin de cerciorarse de que el agua de dilución no interfiere en los resultados del ensayo (por ejemplo, por complejación de la sustancia de ensayo). Cuando se sepa que un agua de dilución es de calidad relativamente constante, conviene proceder, por ejemplo, cada tres meses, a la determinación de los metales pesados (por ejemplo, Cu, Pb, Zn, Hg, Cd y Ni), aniones y cationes principales (por ejemplo, Ca, Mg, Na, K, Cl y SO4), plaguicidas (por ejemplo, organofosforados totales y organoclorados totales), carbono orgánico total y sólidos en suspensión. Si se demuestra que la calidad del agua es constante al menos durante un año, los análisis pueden ser menos frecuentes y espaciarse más (por ejemplo, cada seis meses). En el apéndice 2 se recogen algunas características químicas de un agua de dilución aceptable.
1.6.3. Soluciones de ensayo
Las soluciones de ensayo a las concentraciones elegidas se preparan por dilución de la solución madre.
La solución madre se prepara preferiblemente por simple mezcla o agitación mecánica de la sustancia de ensayo en el agua de dilución (por ejemplo, mediante un agitador o ultrasonidos). Pueden emplearse columnas de saturación (columnas de solubilidad para lograr la concentración adecuada de solución madre.
En algunos casos puede ser necesario usar disolventes o dispersantes (agentes de disolución) para obtener una solución madre a la concentración deseada. Puede entonces utilizarse como disolvente acetona, etanol, meta-nol. dimetilsulfóxido, dimetilformamida o trietílenglicol, y como dispersante Cremophor RH40, Tween 80, metilcelulosa al 0,01 % o HCO-40. Deben tomarse precauciones si se emplean agentes fácilmente biodegrada-bles (como la acetona) y/o muy volátiles, pues estos pueden dar lugar a una proliferación bacteriana en los ensayos dinámicos. Si se emplea un agente de disolución, no debe actuar de forma significativa sobre el crecimiento de los peces ni producir efectos nocivos visibles en los juveniles, lo cual ha de demostrarse en un lote de control que solo contenga disolvente.
Para los ensayos dinámicos se requiere un sistema que aporte y diluya continuamente la solución madre de la sustancia de ensayo (por ejemplo, bomba dosificadora, diluyente proporcional o sistema saturador) para distribuir una serie de concentraciones en los recipientes de ensayo. Los regímenes de flujo de las soluciones madre y del agua de dilución deben supervisarse con regularidad, preferiblemente todos los días, y no debería variar en más del 10 % a lo largo del ensayo. Con arreglo a un ensayo interlaboratorios (3), en el caso de la trucha arco iris se considera adecuado un régimen de renovación del agua durante el ensayo equivalente a 6 1/g de pez/día (véase el punto 1.8.2.2).
En el caso de los ensayos semiestáticos, la frecuencia de renovación del medio dependerá de la estabilidad de la sustancia de ensayo, si bien se recomienda renovarlo todos los días. Si los ensayos preliminares de estabilidad (véase el punto 1.4) ponen de manifiesto que la concentración de la sustancia de ensayo no es estable (es decir, queda fuera del intervalo del 80 al 120 % de la concentración nominal o cae por debajo del 80 % de la concentración medida al inicio) entre dos renovaciones, debe plantearse la realización de un ensayo dinámico.
1.6.4. Selección de la especie
Se recomienda realizar el ensayo con la trucha arco iris (Oncorhynchus mykiss), pues es la especie que más se ha empleado en el ensayo interlaboratorios (1) (3). También es posible emplear otras especies bien estudiadas, aunque entonces podría ser necesario adaptar el procedimiento para que las condiciones de ensayo sean adecuadas. Por ejemplo, se dispone asimismo de experiencia con el pez cebra (Danio rerio) (4) (5) y el medaka (Oryzias latipes) (6) (7) (8). En tal caso, deben justificarse la elección de la especie y el método experimental.
1.6.5. Preparación de los peces
Se seleccionan los peces de ensayo de una misma población, y preferentemente de un mismo desove, que se haya mantenido al menos dos semanas antes del ensayo en condiciones de calidad del agua e iluminación similares a las del ensayo. Debe proporcionárseles una ración alimentaria diaria del 2 % de su peso corporal como mínimo y preferiblemente del 4 % mientras duren la preparación y el ensayo.
Transcurridas 48 horas en esas condiciones, se registra la mortalidad y se aplican los criterios siguientes:
|
— |
si la mortalidad es superior al 10 % de la población en siete días, se rechaza todo el lote, |
|
— |
si la mortalidad se halla entre el 5 y el 10 % de la población, se prolonga otros siete días el período de aclimatación; si durante este segundo período la mortalidad supera el 5 %, se rechaza todo el lote, |
|
— |
si la mortalidad es inferior al 5 % de la población en siete días, se acepta el lote. |
Los peces no deben recibir tratamiento terapéutico alguno durante el ensayo ni en las dos semanas anteriores al mismo.
1.7. DISEÑO DEL ENSAYO
Se entenderá por «diseño del ensayo» la selección del número de concentraciones de ensayo y el intervalo entre las mismas, el número de recipientes por concentración y el número de peces por recipiente. Lo más idóneo es diseñar el ensayo con arreglo a:
|
a) |
el objetivo del estudio; |
|
b) |
el método de análisis estadístico que vaya a emplearse; |
|
c) |
la disponibilidad y el coste de los recursos experimentales. |
En la medida de lo posible, el enunciado del objetivo debe especificar la potencia estadística necesaria para detectar una diferencia determinada (por ejemplo, de tasa de crecimiento) o la precisión con la que ha de proporcionarse la ECx (por ejemplo, x — 10, 20 o 30 y preferiblemente no por debajo de 10) para poder hacer una estimación. Sin ello no puede darse una, indicación precisa de la escala del estudio.
Es importante ser consciente de que un diseño idóneo (que saca mayor partido de los recursos) para un método de análisis estadístico no lo es necesariamente para otro. Así pues, el diseño recomendado para la estimación de una LOEC/NOEC no será el mismo que se recomiende para un análisis por regresión.
En la mayoría de los casos, es preferible el análisis de regresión al análisis de la varianza, por los motivos que exponen Stephan y Rogers (9). Sin embargo, si no se encuentra un modelo de regresión adecuado (r2 < 0,9), debe recurrirse a la NOEC/LOEC.
1.7.1. Diseño para el análisis por regresión
Los aspectos importantes para el diseño de un ensayo que vaya a analizarse por regresión son los siguientes:
|
a) |
Es conveniente que las concentraciones empleadas incluyan la concentración con efecto (EC1O,2O,3O, etc.) y la gama de concentraciones en la sustancia de ensayo. La precisión con la que puede estimarse la concentración con efecto será mayor si dicha concentración se halla en el medio de la gama de concentraciones empleada en el ensayo. Para seleccionar las concentraciones de ensayo adecuadas puede resultar útil un ensayo previo de determinación de gama; |
|
b) |
para que la modelización estadística sea satisfactoria, el ensayo debe incluir al menos un recipiente de control y otros cinco recipientes con distintas concentraciones. En su caso, si se emplea un agente de disolución, debe realizarse, además de las series tratadas con la sustancia de ensayo, un control con agente de disolución a la concentración de ensayo más elevada (véanse los puntos 1.8.3 y 1.8.4); |
|
c) |
puede emplearse una serie geométrica o logarítmica apropiada (10) (véase el apéndice 3). Es preferible un espaciamiento logarítmico entre las concentraciones de ensayo; |
|
d) |
si se dispone de más de seis recipientes, los que sobrepasen dicha cantidad se emplearán como recipientes en paralelo o se distribuirán en la gama de concentraciones para reducir el espaciamiento entre estas.Ambas opciones son igualmente válidas. |
1.7.2. Diseño para la estimación de la NOEC/LOEC mediante análisis de la varianza (ANOVA)
Conviene disponer de varios recipientes en paralelo para cada concentración y realizar el análisis estadístico por recipiente (11). Sin recipientes en paralelo no puede tenerse en cuenta la variabilidad entre recipientes además de la que existe entre los peces. No obstante, la experiencia ha puesto de manifiesto (12) que en el caso estudiado la variabilidad entre recipientes era muy escasa en relación con la variabilidad dentro de un mismo recipiente (es decir, entre los peces). Así pues, una opción relativamente aceptable consiste en efectuar el análisis estadístico a escala individual con cada pez.
Normalmente se emplea una serie geométrica de al menos cinco concentraciones de sustancia de ensayo, espaciadas por un factor que no supere 3,2.
Por lo general, si se realizan ensayos con recipientes en paralelo, el número de recipientes de control en paralelo y, por tanto, el número de peces ha de ser el doble del empleado con cada concentración de ensayo, que debe ser constante (13) (14) (15). De no usarse recipientes en paralelo, el número de peces del grupo de control ha de ser igual al empleado en cada concentración de ensayo.
Si el análisis de la varianza (ANOVA) se realiza en los recipientes y no a nivel individual [lo cual requeriría marcar todos los peces o utilizar las tasas de crecimiento pseudoespecífico (véase el punto 2.1.2)], se necesita una cantidad suficiente de recipientes en paralelo para poder determinar la desviación estándar de los recipientes con la misma concentración, lo cual significa que el error del análisis de la varianza tendrá al menos 5 grados de libertad (11). Si solo se emplean recipientes en paralelo para los controles, se corre el riesgo de que la variabilidad del error esté sesgada, pues podría aumentar con el valor medio de la tasa de crecimiento en cuestión. Dado que es probable que la tasa de crecimiento disminuya cuando aumente la concentración, se tendería a sobrestimar la variabilidad.
1.8. PROCEDIMIENTO
1.8.1. Selección y pesaje de los peces
Es importante que el peso de los peces varíe lo menos posible al inicio del ensayo. En el apéndice I figuran las gamas de peso apropiadas en cada una de las especies recomendadas para el ensayo. Conviene que, al principio del ensayo, la gama de pesos de todo el lote de peces utilizados se mantenga en un intervalo de ± 10 % de la media aritmética y, en cualquier caso, no supere el 25 %. Se recomienda pesar una submuestra de peces antes del ensayo para calcular el peso medio.
No se proporciona alimento alguno a los peces durante las 24 horas anteriores al inicio del ensayo. A continuación, se toman los peces al azar. Se emplea un anestésico general [por ejemplo, una solución acuosa de 100 mg/l de metanosulfonato de tricaína (MS 222) neutralizada mediante adición de dos partes de bicarbonato sódico por parte de MS 222] para pesar cada sujeto en húmedo (secado con material absorbente) con la precisión indicada en el apéndice 1. Se apartan los peces cuyo peso se halla en el intervalo deseado y se reparten al azar entre los recipientes de ensayo. Se registra el peso húmedo total de los peces de cada recipiente. Tanto el uso de anestésico como la manipulación (incluido el secado y pesaje) pueden afectar y herir a los peces juveniles, sobre todo si se trata de especies de pequeño tamaño. Por ello, deben manipularse los animales objeto del ensayo con la mayor precaución para evitar cualquier agresión.
Se vuelven a pesar los peces el 28o día de ensayo (véase el punto 1.8.6). No obstante, si se considera necesario ajustar la ración alimentaria, pueden pesarse los peces el 14o día de ensayo (véase el punto 1.8.2.3). Para valorar las variaciones de tamaño de los peces y, de ahí, ajustar la ración alimentaria, también puede emplearse otro método como el fotográfico.
1.8.2. Condiciones de exposición
1.8.2.1. Duración
La duración del ensayo es > 28 días.
1.8.2.2. Tasa de carga y densidad de población
Es importante que la tasa de carga y la densidad sean apropiadas para la especie de ensayo empleada (véase el apéndice 1). La tensión que genera a los peces una densidad de población demasiado elevada produce una disminución de la tasa de crecimiento y posiblemente la enfermedad. Por el contrario, si la densidad es demasiado baja, puede dar lugar a un comportamiento territorial, lo cual también puede afectar al crecimiento. En cualquier caso, la tasa de carga debe ser suficientemente baja para poder mantener sin aireación una concentración mínima de oxígeno disuelto del 60 % del valor de saturación en el aire. Un ensayo interlaboratorios (3) ha puesto de manifiesto que, en el caso de la trucha arco iris, resulta aceptable una tasa de carga de 16 truchas de 3 a 5 g en un volumen de 40 1. Se recomienda una frecuencia de renovación del agua durante el ensayo de 6 l/g de pez/día.
1.8.2.3. Alimentación
Debe proporcionarse a los peces una dieta suficiente y adecuada (apéndice 1) para permitir una tasa de crecimiento aceptable, al tiempo que se evita la proliferación bacteriana y la turbidez del agua. En el caso de la trucha arco iris, esas condiciones deberían conseguirse con una ración diaria del 4 % de su peso corporal (3) (16) (17) (18). La ración diaria puede dividirse en dos partes iguales y administrarse en dos veces, con un mínimo de 5 horas de intervalo. La ración es función del peso inicial total de los peces de cada recipiente de ensayo. Si los peces se pesan otra vez el 14o día de ensayo, se vuelve a calcular la ración. La alimentación debe suprimirse durante las 24 horas anteriores al pesaje.
Todos los días debe limpiarse cuidadosamente por succión el fondo de los recipientes de ensayo para eliminar los alimentos que no se hayan consumido y la materia fecal.
1.8.2.4. Iluminación y temperatura
El fotoperíodo y la temperatura del agua han de ser adecuados para la especie utilizada (véase el apéndice 1).
1.8.3. Concentraciones de ensayo
Normalmente se necesitan cinco concentraciones de sustancia de ensayo, sea cual sea el diseño del mismo (véase el punto l.7.2). Para seleccionar las concentraciones apropiadas es útil conocer previamente la toxicidad de la sustancia de ensayo (por ejemplo, gracias a un ensayo de toxicidad aguda o un estudio de determinación de gamas). El empleo de menos de cinco concentraciones debe justificarse. Las sustancias no deben someterse a ensayo en concentraciones superiores a su límite de solubilidad en el agua.
Si se emplea un agente de disolución para facilitar la preparación de la solución madre, su concentración final ha de ser preferiblemente la misma en todos los recipientes de ensayo y no debe superar 0,1 ml/l (véase el punto 1.6.3). No obstante, debe evitarse en la medida de lo posible el empleo de tales agentes.
1.8.4. Controles
El número de recipientes de control con agua de dilución dependerá del diseño del ensayo (véanse los puntos 1.7 a 1.7.2). Si se emplea un agente de disolución, se realizará la misma cantidad de controles con agua de dilución que con agente de disolución.
1.8.5. Frecuencia de los análisis y mediciones
Las concentraciones de la sustancia de ensayo deben determinarse periódicamente durante el mismo (véase más adelante).
En los ensayos dinámicos, debe comprobarse periódicamente, a ser posible todos los días, los flujos de diluyente y de solución madre de sustancia tóxica. Dichos flujos no deberían variar en más del 10 % a lo largo del ensayo. Cuando la concentración de la sustancia deba permanecer entre el ± 20 % de la concentración nominal (es decir, en el intervalo del 80 al 120 %; véanse los puntos 1.6.2 y 1.6.3), se recomienda analizar, como mínimo, la concentración de ensayo más baja y la más alta al principio del ensayo y, a continuación, una vez por semana. En los ensayos en que no quepa esperar que la concentración de sustancia de ensayo permanezca en un intervalo de ± 20 % de la concentración nominal (con arreglo a los datos de estabilidad de la sustancia), es preciso analizar todas las concentraciones de ensayo, pero según el mismo régimen.
En los ensayos semiestáticos (con renovación) en que la concentración de la sustancia debe permanecer entre el + 20 % de la concentración nominal, se recomienda analizar, como mínimo, la concentración de ensayo más baja y la más alta en cuanto se preparen y justo antes de renovar el medio al principio del estudio y, a continuación, una vez por semana. En los ensayos en que no quepa esperar que la concentración de sustancia de ensayo permanezca en un intervalo de ± 20 % de la concentración nominal, es preciso analizar todas las concentraciones de ensayo según el mismo régimen que el empleado con las sustancias más estables.
Se recomienda basar los resultados en las concentraciones medidas. No obstante, si los datos disponibles muestran que la concentración de la sustancia de ensayo en la solución se ha mantenido debidamente a lo largo de todo el ensayo en un intervalo de ± 20 % de la concentración nominal o de la concentración medida al inicio, los resultados pueden fundarse en las concentraciones nominales o en las medidas.
En algunos casos puede estar indicado filtrar (por ejemplo, con un filtro de poros de 0,45 μm) o centrifugar las muestras. Aunque la centrifugación es el método recomendado, si el medio de ensayo no se absorbe en los filtros, también puede emplearse la filtración.
Durante el ensayo debe medirse en todos los recipientes de ensayo el oxígeno disuelto, el pH y la temperatura. En los controles y en un uno de los recipientes con la concentración más alta se medirán la dureza total, la alcalinidad y la salinidad, si procede. El oxígeno disuelto y la salinidad, cuando proceda, se medirán como mínimo tres veces: al principio, a la mitad y al final del ensayo. En los ensayos semiestáticos se recomienda medir el oxígeno disuelto más a menudo, preferiblemente antes y después de cada renovación del agua o al menos una vez por semana. El pH ha de medirse al principio y al final de cada período de renovación del agua en los ensayos semiestáticos y al menos una vez por semana en los dinámicos. La dureza y la alcalinidad se medirán una vez en cada ensayo. La temperatura debería someterse a control continuo al menos en un recipiente de ensayo.
1.8.6. Observaciones
Peso: al final del ensayo deben pesarse en húmedo (secados con material absorbente) todos los peces vivos, en grupos por recipiente de ensayo o individualmente. Es preferible pesar los animales por recipiente, pues el pesaje individual requiere marcar todos los peces. Si se determina la tasa de crecimiento específico de cada pez (pesaje individual), se optará por una técnica de marcado que no perturbe a los animales (en lugar del criomar-cado puede emplearse, por ejemplo, hilo fino de pescar de colores).
Deben examinarse los peces todos los días durante el ensayo y registrarse toda anomalía externa (hemorragias, despigmentación, etc.) y todo comportamiento anómalo. Se tomará nota de todas las muertes y se retirarán los peces muertos lo antes posible. Estos no se sustituirán, ya que la tasa de carga y la densidad de población son suficientemente elevadas para evitar que la variación del número de peces en un recipiente afecte al crecimiento. Sin embargo, deberá adaptarse la ración alimentaria.
2. RESULTADOS E INFORME
2.1. TRATAMIENTO DE LOS RESULTADOS
Es aconsejable que participe un estadístico tanto en el diseño como en el análisis del ensayo, pues el presente método da cabida a variaciones considerables en el procedimiento experimental, por ejemplo, en cuanto al número de recipientes y de concentrationes de ensayo, el número de peces, etc. Debido a las diversas posibilidades en el diseño del ensayo, no se proporcionan aqui directrices concretas sobre los métodos estadísticos.
No es preciso calcular la tasa de crecimiento en los recipientes de ensayo en los que la mortalidad es superior al 10 %, si bien debe indicarse la tasa de mortalidad a todas las concetraciones de ensayo.
Con independencia del método empleado par analizar los datos, el concepto central es la tasa de crecimiento específico r entre el momento t1 y el momento t2, que puedo definirse de varias formas según si los peces se han marcado individualmente o no o si se necessita una media por recipiente.

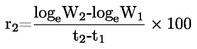

donde:
|
r1 |
= |
tasa de crecimiento específico de cada pez, |
|
r2 |
= |
tasa de crecimiento específico medio por recipiente, |
|
r3 |
= |
tasa de crecimiento pseudoespecífico, |
|
w1, w2 |
= |
peso de un pez determinado en los momentos t1 y t2, respectivamente, |
|
loge w1 |
= |
logaritmo del peso de un pez determinado al inicio del período de estudio, |
|
loge w2 |
= |
logaritmo del peso de un pez determinado al final del período de estudio, |
|
loge W1 |
= |
media de los logaritmos de los valores w1 de los peces del recipiente al inicio del período de estudio, |
|
loge W2 |
= |
media de los logaritmos de los valores w2 de los peces del recipiente al final del período de estudio. |
|
t1, t2 |
= |
tiempo (días) inicial y fin del período de estudio |
r1, r2, r3 pueden calcularse para el período comprendido entr el día 0 y el día 28 y, si procede (es decir, si se han realizado mediciones el día 14), para los períodos entre los días 0 y 14, y 14 y 28.
2.1.1. Análisis de los resultados por regresión (modelización concentración-repuesta)
Este método de análisis establece una relación matemática adecuada entre la tasa de crecimiento específico y la concentración, lo cual permite calcular la «ECx», es decir, todo valor de EC necesario. Con este método no es preciso calcular (r2). Es preferible este ultimo método y, además, resultas más adecuado si se utilizan especies más pequeñas.
Con el fin de estudiar la relación concentratión-respuesta, debe hacerce una representación gráfica de las tasas de crecimiento específico medio por recipiente (r2) en función de la concentración.
Para expresar la relación entre r2 y la concentración, debe elegirse un modelo adecuado y justificarse la elección mediante un razonamiento pertinente.
Si el número de peces que sobreviven varía de un recipiente a otro, debe ponderarse el proceso de ajuste del modelo para tener en cuenta el tamaño desigual de los grupos.
El método de ajuste del modelo debe permitir estimar, por ejemplo, la EC20 y deducir su dispersión (error típico o intervalo de confianza). El gráfico del modelo ajustado debe figurar junto a los datos para poder valorar la adecuación del ajuste del modelo (9) (19) (20) (21).
2.1.2. Análisis de los resultados para calcular la LOEC
Si en el ensayo se han utilizado series en paralelo para todas las concentraciones, la estimación de la LOEC podría basarse en el análisis de la varianza (ANOVA) de la tasa de crecimiento específico medio de cada recipiente (véase el punto 2.1), seguido de un método adecuado [por ejemplo, el de Dunnett o de Williams (13) (14) (15) (22)] para comparar la media r a cada concentración con la media r de los controles con el fin de determinar la concentración mínima a la que dicha diferencia es significativa para p = 0,05. Si las hipótesis necesarias relativas a los métodos paramétricos no se cumplen —distribución no normal (por ejemplo, prueba de Shapiro-Wilk) o varianza heterogénea (prueba de Bartlett)—, deberá estudiarse la posibilidad de transformar los datos para homogeneizar las varianzas antes de efectuar el análisis de la varianza (ANOVA) o el análisis ponderado de la varianza (ANOVA).
Si en el ensayo no se han utilizado series en paralelo para todas las concentraciones, el análisis de la varianza (ANOVA) basado en los recipientes resultará insensible o imposible. En ese caso, puede considerarse aceptable basar el análisis de la varianza (ANOVA) en la tasa de crecimiento pseudoespecífico r3 de cada pez.
A continuación puede compararse la media r, para cada concentración de ensayo con la media r3 de los controles, tras lo cual puede determinarse la LOEC como anteriormente. Debe admitirse que este método no tiene cuenta en absoluto la variabilidad entre recipientes, al margen de la imputable a la variabilidad entre los peces. Sin embargo, la experiencia ha puesto de manifiesto (9) que la variabilidad entre recipientes es muy escasa en relación con la variabilidad dentro de un mismo recipiente (es decir, entre los peces). Si el análisis no incluye los datos relativos a cada pez, deberá indicarse el método de identificación de los valores atípicos y justificarse su utilización.
2.2. INTERPRETACIÓN DE LOS RESULTADOS
Los resultados deben interpretarse con prudencia si las concentraciones medidas de las sustancias tóxicas en las soluciones de ensayo se aproximan a los límites de detección del método de análisis o, en los ensayos semi-estáticos, si la concentración de sustancia de ensayo disminuye entre el momento en que se prepara la solución y el momento previo a la renovación.
2.3. INFORME DEL ENSAYO
El informe del ensayo debe incluir la información siguiente:
2.3.1. Sustancia de ensayo:
|
— |
naturaleza física y propiedades fisicoquímicas pertinentes, |
|
— |
identificación química, incluida la pureza y el método de análisis cuantitativo de la sustancia de ensayo, si procede. |
2.3.2. Especie sometida a ensayo:
|
— |
denominación científica, |
|
— |
cepa, tamaño, proveedor, tratamientos previos, etc. |
2.3.3. Condiciones de ensayo:
|
— |
método empleado (por ejemplo, semiestático/renovación o dinámico, carga, densidad de población, etc.), |
|
— |
diseño del ensayo (por ejemplo, número de recipientes de ensayo, de concentraciones de ensayo y de recipientes en paralelo, número de peces por recipiente), |
|
— |
método de preparación de las soluciones madre y frecuencia de renovación (en su caso, se especificarán el agente de disolución empleado y su concentración), |
|
— |
concentraciones nominales de ensayo, medias de los valores determinados analíticamente en los recipientes de ensayo, desviaciones estándar de estos, método de obtención y datos que muestren que las mediciones se refieren a las concentraciones de la sustancia de ensayo en disolución verdadera, |
|
— |
características del agua de dilución: pH, dureza, alcalinidad, temperatura, concentración de oxígeno disuelto, contenido de cloro residual (si se ha medido), carbono orgánico total, sólidos en suspensión, salinidad del medio de ensayo (si se ha medido) y cualquier otra medición realizada, |
|
— |
calidad del agua en los recipientes de ensayo: pH, dureza, temperatura y concentración de oxígeno disuelto, |
|
— |
información detallada de la alimentación (por ejemplo, tipo de alimento o alimentos, procedencia, cantidad proporcionada y frecuencia). |
2.3.4. Resultados:
|
— |
datos que demuestren que los controles cumplen los criterios de validez relativos a la supervivencia y datos de la mortalidad observada en todas las concentraciones de ensayo, |
|
— |
técnicas de análisis estadístico aplicadas, estadísticas basadas en las series en paralelo o en los peces, tratamiento de los datos y justificación de los métodos empleados, |
|
— |
cuadros que recojan el peso individual y medio de los peces los días 0, 14 (si se han pesado) y 28, valores de las medias por recipiente o tasas de crecimiento pseudoespecífico (si procede) correspondientes a los períodos de 0 a 28 días o, en su caso, de 0 a 14 y de 14 a 28, |
|
— |
resultados del análisis estadístico [análisis de regresión o análisis de la varianza (ANOVA)], preferiblemente nen forma de cuadro y de gráfico. LOEC (p = 0,05) y NOEC o EC con sus errores típicos, si es posible. |
|
— |
incidencia de toda reacción anómala de los peces y de todo efecto visible producido por la sustancia de ensayo. |
3. REFERENCIAS
|
(1) |
Solbe J.F. de LG (1987). Environmental Effects of Chemicals (CFM 93 50 SLD). Report on a UK Ring Test of a Method for Studying the Effects of Chemicals on the Growth rate of Fish. WRc Report No. PRD 1388-M/2. |
|
(2) |
Meyer, A., Bierman, C.H. y Orti, G. (1993). The phylogenetic position of the zebrafish (Danio reno), a model system in developmental biology: an invitation to the comparative method, Proc. R. Soc. Lond. B. 252, 231-236 |
|
(3) |
Ashley S., Mallett M.J. y Grandy N.J. (1990). EEC Ring Test of a Method for Determining the Effects of Chemicals on the Growth Rate of Fish. Final Report to the Commission of the European Communities. WRc Report No EEC 2600-M. Crossland N.O. (1985). A method to evalúate effects of toxic chemicals on fish growth. Chemosphere, 14, pp 1855-1870. |
|
(4) |
Crossland N.O. (1985). A method to evalúate effects of toxic chemicals on fish growth. Chemosphere, 14, pp 1855-1870. |
|
(5) |
Nagel R., Bresh H., Caspers N., Hansen P.D., Market M, Munk R., Scholz N. y Hófte B.B. (1991). Effect of 3.4-dichloroaniline on the early life stages of the Zebrafish (Brachydanio rerio): results of a comparative laboratory study. Ecotox. Environ. Safety. 21. pp 157-164. |
|
(6) |
Yamamoto, Tokio. (1975). Series of stock cultures in biological field. Medaka (killifish) biology and strains. Keigaku Publish. Tokio. Japón. |
|
(7) |
Holcombe . G.W.. Benoit D.A., Hammermeister, D.E., Leonard, E.N. y Johnson, R.D. (1995). Acute and long-term effects of nine chemicals on the Japanese medaka (Oryzias latipes). Arch. Environ. Coma. Toxicol. 28, pp 287-297. |
|
(8) |
Benoit, D.A., Holcombe, GAV. y Spehar, R.L. (199l). Guidelines for conducting early life toxicity tests with Japanese medaka (Oryzias latipes). Ecological Research Series EPA-600:3-91-06 3. U. S. Environmental Protection Agency, Duluth. Minesota. |
|
(9) |
Stephan CE. y Rogers J.W. (1985). Advantages of using regression analysis to calculate results of chronic toxicity tests. Aquatic Toxicology and Hazard Assessment: Eighth Symposium, ASTM STP 891, R C Bahner y D J Hansen. Eds. American Society for Testing and Materials, Filadelfia, pp 328-338. |
|
(10) |
Environment Canada (1992). Biological test method: toxicity tests using early life stages of salmonid fish (rainbow trout, coho salmon, or atlantic salmon). Conservation and Protection, Ontario, Report EPS l/RM/28, 81 p. |
|
(11) |
Cox D.R. (1958). Planning of experiments. Wiley Edt. |
|
(12) |
Pack S. (1991). Statistical issues concerning the design of tests for determining the effects of chemicals on the growth rate of fish. Room Document 4, OECD Ad Hoc Meeting of Experts on Aquatic Toxicology, WRc Medmenham. UK, 10-12 diciembre de 1991. |
|
(13) |
Dunnett C.W. (1955). A Multiple Comparisons Procedure for Comparing Several Treatments with a Control, J. Amer. Statist. Assoc, 50, pp 1096-1121. |
|
(14) |
Dunnett C.W. (1964). New tables for multiple comparisons with a control. Biometrics, 20, pp 482-491. |
|
(15) |
Williams D.A. (1971). A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27, pp 103-117. |
|
(16) |
Johnston, W.L., Atkinson. J.L., Glanville NT. (1994). A technique using sequential feedings of different coloured food to determine food intake by individual rainbow trout, Oncorhynchus mykiss: effect of feeding level. Aquaculture 120, 123-13 3. |
|
(17) |
Quinton, J. C. y Blake, R.W. (1990). The effect of feed cycling and radon level on the compensatory growth response in rainbow trout, Oncorhynhus mykiss. Journal of Fish Biology, 37, 33-41. |
|
(18) |
Post, G. (1987). Nutrition and Nutritional Diseases of Fish. Chapter IX in Testbook of Fish Health. T.F.H. Publications, Inc. Neptune City. Nueva Jersey, E.E.U.U. 288 P. |
|
(19) |
Bruce, R.D. y Vorsteeg D.J. (1992). A statistical procedure for modelling continuous toxicity data . Environ. Toxicol. Chem. 11, 1485-1494. |
|
(20) |
DeGraeve, G.M., Cooney, J.M., Pollock, T.L., Reichenbach, J.H., Dean, Marcus, M.D. y Mclntyre, D.O. (1989). Precision of EPA seven-day fathead minnow larval survival and growth test; intra and interlaboratory study. Report EA-6189 (American Petroleum Institute Publication, n. 4468). Electric Power Research Institute, Palo Alto, CA. |
|
(21) |
Norbert-King T.J. (1988). An interpolation estímate for chronic toxicity: the ICp approach. US Environmental Protection Agency. Environmental Research Lab., Duluth, Minesota. Tech. Rep. No 05-88 of National Effluent Toxicity Assesment Center. Sept. 1988. 12 pp. |
|
(22) |
Williams D.A. (1972). The comparison of several dose levels with a zero dose control. Biometrics 28, pp 510-531. |
Apéndice 1
ESPECIES DE PECES RECOMENDADAS Y CONDICIONES ADECUADAS PARA EL ENSAYO
|
Especie |
Gama de temperatura recomendada ( oC) |
Fotoperíodo (horas) |
Gama recomendada de peso inicial de los peces (g) |
Precisión exigida de la medición |
Tasa de carga (g/l) |
Densidad de población (por litro) |
Alimentación |
Duración del ensayo (días) |
|
Especie recomendada: |
|
|
|
|
|
|
|
|
|
Oncorhynchus mykiss Trucha arco iris |
12,5 - 16,0 |
12-16 |
1-5 |
100 mg |
1,2 - 2,0 |
4 |
Especialidad alimentaria seca para crías de salmónidos |
> 28 |
|
Otras especies bien documentadas: |
|
|
|
|
|
|
|
|
|
Danio rerio Pez cebra |
21-25 |
12-16 |
0,050 - 0,100 |
1 mg |
0,2 - 1,0 |
5-10 |
Alimento vivo (Brachionus artemia) |
> 28 |
|
Oryzias latipes Medaka |
21-25 |
12-16 |
0,050 - 0,100 |
To nearest 1 mg |
0,2 - 1,0 |
5-20 |
Alimento vivo (Brachionus artemia) |
> 28 |
Apéndice 2
ALGUNAS CARACTERÍSTICAS QUÍMICAS DE UN AGUA DE DILUCIÓN ACEPTABLE
|
Sustancia |
Concentraciones |
|
Materia en suspensión |
< 20 mg/l |
|
Carbono orgánico total |
< 2 mg/l |
|
Amoniaco no ionizado |
< 1 μg/l |
|
Cloro residual |
<10 μg/l |
|
Plaguicidas organofosforados totales |
< 50 ng/l |
|
Plaguicidas organoclorados totales y bifenilos policlorados |
< 50 ng/l |
|
Cloro orgánico total |
< 25 ng/l |
Apéndice 3
Series logarítmicas de concentraciones válidas para un ensayo de toxicidad (10)
|
Columna (Número de concentraciones entre 100 y 10, o entre 10 y 1) (18) |
||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
100 |
100 |
100 |
100 |
100 |
100 |
100 |
|
32 |
46 |
56 |
63 |
68 |
72 |
75 |
|
10 |
22 |
32 |
40 |
46 |
52 |
56 |
|
3,2 |
10 |
18 |
25 |
32 |
37 |
42 |
|
1,0 |
4,6 |
10 |
16 |
22 |
27 |
32 |
|
|
2,2 |
5,6 |
10 |
15 |
19 |
24 |
|
|
1,0 |
3,2 |
6,3 |
10 |
14 |
18 |
|
|
|
1,8 |
4,0 |
6,8 |
10 |
13 |
|
|
|
1,0 |
2,5 |
4,6 |
7,2 |
10 |
|
|
|
|
1,6 |
3,2 |
5,2 |
7,5 |
|
|
|
|
1,0 |
2,2 |
3,7 |
5,6 |
|
|
|
|
|
1,5 |
2,7 |
4,2 |
|
|
|
|
|
1,0 |
1,9 |
3,2 |
|
|
|
|
|
|
1,4 |
2,4 |
|
|
|
|
|
|
1,0 |
1,8 |
|
|
|
|
|
|
|
1,3 |
|
|
|
|
|
|
|
1,0 |
C.15. ENSAYO DE TOXICIDAD A CORTO PLAZO EN EMBRIONES DE PEZ Y ALEVINES
1. MÉTODO
El presente método de ensayo de toxicidad a corto plazo reproduce las directrices del documento OCDE TG 212 (1998).
1.1. INTRODUCCIÓN
El presente ensayo de toxicidad a corto plazo en embriones de pez y alevines abarca la exposición del animal desde la fase de huevo recién fecundado hasta el final de la fase de alevín. No se proporciona alimento alguno a embriones y alevines durante el ensayo, que debe, por tanto, concluir cuando los alevines sigan nutriéndose de la vesícula vitelina.
El presente ensayo tiene por objeto determinar los efectos letales y, en menor medida, subletales de sustancias químicas en fases concretas de la vida de las especies sometidas al mismo. Proporciona información útil, pues puede: a) constituir un nexo entre los ensayos letales y subletales; b) servir de ensayo de detección de cara a un ensayo completo en las primeras fases de la vida o a los ensayos de toxicidad crónica, y c) emplearse para estudiar especies en relación con las cuales las técnicas de cría no estén suficientemente avanzadas para abarcar el período de transición de la alimentación endógena a la exógena.
Cabe señalar que los ensayos que abarcan todas las fases de la vida de los peces son los únicos que suelen permitir una estimación precisa de la toxicidad crónica de las sustancias químicas en estos animales y que, si se limita la exposición de manera que no cubra una u otra fase de la vida, puede perderse sensibilidad y, por tanto, subestimarse la toxicidad crónica. Por consiguiente, el ensayo en embriones y alevines resultará menos sensible que un ensayo completo en las primeras fases de la vida, sobre todo si se trata de sustancias muy lipó-filas (log Pow > 4) o sustancias que posean una forma de actuación tóxica particular. Cabe esperar, no obstante, que la diferencia de sensibilidad entre los dos ensayos sea menor con las sustancias de acción narcótica no específica (1).
Hasta la publicación del presente ensayo, el ensayo con embriones y alevines se ha llevado a cabo principalmente con el pez de agua dulce Danio rerio Hamilton-Buchanan (teleósteos, ciprínidos, nombre común: pez cebra); de ahí que en el apéndice 1 figuren indicaciones detalladas para la realización del mismo con esa especie, si bien pueden emplearse otras con las que se disponga de experiencia (cuadros 1A y 1B).
1.2. DEFINICIONES
Concentración mínima con efecto observado (LOEC): concentración más baja de sustancia de ensayo con la que se observa un efecto significativo (para p < 0,05) en comparación con el control. Además, todas las concentraciones de ensayo superiores a la LOEC han de provocar un efecto nocivo superior o igual al que se observa con dicha concentración.
Concentración (máxima) sin efecto observado (NOEC): concentración de ensayo inmediatamente inferior a la LOEC.
1.3. PRINCIPIO DEL ENSAYO
Se exponen los embriones de pez y los alevines a una gama de concentraciones de la sustancia de ensayo disuelta en agua. El protocolo permite elegir entre un procedimiento semiestático o dinámico, según la naturaleza de la sustancia de ensayo. El ensayo comienza cuando se colocan los huevos fecundados en los recipientes de ensayo y finaliza justo antes de que la vesícula vitelina de cualquiera de las larvas de cualquiera de los recipientes sea reabsorbida por completo o antes de que los animales del lote de control empiecen a morir de inanición. Los efectos letales y subletales se evalúan y comparan con los valores del control con el fin de determinar la concentración mínima con efecto observado y, de ahí, la concentración máxima sin efecto observado. Los efectos también pueden analizarse mediante un modelo de regresión a fin de calcular la concentración que provocaría un porcentaje de efecto determinado (CL/ECX, donde x es un porcentaje de efecto definido).
1.4. INFORMACIÓN RELATIVA A LA SUSTANCIA DE ENSAYO
Debe disponerse de los resultados de un ensayo de toxicidad aguda (véase el método C.1) efectuado preferiblemente con la especie objeto del presente ensayo, ya que pueden resultar útiles para seleccionar una gama apropiada de concentraciones para el ensayo en las primeras fases de la vida. Debe conocerse la solubilidad en el agua (incluida la solubilidad en el agua del ensayo) y la presión de vapor de la sustancia de ensayo. Para calcular la cantidad de sustancia en las soluciones de ensayo debe aplicarse un método analítico fiable, cuyo límite de detección y precisión sean conocidos y figuren en el informe.
La información útil para establecer las condiciones de ensayo incluye la fórmula estructural de la sustancia de ensayo, su pureza, estabilidad a la luz, estabilidad en las condiciones de ensayo, pKa, Pow y los resultados de un ensayo de biodegradabilidad fácil (véase el método C.4).
1.5. VALIDEZ DEL ENSAYO
Para que el ensayo sea válido deben darse las condiciones siguientes:
|
— |
la tasa global de supervivencia de los huevos fecundados en los lotes de control y, en su caso, en los recipientes que solo contengan disolvente ha de ser superior o igual a los límites establecidos en los apéndices 2 y 3, |
|
— |
la concentración de oxígeno disuelto ha de situarse entre el 60 y el 100 % del valor de saturación en el aire a lo largo del ensayo, |
|
— |
en ningún momento del ensayo la temperatura del agua debe variar en más de ±1,5 oC entre los recipientes de ensayo ni entre dos días sucesivos y debe mantenerse en las gamas establecidas para la especie sometida a ensayo (apéndices 2 y 3). |
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Recipientes de ensayo
Puede emplearse cualquier recipiente de cristal u otro material inerte químicamente, de dimensiones suficientes para que se cumplan los criterios de carga (véase el punto 1.7.1.2). Se recomienda colocar los recipientes al azar en la zona donde se lleve a cabo el ensayo. Si en el laboratorio hay efectos sistemáticos que pueden paliarse mediante el agrupamiento, es preferible colocar los recipientes según un esquema de agrupamiento aleatorizado (velando por que todos los tratamientos se apliquen en todos los grupos) que según un esquema completamente aleatorizado. En caso de que se agrupen los recipientes, debe tenerse presente en el análisis posterior de los datos. Los recipientes de ensayo han de protegerse de toda perturbación indeseada.
1.6.2. Selección de la especie de peces
Las especies de peces recomendadas figuran en el cuadro 1A, aunque es posible emplear otras (cuadro 1B), si bien entonces puede ser necesario adaptar el procedimiento para que las condiciones de ensayo sean adecuadas. En tal caso, deben justificarse la elección de la especie y el método experimental.
1.6.3. Manipulación de los peces reproductores
Las directrices de ensayo 210 de la OCDE (19) y las referencias (2) (3) (4) (5) (6) de la bibliografía recogen indicaciones detalladas sobre la manipulación de peces reproductores en condiciones satisfactorias.
1.6.4. Manipulación de los embriones y las larvas
Los embriones y las larvas pueden colocarse en receptáculos provistos de paredes o extremos de malla dentro del recipiente principal para permitir el flujo de la solución de ensayo. Puede provocarse un flujo no turbulento a través de los pequeños receptáculos suspendiéndolos de un brazo que los desplace en sentido vertical, pero de manera que los organismos permanezcan siempre sumergidos. Puede emplearse, asimismo, un sistema de sifón. Los huevos fecundados de salmónidos pueden depositarse en rejillas o mallas con aperturas de dimensiones suficientes para que las larvas pasen a través después de la eclosión. En los ensayos semiestáticos con renovación diaria y completa del medio, conviene utilizar pipetas Pasteur para la toma de embriones y larvas (véase el punto 1.6.6).
Si se utilizan cubetas, rejillas o mallas para mantener los huevos en el recipiente principal de ensayo, deben retirarse después de la eclosión de las larvas (19), pero deben conservarse las mallas que impidan que los peces se escapen. Si es necesario trasladar las larvas, no deben exponerse al aire ni emplearse redes para sacar a los peces de los recipientes que contengan los huevos (con algunas especies menos frágiles, como la carpa, estas precauciones pueden resultar superfluas). El momento de dicho traslado varía según la especie y no siempre es necesario. En los ensayos semiestáticos, pueden emplearse vasos de laboratorio o recipientes poco profundos y provistos, en su caso, de una rejilla a escasa distancia del fondo. Si el volumen de esos recipientes es suficiente para que se cumplan los requisitos de carga (véase el punto 1.7.1.2), no es preciso trasladar los embriones o larvas.
1.6.5. Agua
Puede utilizarse para el ensayo toda agua que se ajuste a las características químicas de un agua de dilución aceptable enumeradas en el apéndice 4 y en la que la especie sometida a ensayo muestre una tasa de supervivencia en el lote de control al menos igual que la recogida en los apéndices 2 y 3. Su calidad ha de ser constante a todo lo largo del ensayo. La variación del pH debe permanecer en un intervalo de ±0,5 unidades. Deben tomarse muestras periódicamente para análisis con el fin de cerciorarse de que el agua de dilución no interfiere en los resultados del ensayo (por ejemplo, por complejación de la sustancia de ensayo) ni altera el comportamiento de los peces reproductores. Cuando se sepa que un agua de dilución es de calidad relativamente constante, conviene proceder, por ejemplo, cada 3 meses, a la determinación de los metales pesados (por ejemplo, Cu, Pb, Zn, Hg, Cd y Ni), aniones y cationes principales (por ejemplo, Ca, Mg, Na, K, Cl y SO4), plaguicidas (por ejemplo, organofosforados totales y organoclorados totales), carbono orgánico total y sólidos en suspensión. Si se demuestra que la calidad del agua es constante al menos durante un año, los análisis pueden ser menos frecuentes y espaciarse más (por ejemplo, cada 6 meses).
1.6.6. Soluciones de ensayo
Las soluciones de ensayo a las concentraciones elegidas se preparan por dilución de la solución madre.
La solución madre se prepara preferiblemente por simple mezcla o agitación mecánica de la sustancia de ensayo en el agua de dilución (por ejemplo, mediante un agitador o ultrasonidos). Pueden emplearse columnas de saturación (columnas de solubilidad) para lograr la concentración adecuada de solución madre. Debe evitarse, en la medida de lo posible, el uso de disolventes o dispersantes (agentes de disolución); sin embargo, en algunos casos pueden ser necesarios para obtener una solución madre a la concentración deseada. Puede entonces utilizarse como disolvente acetona, etanol, metanol, dimetilformamida o trietilenglicol, y como dispersante Cremophor RH40, Tween 80, metilcelulosa al 0,01 % o HCO-40. Deben tomarse precauciones si se emplean agentes fácilmente biodegradables (como la acetona) y/o muy volátiles, pues estos pueden dar lugar a una proliferación bacteriana en los ensayos dinámicos. Si se emplea un agente de disolución, no debe actuar de forma significativa sobre la supervivencia ni producir efectos nocivos visibles en las primeras fases de la vida, lo cual ha de demostrarse en un lote de control que solo contenga disolvente. No obstante, debe hacerse todo lo posible por evitar el uso de dichas sustancias.
En el caso de los ensayos semiestáticos, pueden aplicarse dos métodos distintos de renovación: o bien se preparan sustancias de ensayo nuevas en recipientes limpios y se transfieren cuidadosamente los huevos y larvas supervivientes en un pequeño volumen de la solución antigua, evitando exponerlos al aire, o bien se dejan los organismos sometidos a ensayo en su recipiente y se cambian al menos las tres cuartas partes del agua de ensayo. La frecuencia de renovación del medio dependerá de la estabilidad de la sustancia de ensayo, si bien se recomienda renovarlo todos los días. Si los ensayos preliminares de estabilidad (véase el punto 1.4) ponen de manifiesto que la concentración de la sustancia de ensayo no es estable (es decir, queda fuera del intervalo del 80 al 120 % de la concentración nominal o cae por debajo del 80 % de la concentración medida al inicio) entre dos renovaciones, debe plantearse la realización de un ensayo dinámico. En cualquier caso, debe velarse por evitar toda agresión a las larvas durante la renovación del agua.
Para los ensayos dinámicos se requiere un sistema que aporte y diluya continuamente la solución madre de la sustancia de ensayo (por ejemplo, bomba dosificadora, diluyente proporcional o sistema saturador) para distribuir una serie de concentraciones en los recipientes de ensayo. Los regímenes de flujo de las soluciones madre y del agua de dilución deben supervisarse con regularidad, preferiblemente todos los días, y no debería variar en más del 10 % a lo largo del ensayo. Se considera adecuado un régimen de flujo equivalente al menos a 5 veces el volumen del recipiente de ensayo cada 24 horas (2).
1.7. PROCEDIMIENTO
Se ha publicado información útil sobre la realización de los ensayos de toxicidad en embriones de pez y alevines. La bibliografía del presente texto recoge algunos ejemplos (7) (8) (9) de tales publicaciones.
1.7.1. Condiciones de exposición
1.7.1.1. Duración
Conviene iniciar el ensayo en los 30 minutos siguientes a la fecundación de los huevos. Los embriones se sumergen en la solución de ensayo antes o después (pero lo antes posible) de que comience la fase de división del blastodisco y, en cualquier caso, antes de que empiece la fase de gástrula. Si los huevos proceden de un proveedor comercial, puede resultar imposible iniciar el ensayo inmediatamente después de la fecundación. Dado que un retraso en el inicio del ensayo puede afectar seriamente a la sensibilidad del mismo, debería iniciarse en las 8 horas siguientes a la fecundación. Puesto que no se proporciona alimento a las larvas durante el período de exposición, el ensayo debe finalizar justo antes de que se haya reabsorbido por completo la vesícula vitelina de una cualquiera de las larvas de cualquiera de los recipientes de ensayo o antes de que haya muertes por inanición en los lotes de control. La duración del ensayo dependerá de la especie utilizada. Los apéndices 2 y 3 recogen recomendaciones al respecto.
1.7.1.2. Carga
El número de huevos fecundados al principio del ensayo ha de ser suficiente para responder a las necesidades estadísticas. Se reparten al azar entre los distintos tratamientos y se coloca un mínimo de 30 huevos fecundados, repartidos equitativamente (o lo más equitativamente posible, ya que con algunas especies es difícil obtener lotes iguales) entre, al menos, tres recipientes de ensayo en paralelo por cada concentración. La tasa de carga (biomasa por volumen de ensayo de solución) debe ser suficientemente baja para poder mantener sin aireación una concentración mínima de oxígeno disuelto del 60 % del valor de saturación en el aire. En los ensayos dinámicos, se recomienda una tasa de carga que no supere 0,5 g/l en 24 horas ni 5 g/l de solución en cualquier momento (2).
1.7.1.3. Iluminación y temperatura
El fotoperíodo y la temperatura del agua han de ser adecuados para la especie utilizada (apéndices 2 y 3). Para vigilar la temperatura puede ser conveniente emplear un recipiente de ensayo adicional.
1.7.2. Concentraciones de ensayo
Normalmente se necesitan 5 concentraciones de sustancia de ensayo espaciadas por un factor constante que no supere 3.2. Para seleccionar la gama de concentraciones de ensayo debe tenerse presente la curva CL50/período de exposición del estudio de toxicidad aguda. En algunos casos puede estar indicado utilizar menos de 5 concentraciones, por ejemplo, en los ensayos límite, y un intervalo más pequeño entre concentraciones. El empleo de menos de 5 concentraciones debe justificarse. No es preciso someter a ensayo concentraciones superiores a la CL50 tras 96 horas, o a 100 mg/l, si esta concentración es inferior a dicha CL50. Las sustancias no deben someterse a ensayo en concentraciones superiores a su límite de solubilidad en el agua de ensayo.
Si se emplea un agente de disolución para facilitar la preparación de las soluciones de ensayo (véase el punto 1.6.6), su concentración final ha de ser la misma en todos los recipientes de ensayo y no debe superar 0,1 ml/l.
1.7.3. Controles
Paralelamente a las series tratadas con la sustancia de ensayo, debe hacerse un lote de control con agua de dilución (en tantos ejemplares como sea necesario) y, si procede, otro control con agente de disolución (en tantos ejemplares como sea necesario).
1.7.4. Frecuencia de los análisis y medidas
Las concentraciones de la sustancia de ensayo deben determinarse periódicamente durante el mismo.
En los ensayos semiestáticos en que la concentración de la sustancia debe permanecer entre el ± 20 % de la concentración nominal (es decir, en el intervalo del 80 al 120 %; véanse los puntos 1.4 y 1.6.6), se recomienda analizar, como mínimo, la concentración de ensayo más baja y la más alta en cuanto se preparen y justo antes de renovar el medio, al menos en tres ocasiones regularmente espaciadas a lo largo del ensayo (los análisis deben practicarse en muestras de la misma solución, tomadas justo después de su preparación y en el momento de la renovación).
En los ensayos en que no quepa esperar que la concentración de sustancia de ensayo permanezca en un intervalo de ± 20 % de la concentración nominal (con arreglo a los datos de estabilidad de la sustancia), es preciso analizar todas las concentraciones de ensayo cuando se acaben de preparar y en el momento de la renovación, pero según el mismo régimen, es decir, al menos tres veces regularmente espaciadas a lo largo del ensayo. Es suficiente con determinar las concentraciones de la sustancia de ensayo antes de la renovación del medio en un solo recipiente por concentración. Las determinaciones no han de espaciarse más de 7 días. Se recomienda basar los resultados en las concentraciones medidas. No obstante, si los datos disponibles muestran que la concentración de la sustancia de ensayo en la solución se ha mantenido debidamente a lo largo de todo el ensayo en un intervalo de ± 20 % de la concentración nominal o de la concentración medida al inicio, los resultados pueden fundarse en estas.
En los ensayos dinámicos, conviene aplicar un régimen de muestreo similar al descrito para los ensayos semiestáticos (si bien en este caso no cabe hacer determinaciones antes de renovar las soluciones). No obstante, si el ensayo dura más de 7 días, puede ser oportuno incrementar el número de tomas de muestras durante la primera semana (por ejemplo, tres series de medidas) para cerciorarse de que las concentraciones de ensayo permanecen estables.
En algunos casos puede estar indicado filtrar (por ejemplo, con un filtro de poros de 0,45 μm) o centrifugar las muestras, pero, como ni la filtración ni la centrifugación permiten siempre separar la fracción no biodispo-nible de la sustancia de ensayo de su fracción biodisponible, no es indispensable efectuarlas.
Durante el ensayo debe medirse en todos los recipientes de ensayo el oxígeno disuelto, el pH y la temperatura. En los controles y en un uno de los recipientes con la concentración más alta se medirá la dureza total y la salinidad, si procede. El oxígeno disuelto y la salinidad, cuando proceda, se medirán como mínimo tres veces: al principio, a la mitad y al final del ensayo. En los ensayos semiestáticos se recomienda medir el oxígeno disuelto más a menudo, preferiblemente antes y después de cada renovación del agua o al menos una vez por semana. El pH ha de medirse al principio y al final de cada período de renovación del agua en los ensayos semiestáticos y al menos una vez por semana en los dinámicos. La dureza se medirá una vez en cada ensayo. La temperatura debería medirse todos los días y lo ideal sería un control continuo al menos en un recipiente de ensayo.
1.7.5. Observaciones
1.7.5.1. Fase de desarrollo embrionario
Al principio de la exposición a la sustancia de ensayo debe comprobarse con tanta precisión como sea posible la fase embrionaria (gástrula) en una muestra representativa de huevos debidamente conservados y limpiados. Puede asimismo consultarse la bibliografía, que recoge descripciones e ilustraciones de las fases embrionarias (2) (5) (10) (11).
1.7.5.2. Eclosión y supervivencia
Al menos una vez al día deben observarse y contarse las eclosiones y los supervivientes. Puede ser conveniente efectuar observaciones más frecuentes al principio del ensayo (por ejemplo, cada 30 minutos durante las tres primeras horas), pues en algunos casos el tiempo de supervivencia puede ser más significativo que solo el número de muertes (por ejemplo, si se producen efectos tóxicos agudos). Las larvas y embriones muertos deben retirarse en cuanto se localicen, ya que pueden descomponerse rápidamente. Dicha retirada ha de hacerse con sumo cuidado para no tocar ni dañar físicamente los huevos y larvas adyacentes, pues son extremadamente delicados y sensibles. Los criterios para considerar que un organismo está muerto varían según la fase del desarrollo:
|
— |
huevos: sobre todo en la fase más temprana, disminución acusada de translucidez y cambio de la coloración debido a la coagulación y/o precipitación de proteínas, que les dan un aspecto blanco opaco, |
|
— |
embriones: ausencia de movimientos corporales y/o ausencia de latido cardíaco y/o decoloración opaca en las especies cuyos embriones son translúcidos normalmente, |
|
— |
larvas: inmovilidad y/o ausencia de movimiento respiratorio y/o ausencia de latido cardíaco y/o coloración blanca opaca del sistema nervioso central y/o ausencia de reacción a los estímulos mecánicos. |
1.7.5.3. Aspecto anómalo
El número de larvas que presenten anomalías corporales y/o pigmentarias, así como la fase de reabsorción de la vesícula vitelina deben registrarse a intervalos adecuados con arreglo a la duración del ensayo y la naturaleza de la anomalía observada. Cabe señalar que la aparición de larvas y embriones anómalos ocurre en condiciones normales y que su proporción puede alcanzar varias unidades por cada 100 en los controles de algunas especies. Los animales que presenten anomalías solo deben retirarse de los recipientes de ensayo cuando estén muertos.
1.7.5.4. Comportamiento anómalo
Las anomalías como la hiperventilación, natación descoordinada o inactividad atípica deben registrarse a intervalos adecuados según la duración del ensayo. Pese a ser difíciles de cuantificar, estos efectos pueden contribuir a interpretar los datos de mortalidad proporcionando información sobre el modo de actuación tóxica de la sustancia.
1.7.5.5. Longitud
Se recomienda medir la longitud de cada sujeto al final del ensayo utilizando la longitud estándar, la longitud a la horquilla o la longitud total. Si se observa una descomposición de la aleta caudal o un desgaste de las aletas, debe emplearse la longitud estándar. Por lo general, en un ensayo realizado correctamente, el coeficiente de variación de la longitud entre los distintos recipientes de los controles ha de ser 20 %.
1.7.5.6. Peso
Puede pesarse cada sujeto al final del ensayo; el peso seco (24 horas a 60 oC) es preferible al peso húmedo (secados con material absorbente). Por lo general, en un ensayo realizado correctamente, el coeficiente de variación del peso entre los distintos recipientes de los controles ha de ser ≤ 20 %.
Gracias a las observaciones anteriores, se obtienen algunos o todos los datos siguientes para el análisis estadístico:
|
— |
mortalidad acumulativa, |
|
— |
número de larvas sanas al final del ensayo, |
|
— |
momento en que empieza y acaba la eclosión (90 % de eclosiones en cada recipiente en paralelo), |
|
— |
número de larvas que eclosionan cada día, |
|
— |
longitud (y peso) de los animales supervivientes al final del ensayo, |
|
— |
número de larvas con deformidades o aspecto anómalo, |
|
— |
número de larvas que presenten un comportamiento anómalo. |
2. RESULTADOS E INFORME
2.1. TRATAMIENTO DE LOS RESULTADOS
Es aconsejable que participe un estadístico tanto en el diseño como en el análisis del ensayo, pues el presente método da cabida a variaciones considerables en el procedimiento experimental, por ejemplo, en cuanto al número de recipientes y de concentraciones de ensayo, al número inicial de huevos fecundados y a los parámetros medidos. Debido a las diversas posibilidades en el diseño del ensayo, no se proporcionan aquí directrices concretas sobre los métodos estadísticos.
Si tienen que calcularse la LOEC y la NOEC, será preciso analizar las variaciones en cada serie en paralelo mediante el análisis de la varianza (ANOVA) o las tablas de contingencia. El método de Dunnett puede resultar útil para realizar comparaciones múltiples entre los resultados obtenidos con cada concentración y los de los controles (12) (13). Las referencias (14) y (15) de la bibliografía recogen otros ejemplos útiles. Debe calcularse y registrarse la magnitud del efecto detectable mediante el análisis de la varianza (ANOVA) u otros métodos (es decir, la potencia del ensayo). Cabe señalar que no todas las observaciones relacionadas en el punto 1.7.5.6 se prestan al tratamiento estadístico por análisis de la varianza (ANOVA). Así, por ejemplo, la mortalidad acumulativa y el número de larvas sanas al final del ensayo pueden analizarse por métodos de probita.
Si procede calcular la CL y la ECX, una o varias curvas adecuadas, como puede ser la curva logística, deben ajustarse a los resultados pertinentes mediante un método estadístico como el de los mínimos cuadrados o los mínimos cuadrados no lineales. Las curvas deben parametrarse de manera que puedan estimarse directamente la CL y la ECX que interese y su error estándar. Así se facilitará en gran medida el cálculo de los límites de confianza en torno a la CL y la ECX. Salvo que haya razones de peso para preferir otros niveles de confianza, se seleccionarán límites del 95 % en ambas direcciones. Es preferible que el método de ajuste permita evaluar la significación de la falta de ajuste. Pueden emplearse métodos gráficos para ajustar las curvas. Todas las observaciones enumeradas en el punto 1.7.5.6 se prestan al análisis de regresión.
2.2. INTERPRETACIÓN DE LOS RESULTADOS
Los resultados deben interpretarse con prudencia cuando las concentraciones medidas de las sustancias tóxicas en las soluciones de ensayo se aproximen a los límites de detección del método de análisis. Asimismo, los resultados relativos a concentraciones superiores a la solubilidad de la sustancia en el agua han de interpretarse con precaución.
2.3. INFORME DEL ENSAYO
El informe del ensayo debe incluir la información siguiente:
2.3.1. Sustancia de ensayo:
|
— |
naturaleza física y propiedades fisicoquímicas pertinentes, |
|
— |
identificación química, incluida la pureza y el método de análisis cuantitativo de la sustancia de ensayo, si procede. |
2.3.2. Especie sometida a ensayo:
|
— |
denominación científica, variedad, número de peces parentales (es decir, número de hembras empleadas para obtener la cantidad de huevos necesarios para el ensayo), fuente y método de recogida de los huevos fecundados y manipulaciones posteriores. |
2.3.3. Condiciones de ensayo:
|
— |
método empleado (por ejemplo, semiestático o dinámico, tiempo transcurrido entre la fecundación y el inicio del ensayo, carga, etc.), |
|
— |
fotoperíodo(s), |
|
— |
diseño del ensayo (por ejemplo, número de recipientes de ensayo y número de recipientes en paralelo, número de embriones por recipiente en paralelo), |
|
— |
método de preparación de las soluciones madre y frecuencia de renovación (en su caso, se especificarán el agente de disolución empleado y su concentración), |
|
— |
concentraciones nominales de ensayo, valores medidos en los recipientes de ensayo, medias y desviaciones estándar de estos y método de obtención; si la sustancia de ensayo es soluble en el agua a concentraciones inferiores a las de ensayo, debe demostrarse que las mediciones se refieren a las concentraciones de la sustancia de ensayo en disolución, |
|
— |
características del agua de dilución: pH, dureza, temperatura, concentración de oxígeno disuelto, contenido de cloro residual (si se ha medido), carbono orgánico total, sólidos en suspensión, salinidad del medio de ensayo (si se ha medido) y cualquier otra medición realizada, |
|
— |
calidad del agua en los recipientes de ensayo: pH, dureza, temperatura y concentración de oxígeno disuelto. |
2.3.4. Resultados:
|
— |
resultados de los posibles estudios preliminares relativos a la estabilidad de la sustancia de ensayo, |
|
— |
datos que demuestren que los controles cumplen la norma general de aceptabilidad en materia de supervivencia de la especie sometida a ensayo (apéndices 2 y 3), |
|
— |
datos de la mortalidad y la supervivencia en las fases de embrión y de larva y tasas globales de mortalidad y supervivencia, |
|
— |
días transcurridos hasta la eclosión y número de eclosiones, |
|
— |
longitud (y peso), |
|
— |
incidencia y descripción de las anomalías morfológicas, en su caso, |
|
— |
incidencia y descripción de los efectos sobre el comportamiento, en su caso, |
|
— |
análisis estadístico y tratamiento de los datos, |
|
— |
en el caso de los ensayos sometidos al análisis de la varianza, la concentración mínima con efecto observado (LOEC) para p = 0,05 y la concentración máxima sin efecto observado (NOEC) para cada respuesta evaluada, así como una descripción de los métodos estadísticos empleados y una indicación de la magnitud del efecto que puede detectarse, |
|
— |
en el caso de los ensayos analizados mediante técnicas de regresión, la CL/ECx, e intervalos de confianza, y un gráfico del modelo ajustado que se haya utilizado para su cálculo, |
|
— |
justificación de toda desviación respecto al presente método de ensayo. |
3. REFERENCIAS
|
(1) |
Kristensen P. (1990) Evaluation of the Sensitivity of Short Term Fish Early Life Stage Tests in Relation to other FELS Test Methods. Final report to the Commission of the European Communities, pp. 60. Junio de 1990. |
|
(2) |
ASTM (1988). Standard Guide for Conducting Early Life-Stage Toxicity Tests with Fishes. American Society for Testing and Materials. E 1241-88. 26 pp. |
|
(3) |
Brauhn J.L. y Schoettger R.A. (1975). Acquisition and Culture of Research Fish: Rainbow trout, Fathead minnows, Channel Catfish and Bluegills. p. 54, Ecological Research Series, EPA-660/3-75-011, Duluth, Minnesota. |
|
(4) |
Brungs W.A. y Jones B.R. (1977) Temperature Criteria for Freshwater Fish: Protocol and Procedures p. 128, Ecological Research Series EPA-600/3-77-061. Duluth, Minnesota. |
|
(5) |
Laale H.W. (1977) The Biology and Use of the Zebrafish (Brachydanio rerio) in Fisheries Research. A Literature Review. J. Biol. 10, 121-173. |
|
(6) |
Legault R. (1958) A Technique for Controlling the Time of Daily Spawning and Collecting Eggs of the Zebrafish, Brachydanio rerio (Hamilton-Buchanan) Copeia, 4, pp. 328-330. |
|
(7) |
Dave G., Damgaard B., Grande M., Martelin J.E., Rosander B. y Viktor T. (1987). Ring Test of an Embryo-larval Toxicity Test with Zebrafish (Brachydanio rerio) Using Chromium and Zinc as Toxicants. Environmental Toxicology and Chemistry, 6, pp. 61-71. |
|
(8) |
Birge J.W., Black J.A. y Westerman A.G. (1985). Short-term Fish and Amphibian Embryo-larval Tests for Determinmg the Effects of Toxicant Stress on Early Life Stages and Estimating Chronic Values for Single Compounds and Complex Effluents. Environmental Toxicology and Chemistry 4, pp. 807-821. |
|
(9) |
Van Leeuwen C.J., Espeldoorn A. y Mol F. (1986) Aquatic Toxicological Aspects of Dithiocarbamates and Related Compounds. III. Embryolarval Studies with Rainbow Trout (Salmo gairdneri). J. Aquatic Toxicology, 9, 129-145. |
|
(10) |
Kirchen R.V. y W. R. West (1969). Teleostean Development. Carolina Tips 32(4): 1-4. Carolina Biological Supply Company. |
|
(11) |
Kirchen R.V. y W. R. West (1976). The Japanese Medaka. Its care and Development. Carolina Biological Supply Company, Carolina del Norte. 36 pp. |
|
(12) |
Dunnett C.W. (1955) A Multiple Comparisons Procedure for Comparing Several Treatments with Control. J. Amer. Statist. Assoc, 50, pp 1096-1121. |
|
(13) |
Dunnett C.W. (1964). New Tables for Multiple Comparisons with a Control. Biometrics, 20, 482-491. |
|
(14) |
Mc Clave J.T., Sullivan J.H. y Pearson J.G. (1980). Statistical Analysis of Fish Chronic Toxicity Test Data. Proceedings of 4th Aquatic Toxicology Symposium, ASTM, Filadelfia. |
|
(15) |
Van Leeuwen C.J., Adema D.M.M. y Hermes J. (1990). Quantitative Structure-Activity Relationships for Fish Early Life Stage Toxicity. Aquatic Toxicology, 16, pp. 321-334. |
|
(16) |
Environment Canada. (1992). Toxicity Tests Using Early Life Stages of Salmonid Fish (Rainbow Trout, Coho Salmon or Atlantic Salmon). Biological Test Method Series. Report EPS l/RM/28, Diciembre de 1992, pp. 81. |
|
(17) |
Dave G. y Xiu R. (1991). Toxicity of Mercury. Nickel, Lead and Cobalt to Embryos and Larvae of Zebrafish, Brachydanio rerio. Arch. of Environmental Contamination and Toxicology, 21, 126-134. |
|
(18) |
Meyer A., Bierman C.H. y Orti G. (199 3). The phylogenetic position of the Zebrafish (Danio rerio). A model system in developmental biology — an invitation to the comperative methods. Proc. Royal Society of London, Series B, 252: 231-236. |
|
(19) |
Ghillebaert F. Chaillou C, Deschamps F. y Roubaud P. (1995). Toxic Effects, at Three pH Levels, of Two Reference Molecules on Common Carp Embryo. Ecotoxicology and Environmental Safety 32, 19-28. |
|
(20) |
US EPA, (1991). Guidelines for Culturing the Japanese Medaka, Oryzias latipes. EPA repon EPA/600/3-91/064, Dic. 1991. EPA, Duluth. |
|
(21) |
US EPA, (1991). Guidelines for Conducting Early Life Stage Toxicity Tests with Japanese Medaka, (Oryzias latipes). EPA report EPA/600/3-91/063, Dic. 1991, EPA, Duluth. |
|
(22) |
De Graeve G.M., Cooney J.D., McIntyre D.O., Poccocic T.L., Reichenbach N.G. Dean J.H. y Marcus M.D. (1991). Validity in the performance of the seven-day Fathead minnow (Pimephales promelas) larval survival and growth test: an intra- and interlaboratory study. Environ. Tox. Chem. 10, 1189-1203. |
|
(23) |
Calow P. (1993). Handbook of Ecotoxicology, Blackwells. Oxford. Vol. 1, capítulo 10: Methods for spawning, culturing and conducting toxicity tests with Early Life stages of Estuarine and Marine fish. |
|
(24) |
Balon E.K. (1985). Early life history of fishes: New developmental, ecological and evolutionary perspectives, Junk Publ., Dordrecht, 280 pp. |
|
(25) |
Blaxter J.H.S. (1988). Pattern and variety in development, In: W.S. Hoar y D.J. Randall Eds., Fish Physiology, vol. XIA, Academic press, pp. 1-58. |
Cuadro 1A
Especies de peces recomendadas para el ensayo
|
Agua dulce |
|
Oncorhynchus mykiss Trucha arco iris (9) (16) |
|
Danio rerio Pez cebra (7) (17) (18) |
|
Cyprinus caprio Carpa común (8) (19) |
|
Oryzias latipes Medaka (20) (21) |
|
Pimephales promelas Piscardo (8) (22) |
Cuadro 1B
Ejemplos de otras especies bien documentada que también han sido utilizadas
|
Agua dulce |
Agua salada |
|
Carassius auratus Carpa dorada (8) |
Menidia peninsulae Pejerrey del Atlántico Occidental (23) (24) (25) |
|
Lepomis macrochirus Pez sol de agallas azules (8) |
Clupea harengus Arenque (24) (25) |
|
Gadus morhua Bacalao (24) (25) |
|
|
Cyprinodon variegatus Pelota (23) (24) (25) |
Apéndice 1
DIRECTRICES SOBRE LA REALIZACIÓN DE UN ENSAYO DE TOXICIDAD EN EMBRIONES Y ALEVINES DE PEZ CEBRA (BRACHYIANIO RERIO)
INTRODUCCIÓN
El pez cebra es originario de la costa de Coromandel en la India, donde vive en ríos de corriente rápida. Es una especie común de acuario, que pertenece a la familia de las carpas. La información relativa a los métodos de cría y los cuidados que requieren figuran en los manuales de referencia sobre peces tropicales. Laale ha estudiado su biología y su empleo en la investigación relacionada con la pesca (1).
Este pez rara vez supera los 45 mm de longitud. Su cuerpo cilíndrico posee entre 7 y 9 rayas horizontales de color azul oscuro y plateado, que se prolongan hasta las aletas caudal y anal. El dorso es verde aceituna. Los machos son más estrechos que las hembras, y estas son más plateadas y presentan un abdomen más distendido, en particular antes del desove.
Los peces adultos soportan grandes variaciones de temperatura, pH y dureza. Sin embargo, para obtener peces sanos que produzcan huevos de buena calidad debe mantenérselos en condiciones óptimas.
Durante el desove, el macho persigue a la hembra y le da golpes con la cabeza. Los huevos son fecundados apenas expulsados. Los huevos, que son transparentes y no adherentes, caen al fondo, donde pueden ser ingeridos por los padres. La luz influye en el desove. Si la luz de la mañana es suficiente, los peces suelen desovar en las horas siguientes al amanecer.
Una hembra puede realizar desoves de varios centenares de huevos a intervalos de una semana.
ESTADO DE LOS PECES PARENTALES, REPRODUCCIÓN Y PRIMERAS FASES DE LA VIDA
Se selecciona un número adecuado de peces sanos y se mantienen en un agua apropiada (véase el apéndice 4, por ejemplo) al menos durante dos semanas antes de la fecha prevista para el desove. Los peces han de reproducirse al menos una vez antes de producir los huevos que vayan a emplearse en el ensayo. La densidad en este período no debe superar 1 gramo de peces por litro. La renovación regular del agua y la utilización de sistemas de purificación pueden dar lugar a densidades mayores. La temperatura en los acuarios debe mantenerse a 25 ± 2 °C y debe proporcionarse a los peces un régimen alimentario variado, que puede consistir, por ejemplo, en alimentos deshidratados adecuados que se encuentran en el comercio, Artemia vivos recién eclosionados, quironómidos, Dafnia o gusanos blancos (Enquitreidos).
A continuación se recogen dos métodos con los que se han obtenido en la práctica camadas suficientes de huevos fecundados sanos de cara a la realización de un ensayo:
|
i) |
se colocan 8 hembras y 16 machos en un acuario con 50 litros de agua de dilución y protegido de la luz directa. Debe evitarse en la mayor medida posible perturbarlos al menos durante 48 horas. En la tarde del día anterior al inicio del ensayo se coloca en el fondo del acuario un soporte para el desove, formado por un bastidor (de plexiglás u otro material adecuado) de 5 a 7 cm de alto, con una red de malla gruesa (2-5 mm) en la parte superior y una red de malla fina (10-30 μm) en la parte inferior. Se atan a la malla gruesa del soporte varios «árboles de desove», consistentes en una cuerda de nailon sin retorcer. Se deja a los peces 12 horas en la oscuridad y, a continuación, se enciende una luz tenue para que empiece el desove. Entre 2 y 4 horas después de este, se retira el soporte y se recogen los huevos. El soporte impide que los peces ingieran los huevos, al tiempo que facilita la recogida de estos. Los peces deben haber desovado al menos una vez antes de recoger los huevos que vayan a emplearse en el ensayo; |
|
ii) |
se mantienen de 5 a 10 peces machos y hembras en acuarios individuales durante un mínimo de 2 semanas antes de la fecha prevista para el desove. Entre 5 y 10 días después, el abdomen de las hembras se distiende y sus papilas genitales se hacen visibles. Los machos no tienen papilas. El desove se lleva a cabo en recipientes provistos de un falso fondo de malla (véase más arriba). El recipiente se llena con agua de dilución hasta una altura de 5 a 10 cm por encima de la malla. Se colocan una hembra y dos machos en el recipiente la víspera de la fecha prevista para el desove. Se aumenta progresivamente la temperatura del agua hasta un grado por encima de la temperatura de aclimatación. Se apaga la luz y se evita en la mayor medida posible toda perturbación de los peces. Por la mañana, se enciende una luz tenue para que empiece el desove. Entre 2 y 4 horas más tarde, se retiran los peces y se recogen los huevos. Si se necesitan más huevos de los que puede poner una hembra, pueden disponerse en paralelo los acuarios que sean necesarios. Si se registra la capacidad de reproducción de cada hembra antes del ensayo (volumen de la camada y calidad de los huevos), podrán seleccionarse para este las que tengan mayor capacidad reproductora. |
Los huevos se trasladan a los recipientes de ensayo en tubos de vidrio, de diámetro interno superior o igual a 4 mm y provistos de una pera de succión. El trasvase de huevos debe realizarse con la menor cantidad de agua posible. Los huevos pesan más que el agua y caen fuera del tubo. Debe evitarse que los huevos (y las larvas) estén en contacto con el aire. Se practica un examen microscópico de una o varias muestras del lote o lotes para cerciorarse de que no hay anomalías en las primeras fases del desarrollo. No está permitido desinfectar los huevos.
La tasa de mortalidad de los huevos más elevada se da durante las 24 horas siguientes a la fecundación. A menudo se observa durante ese período una tasa de mortalidad del 5 al 40 %. La degeneración de los huevos se debe a una fertilización fallida o a problemas en el desarrollo. Al parecer, la calidad de los huevos depende de la hembra, ya que algunas producen siempre huevos de buena calidad, mientras que otras no lo consiguen nunca. Además, el ritmo de desarrollo y de eclosión varía de una camada a otra. La tasa de supervivencia de los huevos correctamente fecundados y de las larvas suele ser elevada, por lo general superior al 90 %. A 25 oC, los huevos eclosionan entre 3 y 5 días después de la fecundación y la vesícula vitelina se reabsorbe unos 13 días después de esta.
Hisaoka y Battle han estudiado a fondo el desarrollo embrionario (2). La transparencia de los huevos y de las larvas eclosionadas permite seguir el desarrollo de los peces y detectar la presencia de malformaciones. Unas 4 horas después del desove ya pueden distinguirse los huevos fecundados de los no fecundados (3). Para ello, se colocan los huevos y las larvas en recipientes de ensayo pequeños y se estudian al microscopio.
Las condiciones de ensayo aplicables a las primeras fases de la vida figuran en el apéndice 2. Los valores óptimos de pH y dureza del agua de dilución son de 7,8 y de 250 mg CaCO3/l, respectivamente.
CÁLCULOS Y ESTADÍSTICAS
Se propone un procedimiento en dos etapas. En primer lugar, se hace un análisis estadístico de los datos relativos a la mortalidad, las anomalías en el desarrollo y el momento de la eclosión. A continuación, se hace una evaluación estadística de la longitud corporal de los peces con las concentraciones a las que no se haya observado ningún efecto adverso sobre esos tres parámetros. Se recomienda proceder así porque las sustancias tóxicas pueden matar selectivamente a los peces más pequeños, retrasar el momento de la eclosión e inducir malformaciones macroscópicas, lo cual puede sesgar las mediciones de longitud. Además de ello, la cantidad de peces por medir con cada tratamiento será más o menos la misma, lo cual garantiza la validez del análisis estadístico del ensayo.
DETERMINACIÓN DE LA CL50 Y LA EC50
Se calcula el porcentaje de huevos y larvas supervivientes y se corrige en función de la mortalidad en los controles mediante la fórmula de Abbott (4):

donde:
|
P |
= |
porcentaje de supervivientes corregido, |
|
P' |
= |
porcentaje de supervivientes observado en la concentración de ensayo, |
|
C |
= |
porcentaje de supervivientes en los controles. |
Si es posible, la Cl 50 se determina por un método apropiado al final del ensayo.
Si se desea incluir las anomalías morfológicas en el tratamiento estadístico de la EC50 pueden encontrarse indicaciones al respecto en el artículo de Stephan (5).
ESTIMACIÓN DE LA LOEC Y LA NOEC
Uno de los objetivos del ensayo con huevos y alevines es comparar los lotes sometidos a concentraciones positivas con los controles para determinar la LOEC, para lo cual habrá que emplear métodos de comparaciones múltiples (6) (7) (8) (9) (10).
REFERENCIAS
|
(l) |
Laale H.W. (1977) The Biology and Use of the Zebrafish (Brachyianio rerio) in Fisheries Research. A Literature Review. J. Fish Biol. 10, 121-173. |
|
(2) |
Hisaoka K.K. y Battle H.L. (1958). The Normal Development Stages of the Zebrafish Brazhydanio rerio (Hamilton-Buchanan) I. Morph., 102, pp. 311. |
|
(3) |
Nagel R. (1986). Untersuchungen zur Eiproduktion beim Zebrabärbling (Brachydanio rerio Hamilton-Buchanan). Journal of Applied Ichthyology. 2. pp. 173-181. |
|
(4) |
Finney D.J. (1971). Probit Analysis, 3a ed., Cambridge University Press, Gran Bretaña, pp. 1-333. |
|
(5) |
Stephan CE. (1982). Increasing the Usefulness of Acute Toxicity Tests. Aquatic Toxicology and Hazard Assessment: Fifth Conference, ASTM STP 766, J.G. Pearson, R.B. Foster y W.E. Bishop, Eds., American Society for Testing and Materials, pp. 69-81. |
|
(6) |
Dunnett C.W. (1955). A Multiple Comparisons Procedure for Comparing Several Treatments with a Control. J. Amer. Statist. Assoc, 50, pp 1096-1121. |
|
(7) |
Dunnett C.W. (1964) New Tables for Multiple Comparisons with a Control. Biometrics, 20, pp. 482-491. |
|
(8) |
Williams D.A. (1971). A Test for Differences Between Treatment Means when Several Dose Levels are Compared with a Zero Dose Control. Biometrics, 27, pp. 103-117. |
|
(9) |
Williams D.A. (1972). The Comparison of Several Dose Levels with a Zero Dose Control. Biometrics 28, pp. 519-531. |
|
(10) |
Sokal R.R. y Rohlf F.J. (1981). Biometry, the Principies and Practice of Statistics in Biological Research, W.H. Freeman and Co., San Francisco. |
Apéndice 2
CONDICIONES Y DURACIÓN DEL ENSAYO Y CRITERIOS DE SUPERVIVENCIA PARA LAS ESPECIES RECOMENDADAS
|
Especie |
Temperaturea ( oC) |
Salinidad (0/00) |
Fotoperíodo (horas) |
Duración de las fases (días) |
Duración habitual del ensayo |
Supervivencia en los controles ( % mínimo) |
||
|
Embrión |
Alevín |
Tasa de eclosiones satisfactorias |
Tras la eclosión |
|||||
|
Agua dulce |
||||||||
|
Brachydanio rerio Pez cebra |
25 ±1 |
— |
12-16 |
3-5 |
8-10 |
Desde lo antes posible tras la fecundación (fase precoz de gástrula) hasta 5 días después de la eclosión (8-10 días) |
80 |
90 |
|
Onicorhynchus mykiss Trucha arco iris |
10 ± 1 (20) 12 ± 1 (21) |
— |
0 (22) |
30-35 |
25-30 |
Desde lo antes posible tras la fecundación (fase precoz de gástrula) hasta 20 días después de la eclosión (50-55 días) |
66 |
70 |
|
Cyprinus carpio Carpa común |
21-25 |
— |
12-16 |
5 |
> 4 |
Desde lo antes posible tras la fecundación (fase precoz de gástrula) hasta 4 días después de la eclosión (8-9 días) |
80 |
75 |
|
Oryzias latipes Medaka |
24 ± 1 (20) 23 ± 1 (21) |
— |
12-16 |
8-11 |
4-8 |
Desde lo antes posible tras la fecundación (fase precoz de gástrula) hasta 5 días después de la eclosión (13-16 días) |
80 |
80 |
|
Pimephales promelas Piscardo |
25 ± 2 |
— |
16 |
4-5 |
5 |
Desde lo antes posible tras la fecundación (fase precoz de gástrula) hasta 4 días después de la eclosión (8-9 días) |
60 |
70 |
Apéndice 3
Condiciones y duración del ensayo y criterios de supervivencia para otras especies bien documentadas
|
Especie |
Temperatura ( oC) |
Salinidat(0/00) |
Fotoperíodo(horas) |
Duración de las fases (días) |
Duración habitual del ensayo con embriones y alevines |
Supervivencia en los controles (% minimo) |
||
|
|
|
|
|
Embriones |
Alevín |
Tasa de eclosiones satisfactorias |
Tras la eclosión |
|
|
AGUA DULCE |
||||||||
|
Carassius auratus Carpa dorada |
24 ± 1 |
— |
— |
3 — 4 |
> 4 |
Desde lo antes posible tras la fecundación (fase precoz de gástrula) hasta 4 días después de la eclosión (7 días) |
— |
80 |
|
Leopomis macrochirus Pez sol do agallas azules |
21 ± 1 |
— |
16 |
3 |
> 4 |
Desde lo antes posible tras la fecundación (fase precoz de gástrula) hasta 4 días después de la eclosión (7 días) |
— |
75 |
|
AGUA SALADA |
||||||||
|
Menidia peninsulae Pejerrey del Atlántico Occidental |
22-25 |
15-22 |
12 |
1,5 |
10 |
Desde lo antes posible tras la fecundación (fase precoz de gástrula) hasta 5 días después de la eclosión (6-7 días) |
80 |
60 |
|
Clupea harengus Arenque |
10 ± 1 |
8-15 |
12 |
20-25 |
3-5 |
Desde lo antes posible tras la fecundación (fase precoz de gástrula) hasta 3 días después de la eclosión (23-27 días) |
60 |
80 |
|
Gadus morhua Bacalao |
5 ± 1 |
5-30 |
12 |
14-16 |
3-5 |
Desde lo antes posible tras la fecundación (fase precoz de gástrula) hasta 3 días después de la eclosión (18 días) |
60 |
80 |
|
Cyprinodon variegatus Petota |
25 ± 1 |
15-30 |
12 |
— |
— |
Desde lo antes posible tras la fecundación (fase precoz de gástrula) hasta 4-7 días después de la eclosión (28 días) |
> 75 |
80 |
APÉNDICE 4
ALGUNAS CARACTERÍSTICAS QUÍMICAS DE UN AGUA DE DILUCIÓN ACEPTABLE
|
Sustancia |
Concentraciones |
|
Materia en partículas |
< 20 mg/l |
|
Carbono orgánico total |
< 2 mg/l |
|
Amoniaco no ionizado |
< 1 μg/l |
|
Cloro residual |
< 10 μg/l |
|
Plaguicidas organofosforados totales |
< 50 ng/l |
|
Plaguicidas organoclorados totales y bifenilos policlorados |
< 50 ng/l |
|
Cloro orgánico total |
< 25 ng/l |
C.16. ENSAYO DE TOXICIDAD ORAL AGUDA EN ABEJAS
1. MÉTODO
El presente ensayo de toxicidad aguda reproduce las directrices del documento OCDE TG 213 (1998).
1.1. INTRODUCCIÓN
El presente ensayo de toxicidad consiste en un método de laboratorio diseñado para determinar la toxicidad oral aguda de los productos fitosanitanos y otros productos químicos en abejas obreras adultas.
Para determinar y evaluar las características tóxicas de una sustancia puede ser preciso estudiar la toxicidad oral aguda en abejas, por ejemplo, cuando es probable que estas queden expuestas a un producto químico determinado. El ensayo de toxicidad oral aguda se realiza para determinar la toxicidad inherente de los plaguicidas y otras sustancias para las abejas. Sus resultados se emplean para saber si es necesario profundizar en la evaluación. En particular, el método puede emplearse en programas de evaluación del peligro de los plaguicidas para las abejas, basados en una progresión secuencial desde los ensayos de toxicidad en laboratorio hasta los experimentos de semicampo y de campo (1). Los plaguicidas pueden someterse a ensayo en forma de principios activos (p.a.) o de productos formulados.
Debe emplearse un tóxico de referencia para comprobar la sensibilidad de las abejas y la precisión del procedimiento de ensayo.
1.2. DEFINICIONES
Toxicidad oral aguda: efectos nocivos que se manifiestan durante un período máximo de 96 horas tras la administración oral de una dosis única de la sustancia de ensayo.
Dosis: cantidad de sustancia de ensayo consumida. Se expresa en peso de la sustancia por animal de ensayo (μg/abeja). Dado que la alimentación es colectiva, no puede calcularse la dosis real por abeja, pero puede hacerse una estimación de la dosis media (sustancia de ensayo consumida en su totalidad/número de abejas en una jaula).
DL 50 (dosis letal media) oral: dosis única, obtenida por estadística, de una sustancia que puede provocar la muerte del 50 % de los animales a los que se haya administrado por vía oral. La DL50 se expresa en μg de sustancia de ensayo por abeja. En el caso de los plaguicidas, la sustancia de ensayo puede ser un principio activo (p.a.) o un producto formulado que contenga uno o varios principios activos.
Mortalidad: número de animales muertos, es decir, completamente inmóviles.
1.3. PRINCIPIO DEL MÉTODO DE ENSAYO
Se expone a las abejas obreras adultas (Apis mellifera) a una gama de dosis de la sustancia de ensayo dispersada en una solución de sacarosa. A continuación, se alimenta a las abejas de la misma forma, pero sin sustancia de ensayo. Se registra la mortalidad todos los días, al menos durante 48 horas, y se compara con las cifras del control. Si la tasa de mortalidad aumenta entre las 24 y las 48 horas y la mortalidad del control permanece a un nivel aceptable, es decir, ≤ 10 %, está indicado prolongar el ensayo hasta un máximo de 96 horas. Se analizan los resultados para calcular la DL50 a las 24 y a las 48 horas y, en caso de que se haya prolongado el estudio, a las 72 y a las 96 horas.
1.4. VALIDEZ DEL ENSAYO
Para que el ensayo sea válido han de darse las condiciones siguientes:
|
— |
la mortalidad media del total de los controles no ha de superar el 10 % al final del ensayo, |
|
— |
la DL50 del tóxico de referencia debe ajustarse a la gama especificada. |
1.5. DESCRIPCIÓN DEL MÉTODO
1.5.1. Recolección de las abejas
Deben emplearse ejemplares adultos jóvenes de abejas obreras de la misma raza, edad, alimentación, etc. Las abejas han de proceder de colonias sanas y bien alimentadas, a ser posible sin enfermedades y con reina, y con antecedentes y estado fisiológico conocidos. Pueden recolectarse la mañana del día en que vaya a realizarse el ensayo, o la víspera por la tarde y mantenerse en condiciones de ensayo hasta el día siguiente. Pueden emplearse abejas procedentes de cuadros sin cría, pero debe evitarse la recolección a principios de la primavera o a finales del otoño, pues en esas épocas las abejas modifican su fisiología. Si es preciso realizar el ensayo en esas épocas, las abejas pueden colocarse en una incubadora y criarse durante una semana con pan de abejas (polen tomado del panal) y solución de sacarosa. Si se utilizan abejas tratadas con productos químicos como antibióticos, anti-varroa, etc., antes de iniciar el ensayo de toxicidad habrá que esperar 4 semanas desde el final del último tratamiento.
1.5.2. Alojamiento y alimentación
Se emplean jaulas fáciles de limpiar, bien ventiladas, de cualquier material apropiado, como acero inoxidable, tela metálica, plástico, madera desechable, etc., y de tamaño apropiado, es decir, suficientemente espaciosas para el número de abejas. Es preferible colocar grupos de 10 abejas en cada jaula.
Las abejas se mantienen en la oscuridad en un cuarto de experimentación a 25 ± 2 oC. Debe registrarse a lo largo del ensayo la humedad relativa, que será por lo general del 50-70 %. Tanto el manejo como el tratamiento y las observaciones pueden realizarse con luz (del día). Se alimenta a las abejas con solución acuosa de sacarosa con una concentración final de 500 g/l (50 % w/v). Tras las dosis de ensayo, se las alimenta ad libi-tum. El sistema de alimentación debe permitir registrar la ingesta de cada jaula (véase el punto 1.6.3.1). Puede emplearse un tubo de vidrio de unos 50 mm de largo por 10 mm de ancho, con un extremo abierto de unos 2 mm de diámetro.
1.5.3. Preparación de las abejas
Se asignan las abejas de forma aleatoria a las jaulas, que se colocan, también de forma aleatoria, en el cuarto de experimentación.
Conviene suprimir la alimentación de las abejas 2 horas antes de iniciar el ensayo. Se recomienda esta práctica con el fin de que el contenido intestinal de todos los animales sea el mismo cuando comience el ensayo. Deben retirarse las abejas moribundas y sustituirse por otras sanas antes de empezar el ensayo.
1.5.4. Preparación de las dosis
Si la sustancia de ensayo puede mezclarse con agua, se añade directamente a una solución de sacarosa al 50 %. Si se trata de productos de calidad técnica o de sustancias escasamente solubles en agua, pueden emplearse disolventes orgánicos, emulgentes o dispersantes poco tóxicos para las abejas (por ejemplo, acetona, dimetilformamida o dimetilsulfóxido). La concentración del excipiente dependerá de la solubilidad de la sustancia de ensayo y ha de ser la misma para todas las concentraciones que se empleen en el ensayo. Por lo general, resulta adecuada una concentración de excipiente del 1 %, que no debe sobrepasarse.
Se preparan soluciones de control adecuadas, lo cual significa que, cuando se utilice un disolvente o dispersante para solubilizar la sustancia de ensayo, han de emplearse dos grupos de control distintos: una solución acuosa y una solución de sacarosa con el disolvente o excipiente a la concentración empleada en las dosis de ensayo.
1.6. PROCEDIMIENTO
1.6.1. Grupos de ensayo y controles
El número de dosis y pruebas en paralelo ha de ser suficiente desde el punto de vista estadístico para poder determinar la DL50 con un límite de confianza del 95 %. Suele ser necesaria una serie geométrica de 5 dosis con un factor que no sobrepase el 2,2 y que incluya la gama de la DL50. No obstante, el factor de dilución y el número de concentraciones han de determinarse con arreglo a la pendiente de la curva de toxicidad (dosis/mortalidad) y al método estadístico que se haya elegido para analizar los resultados. Las concentraciones apropiadas para el tratamiento pueden establecerse tras un experimento de determinación de gamas.
Con cada concentración de ensayo deben tratarse al menos 3 grupos en paralelo, de 10 abejas cada uno. Además de la serie de ensayo, deben efectuarse al menos 3 lotes de control, de 10 abejas cada uno. También deben efectuarse lotes de control para los disolventes/excipientes que se utilicen (véase el punto 1.5.4).
1.6.2. Tóxico de referencia
Debe incluirse un tóxico de referencia en la serie de ensayo. Se utilizarán al menos 3 dosis que abarquen la DL50, prevista. Para cada dosis de ensayo se emplearán al menos 3 jaulas en paralelo, con 10 abejas cada una. El tóxico de referencia idóneo es el dimetoato, cuya DL50-24 horas tras administración oral es del orden de 0,10-0,35 μg p.a./abeja (2). También pueden emplearse otros tóxicos si se dispone de datos suficientes para comprobar que la relación dosis/respuesta es la esperada (por ejemplo, paratión).
1.6.3. Exposición
1.6.3.1. Administración de las dosis
Debe administrarse a cada grupo de abejas 100-200 μl de solución acuosa de sacarosa al 50 % con la sustancia de ensayo a la concentración adecuada. Si la sustancia es poco soluble, poco tóxica o su concentración en la formulación es escasa, ha de administrarse un volumen mayor, pues se necesita una mayor cantidad de sustancia en la solución de sacarosa. Se controla la cantidad de alimento tratado que consume cada grupo. Una vez consumido el alimento (por lo general tras 3-4 horas), se retira el comedero de la jaula y se sustituye por otro con solución de sacarosa únicamente, que se proporciona ad libitum. El rechazo de la dosis de ensayo de algunos productos a concentraciones superiores puede dar lugar a un consumo escaso o nulo de alimento. Tras un máximo de 6 horas, el alimento tratado que no haya sido consumido se sustituye por solución de sacarosa sola. Se calcula el consumo de alimento tratado (por ejemplo, midiendo el volumen/peso del alimento tratado que haya sobrado).
1.6.3.2. Duración
Conviene que el ensayo dure hasta 48 horas después de la sustitución de la solución de ensayo por la de sacarosa sola. Si la mortalidad sigue aumentando en más del 10 % tras las primeras 24 horas, debe prolongarse el ensayo hasta un máximo de 96 horas, siempre y cuando la mortalidad del control no supere el 10 %.
1.6.4. Observaciones
Se registra la mortalidad a las 4, las 24 y las 48 horas del inicio del ensayo (es decir, de la administración de la dosis). Si es necesario prolongar el período de observación, se harán registros cada 24 horas, hasta un máximo de 96 horas, siempre y cuando la mortalidad del control no supere el 10 %.
Se calcula la cantidad de alimento consumido en cada grupo. Para saber si el alimento tratado es apetecible puede compararse el consumo de este y el de alimento sin tratar, durante un período de 6 horas.
Debe registrarse todo comportamiento anormal observado durante el período de ensayo.
1.6.5. Ensayo límite
En algunos casos (por ejemplo, cuando se piense someter a ensayo una sustancia escasamente tóxica), puede realizarse un ensayo límite con 100 μg p.a./abeja para demostrar que la DL50 es superior a ese valor. Debe seguirse el mismo procedimiento, incluidos los 3 grupos de ensayo en paralelo, con la dosis de ensayo, los controles pertinentes, el tóxico de referencia y para el cálculo de la cantidad de alimento tratado que se ha consumido. Si mueren abejas debe realizarse un estudio completo. Deben registrarse los efectos subletales, en caso de producirse (véase el punto 1.6.4).
2. RESULTADOS E INFORME
2.1. RESULTADOS
Se resumen los resultados en un cuadro que recoja, para cada grupo tratado, grupo de control y grupo con el tóxico de referencia, el número de abejas empleadas, la mortalidad en cada una de las observaciones y el número de abejas que presenten alteraciones del comportamiento. Se analizan los datos relativos a la mortalidad con métodos estadísticos apropiados (por ejemplo, análisis por probitas, media móvil, probabilidad bino-mial) (3) (4). Se trazan las curvas de dosis-respuesta correspondientes a todas las observaciones recomendadas y se calculan las pendientes de las mismas y las dosis letales medias (DL50) con un límite de confianza del 95 %. La mortalidad de los controles puede corregirse mediante la corrección de Abbott (4) (5). Si el alimento tratado no se ha consumido en su totalidad, se calcula la cantidad de sustancia de ensayo consumida en cada grupo. La DL50 se expresa en μg de sustancia de ensayo por abeja.
2.2. INFORME DEL ENSAYO
El informe del ensayo debe incluir la información siguiente:
2.2.1. Sustancia de ensayo:
|
— |
naturaleza física y propiedades fisicoquímicas pertinentes (estabilidad en el agua, presión de vapor, etc.), |
|
— |
identificación química: fórmula estructural, pureza, etc. (en el caso de los plaguicidas, identidad y concentración del principio o principios activos). |
2.2.2. Especie sometida a ensayo:
|
— |
denominación científica, raza, edad aproximada (en semanas), método y fecha de recolección, |
|
— |
información sobre las colonias de donde proceden las abejas de ensayo: estado de salud, enfermedades de los sujetos adultos, tratamientos previos, etc. |
2.2.3. Condiciones de ensayo:
|
— |
temperatura y humedad relativa en el cuarto de experimentación, |
|
— |
condiciones de alojamiento, incluido el tipo, tamaño y material de las jaulas, |
|
— |
método de preparación de la solución madre y de ensayo (en su caso, el disolvente y su concentración), |
|
— |
diseño del ensayo, por ejemplo, número de concentraciones y concentraciones de ensayo, número de controles, etc., |
|
— |
para cada concentración de ensayo y control, número de jaulas utilizadas en paralelo y número de abejas por jaula, fecha del ensayo. |
2.2.4. Resultados:
|
— |
resultados del estudio previo de determinación de gamas, si procede, |
|
— |
datos en bruto: mortalidad con cada dosis de ensayo en cada período de observación, |
|
— |
gráfico de las curvas de dosis-respuesta al final del ensayo, |
|
— |
valores de la DL50 con un límite de confianza del 95 % en cada período de observación recomendado, con la sustancia de ensayo y el tóxico de referencia, |
|
— |
métodos estadísticos empleados para calcular la DL50, |
|
— |
mortalidad en los controles, |
|
— |
otros efectos biológicos observados o medidos en las abejas, como alteraciones del comportamiento (incluido el rechazo de la sustancia de ensayo), tasa de consumo del alimento tratado y sin tratar, etc., |
|
— |
toda desviación de los protocolos de ensayo aquí descritos y cualquier otra información de interés. |
3. REFERENCIAS
|
(1) |
EPPO/Council of Europe (1993). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Products — Honeybees. EPPO bulletin, vol. 23, N.1, 151-165. Marzo de 1993. |
|
(2) |
Gough, H. J., McIndoe, E.C., Lewis, G.B. (1994). The use of dimethoate as a reference compound in laboratory acute toxicity tests on honeybees (Apis mellifera L.) 1981-1992. Journal of Apicultural Research, 22, 119-125. |
|
(3) |
Litchfield, J.T. y Wilcoxon, F. (1949). A simplified method of evaluating dose-effect experiments. Jour, Pharmacol. and Exper. Ther., 96, 99-113. |
|
(4) |
Finney, D.J. (1971). Probit Analysis. 3a ed., Cambridge, Londres y Nueva York. |
|
(5) |
Abbott, W.S. (1925). A method for computing the effectiveness of an insecticide. Jour. Econ. Entomol., 18, 265-267. |
C.17. ENSAYO DE TOXICIDAD AGUDA POR CONTACTO EN ABEJAS
1. MÉTODO
El presente ensayo de toxicidad aguda reproduce las directrices del documento OCDE TG 214 (1998).
1.1. INTRODUCCIÓN
El presente ensayo de toxicidad consiste en un método de laboratorio diseñado para determinar la toxicidad aguda por contacto de los productos fitosanitarios y otros productos químicos en abejas obreras adultas.
Para determinar y evaluar las características tóxicas de una sustancia puede ser preciso estudiar la toxicidad aguda por contacto en abejas, por ejemplo, cuando es probable que estas queden expuestas a un producto químico determinado. El ensayo de toxicidad aguda por contacto se realiza para determinar la toxicidad inherente de los plaguicidas y otras sustancias para las abejas. Sus resultados se emplean para saber si es necesario profundizar en la evaluación. En particular, el método puede emplearse en programas de evaluación del peligro de los plaguicidas para las abejas, basados en una progresión secuencial desde los ensayos de toxicidad en laboratorio hasta los experimentos de semicampo y de campo (1). Los plaguicidas pueden someterse a ensayo en forma de principios activos (p.a.) o de productos formulados.
Debe emplearse un tóxico de referencia para comprobar la sensibilidad de las abejas y la precisión del procedimiento de ensayo.
1.2. DEFINICIONES
Toxicidad aguda por contacto: efectos nocivos que se manifiestan durante un período máximo de 96 horas tras la aplicación tópica de una dosis única de una sustancia.
Dosis: cantidad de sustancia de ensayo aplicada. Se expresa en peso de la sustancia por animal de ensayo (μg/abeja).
DL50 (dosis letal media) por contacto: dosis única, obtenida por estadística, de una sustancia que puede provocar la muerte del 50 % de los animales a los que se haya aplicado. La DL50 se expresa en μg de sustancia de ensayo por abeja. En el caso de los plaguicidas, la sustancia de ensayo puede ser un principio activo (p.a.) o un producto formulado que contenga uno o varios principios activos.
Mortalidad: número de animales muertos, es decir, completamente inmóviles.
1.3. PRINCIPIO DEL MÉTODO DE ENSAYO
Se expone a las abejas obreras adultas (Apis mellifera), por aplicación directa en el tórax (gotitas), a una gama de dosis de la sustancia de ensayo disuelta en un excipiente adecuado. El ensayo dura 48 horas. Si la tasa de mortalidad aumenta entre las 24 y las 48 horas y la mortalidad del control permanece a un nivel aceptable, es decir, <10 %, está indicado prolongar el ensayo hasta un máximo de 96 horas. Se registra la mortalidad todos los días y se compara con las cifras del control. Se analizan los resultados para calcular la DL50 a las 24 y a las 48 horas y, en caso de que se haya prolongado el estudio, a las 72 y a las 96 horas.
1.4. VALIDEZ DEL ENSAYO
Para que el ensayo sea válido han de darse las condiciones siguientes:
|
— |
la mortalidad media del total de los controles no ha de superar el 10 % al final del ensayo, |
|
— |
la DL50 del tóxico de referencia debe ajustarse a la gama especificada. |
1.5. DESCRIPCIÓN DEL MÉTODO
1.5.1. Recolección de las abejas
Deben emplearse ejemplares adultos jóvenes de abejas obreras de la misma raza, edad, alimentación, etc. Las abejas han de proceder de colonias sanas y bien alimentadas, a ser posible sin enfermedades y con reina, y con antecedentes y estado fisiológico conocidos. Pueden recolectarse la mañana del día en que vaya a reali-zarse el ensayo, o la víspera por la tarde y mantenerse en condiciones de ensayo hasta el día siguiente. Pueden emplearse abejas procedentes de cuadros sin cría, pero debe evitarse la recolección a principios de la primavera o finales del otoño, pues en esas épocas las abejas modifican su fisiología. Si es preciso realizar el ensayo en esas épocas, las abejas pueden colocarse en una incubadora y criarse durante una semana con pan de abejas (polen tomado del panal) y solución de sacarosa. Si se utilizan abejas tratadas con productos químicos como antibióticos, anti-varroa, etc., antes de iniciar el ensayo de toxicidad habrá que esperar 4 semanas desde el final del último tratamiento.
1.5.2. Alojamiento y alimentación
Se emplean jaulas fáciles de limpiar, bien ventiladas, de cualquier material apropiado, como acero inoxidable, tela metálica, plástico, madera desechable, etc., y de tamaño apropiado, es decir, suficientemente espaciosas para el número de abejas. Es preferible colocar grupos de 10 abejas en cada jaula.
Las abejas se mantienen en la oscuridad en un cuarto de experimentación a 25 ± 2 oC Debe registrarse a lo largo del ensayo la humedad relativa, que será por lo general del 50-70 %. Tanto el manejo como el tratamiento y las observaciones pueden realizarse con luz (del día). Se alimenta a las abejas con solución acuosa de sacarosa con una concentración final de 500 g/l (50 % w/v). Esta se proporciona ad libitum mientras dure el ensayo en un comedero de abejas, que puede ser un tubo de vidrio de unos 50 mm de largo por 10 mm de ancho, con un extremo abierto de unos 2 mm de diámetro.
1.5.3. Preparación de las abejas
Antes de aplicar la sustancia de ensayo, pueden anestesiarse las abejas con anhídrido carbónico o nitrógeno. Debe reducirse al mínimo la cantidad de anestésico aplicado y el tiempo de exposición. Se retiran las abejas moribundas y se sustituyen por otras sanas antes de empezar el ensayo.
1.5.4. Preparación de las dosis
La sustancia de ensayo se diluye en un excipiente, que puede ser un disolvente orgánico o una solución acuosa con un humectante. La acetona es el disolvente orgánico más conveniente, si bien pueden emplearse otros escasamente tóxicos para las abejas (por ejemplo, dimetilformamida o dimetilsulfóxido). Si se trata de formulados dispersados en agua o de sustancias orgánicas de gran polaridad insolubles en disolventes orgánicos, puede resultar más fácil aplicarlos en una solución débil de un humectante de una marca comercial (Agral, Cittowett, Lubrol, Triton, Tween, etc.).
Se preparan soluciones de control adecuadas, lo cual significa que, cuando se utilice un disolvente o dispersante para solubilizar la sustancia de ensayo, han de emplearse dos grupos de control distintos: uno con agua y otro con disolvente/dispersante.
1.6. PROCEDIMIENTO
1.6.1. Grupos de ensayo y controles
El número de dosis y pruebas en paralelo ha de ser suficiente desde el punto de vista estadístico para poder determinar la DL50 con un límite de confianza del 95 %. Suele ser necesaria una serie geométrica de 5 dosis con un factor que no sobrepase el 2,2 y que incluya la gama de la DL50. No obstante, el número de concentraciones ha de determinarse con arreglo a la pendiente de la curva de toxicidad (dosis/mortalidad) y al método estadístico que se haya elegido para analizar los resultados. Las concentraciones apropiadas para el tratamiento pueden establecerse tras un experimento de determinación de gamas.
Con cada concentración de ensayo deben tratarse al menos 3 grupos en paralelo, de 10 abejas cada uno.
Además de la serie de ensayo, deben efectuarse al menos 3 lotes de control, de 10 abejas cada uno. En su caso, deben hacerse 3 lotes de control adicionales, de 10 abejas cada uno, con el disolvente orgánico o humectante.
1.6.2. Tóxico de referencia
Debe incluirse un tóxico de referencia en la serie de ensayo. Se utilizarán al menos 3 dosis que abarquen la DL50 prevista. Para cada dosis de ensayo se emplearán al menos 3 jaulas en paralelo, con 10 abejas cada una. El tóxico de referencia idóneo es el dimetoato, cuya DL50-24 horas por contacto es del orden de 0,10-0,35μg p.a./abeja (2). También pueden emplearse otros tóxicos si se dispone de datos suficientes para comprobar que la relación dosis/respuesta es la esperada (por ejemplo, paratión).
1.6.3. Exposición
1.6.3.1. Administración de las dosis
Se hace una aplicación local a cada una de las abejas anestesiadas. Las abejas se asignan de forma aleatoria para las distintas dosis de ensayo y los controles. Con un microaplicador, se aplica a cada abeja en la cara dorsal del tórax 1 μl de solución con sustancia de ensayo a la concentración adecuada. Si procede, pueden aplicarse otros volúmenes. Después de la aplicación, se colocan las abejas en las jaulas de ensayo y se les proporciona solución de sacarosa.
1.6.3.2. Duración
Conviene que el ensayo dure 48 horas. Si la mortalidad sigue aumentando en más del 10 % entre las 24 y las 48 horas, debe prolongarse el ensayo hasta un máximo de 96 horas, siempre y cuando la mortalidad del control no supere el 10 %.
1.6.4. Observaciones
Se registra la mortalidad a las 4, las 24 y las 48 horas de la administración de la dosis. Si es necesario prolongar el período de observación, se harán registros cada 24 horas, hasta un máximo de 96 horas, siempre y cuando la mortalidad del control no supere el 10 %.
Debe registrarse todo comportamiento anormal observado durante el período de ensayo.
1.6.5. Ensayo límite
En algunos casos (por ejemplo, cuando se piense someter a ensayo una sustancia escasamente tóxica), puede realizarse un ensayo límite con 100 μg p.a./abeja para demostrar que la DL50 es superior a ese valor. Debe seguirse el mismo procedimiento, incluidos los 3 grupos de ensayo en paralelo, con la dosis de ensayo, los controles pertinentes y el tóxico de referencia. Si mueren abejas debe realizarse un estudio completo. Deben registrarse los efectos subletales, en caso de producirse (véase el punto 1.6.4).
2. RESULTADOS E INFORME
2.1. RESULTADOS
Se resumen los resultados en un cuadro que recoja, para cada grupo tratado, grupo de control y grupo con el tóxico de referencia, el número de abejas empleadas, la mortalidad en cada una de las observaciones y el número de abejas que presenten alteraciones del comportamiento. Se analizan los datos relativos a la mortalidad con métodos estadísticos apropiados (por ejemplo, análisis por probitas, media móvil, probabilidad bino-mial) (3) (4). Se trazan las curvas de dosis-respuesta correspondientes a todas las observaciones recomendadas (a las 24, 48 y, en su caso, 72 horas y 96 horas) y se calculan las pendientes de las mismas y las dosis letales medias (DL50) con un límite de confianza del 95 %. La mortalidad de los controles puede corregirse mediante la corrección de Abbott (4) (5). La DL50, se expresa en μg de sustancia de ensayo por abeja.
2.2. INFORME DEL ENSAYO
El informe del ensayo debe incluir la información siguiente:
2.2.1. Sustancia de ensayo:
|
— |
naturaleza física y propiedades fisicoquímicas (estabilidad en el agua, presión de vapor, etc.), |
|
— |
identificación química: fórmula estructural, pureza, etc. (en el caso de los plaguicidas, identidad y concentración del principio o principios activos). |
2.2.2. Especie sometida a ensayo:
|
— |
denominación científica, raza, edad aproximada (en semanas), método y fecha de recolección, |
|
— |
información sobre las colonias de donde proceden las abejas de ensayo: estado de salud, enfermedades de los sujetos adultos, tratamientos previos, etc. |
2.2.3. Condiciones de ensayo:
|
— |
temperatura y humedad relativa en el cuarto de experimentación, |
|
— |
condiciones de alojamiento, incluido el tipo, tamaño y material de las jaulas, |
|
— |
método de administración de la sustancia de ensayo: disolvente, volumen de solución de ensayo, anestésicos empleados, etc., |
|
— |
diseño del ensayo, por ejemplo, número de concentraciones y concentraciones de ensayo, número de controles, etc.; para cada concentración de ensayo y control, número de jaulas utilizadas en paralelo y número de abejas por jaula, |
|
— |
fecha del ensayo. |
2.2.4. Resultados:
|
— |
resultados del estudio previo de determinación de gamas, si procede, |
|
— |
datos en bruto: mortalidad con cada concentración de ensayo en cada período de observación, |
|
— |
gráfico de las curvas de dosis-respuesta al final del ensayo, |
|
— |
valores de la DL50 con un límite de confianza del 95 % en cada período de observación recomendado, con la sustancia de ensayo y el tóxico de referencia, |
|
— |
métodos estadísticos empleados para calcular la DL50, |
|
— |
mortalidad en los controles, |
|
— |
otros efectos biológicos observados o medidos en las abejas, así como cualquier reacción anormal, |
|
— |
toda desviación de los protocolos de ensayo aquí descritos y cualquier otra información de interés. |
3. REFERENCIAS
|
(1) |
EPPO/Council of Europe (1993). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Products — Honeybees. EPPO bulletin, vol. 23, N. 1, 151-165. Marzo de 1993. |
|
(2) |
Gough, H. J., McIndoe, E.C., Lewis, G.B. (1994). The use of dimethoate as a reference compound in laboratory acute toxicity tests on honeybees (Apis mellifera L. ,1981-1992. Journal of Apicultural Research 22119-125. |
|
(3) |
Litchfield, J.T. y Wilcoxon, F. (1949). A simplified method of evaluating dose-effect experiments. Jour. Pharmacol. and Exper. Ther., 96, 99-113. |
|
(4) |
Finney, D.J. (1971). Probit Analysis. 3a ed., Cambridge, Londres y Nueva York. |
|
(5) |
Abbott, W.S. (1925). A method for computing the effectiveness of an insecticide. jour. Econ. Entomol. 18, 265-267. |
---
C.18. ADSORCIÓN/DESORCIÓN SEGÚN UN MÉTODO DE EQUILIBRIO POR LOTES
1. MÉTODO
Este método reproduce las directrices de la OCDE TG 106 para la determinación de la adsorción/desorción en el suelo, según un método de equilibrio por lotes (2000).
1.1. INTRODUCCIÓN
El método tiene en cuenta una prueba de anillo y un taller para la selección de suelo con vistas al desarrollo de una prueba de adsorción (1) (2) (3) (4), así como distintas directrices nacionales vigentes (5) (6) (7) (8) (9) (10) (11).
Los estudios de adsorción/desorción son útiles para proporcionar información esencial sobre la movilidad de las sustancias químicas y su distribución en los compartimentos edáfico, acuático y aéreo de la biosfera (12) (13) (14) (15) (16) (17) (18) (19) (20) (21). Esta información puede ser utilizada en la predicción o estimación, por ejemplo, de la capacidad de una sustancia química para su degradación (22) (23), transformación y absorción por organismos (24), lixiviación a través del perfil edáfico (16) (18) (19) (21) (25) (26) (27) (28), volatilidad desde el suelo (21) (29) (30) o arrastre desde la tierra hasta las aguas naturales (18) (31) (32). Los datos de adsorción pueden utilizarse para establecer comparaciones y modelos (19) (33) (34) (35).
La distribución de una sustancia química entre las fases edáfica y acuosa es un proceso complejo que depende de diversos factores: la naturaleza química de la sustancia (12) (36) (37) (38) (39) (40), las características del suelo (4) (12) (13) (14) (41) (42) (43) (44) (45) (46) (47) (48) (49) y factores climáticos tales como las precipitaciones, la temperatura, la luz solar y el viento. Así pues, los numerosos fenómenos y mecanismos implicados en el proceso de adsorción de una sustancia química por el suelo no pueden definirse completamente con un modelo simplificado de laboratorio como es el presente método. Sin embargo, esta tentativa, aunque no puede cubrir todos los casos ambientalmente posibles, proporciona información valiosa sobre la importancia ambiental de la adsorción de una sustancia química.
Véase también la Introducción general.
1.2. OBJETO
El objeto del método es estimar el comportamiento de adsorción/desorción de una sustancia en los suelos. Lo que se pretende es obtener un valor de sorción que pueda utilizarse para predecir el reparto en diversas condiciones ambientales; con este fin, se determinan los coeficientes de adsorción en el equilibrio de una sustancia química en diversos suelos en función de las características de estos (por ejemplo, contenido de carbono orgánico, contenido de arcilla y textura del suelo y pH). Hay que utilizar diversos tipos de suelo para cubrir lo más ampliamente posible las interacciones de una sustancia dada con suelos naturales.
En este método, la adsorción representa el proceso de enlace de una sustancia química a las superficies de los suelos; no se distingue entre diversos procesos de adsorción (adsorción física y química) y procesos tales como la degradación catalizada en superficie, la adsorción en la masa o las reacciones químicas. La adsorción que pueda darse en las partículas coloidales (diámetro < 0,2 μm) generadas por los suelos no se tiene en cuenta completamente.
Los parámetros del suelo que se consideran más importantes para la adsorción son los siguientes: contenido de carbono orgánico (3) (4) (12) (13) (14) (41) (43) (44) (45) (46) (47) (48); contenido de arcilla y textura del suelo (3) (4) (41) (42) (43) (44) (45) (46) (47) (48) y pH para los compuestos ionizables (3) (4) (42). Otros parámetros del suelo que pueden influir en la adsorción/desorción de determinadas sustancias son la capacidad efectiva de intercambio catiónico (ECEC), el contenido de óxidos amorfos de hierro y aluminio, particularmente en caso de suelos volcánicos y tropicales (4), y la superficie específica (49).
La prueba se diseña para evaluar la adsorción de una sustancia química en distintos tipos de suelo con una amplia gama de contenido de carbono orgánico, contenido de arcilla y textura del suelo, y pH. Comprende tres etapas:
|
Etapa 1: |
Estudio preliminar para determinar:
|
|
Etapa 2: |
Prueba de barrido: se estudia la adsorción en 5 tipos diversos de suelo mediante cinética de adsorción con una sola concentración y determinación del coeficiente de distribución Kd y Koc. |
|
Etapa 3: |
Determinación de las isotermas de adsorción de Freundlich para determinar la influencia de la concentración sobre el grado de adsorción en los suelos. Estudio de la desorción mediante cinética de desorción/isotermas de desorción de Freundlich (apéndice 1). |
1.3. DEFINICIONES Y UNIDADES
|
Símbolo |
Definición |
Unidad |
|
|
Adsorción porcentual al tiempo ti |
% |
|
Aeq |
Adsorción porcentual en el equilibrio de adsorción |
% |
|
|
Masa de la sustancia problema adsorbida en el suelo al tiempo ti |
μg |
|
|
Masa de la sustancia problema adsorbida en el suelo durante el intervalo de tiempo Δti |
μg |
|
|
Masa de la sustancia problema adsorbida en el suelo en el equilibrio de adsorción |
μg |
|
m0 |
Masa de la sustancia problema en el tubo de ensayo, al principio de la prueba de adsorción |
μg |
|
|
Masa de la sustancia problema medida en una alícuota (
|
μg |
|
|
Masa de la sustancia en la solución en el equilibrio de adsorción |
μg |
|
msoil |
Cantidad de la fase edáfica, expresada en masa seca de suelo |
g |
|
Cst |
Concentración en masa de la solución madre de la sustancia |
μg cm-3 |
|
C0 |
Concentración inicial en masa de la solución problema en contacto con el suelo |
μg cm-3 |
|
|
Concentración en masa de la sustancia en la fase acuosa al tiempo ti en que se realiza el análisis |
μg cm-3 |
|
|
Contenido de la sustancia adsorbida en el suelo en el equilibrio de adsorción |
μg g-1 |
|
|
Concentración en masa de la sustancia en la fase acuosa en el equilibrio de adsorción |
μg cm-3 |
|
V0 |
Volumen inicial de la fase acuosa en contacto con el suelo durante la prueba de adsorción |
cm3 |
|
|
Volumen de la alícuota en que se mide la sustancia problema |
cm3 |
|
Kd |
Coeficiente de distribución de la adsorción |
cm-3 g-1 |
|
Koc |
Coeficiente de adsorción normalizado para tener en cuenta el carbono orgánico |
cm3 |
|
Kom |
Coeficiente de distribución, normalizado para tener en cuenta la materia orgánica |
cm3g-1 |
|
|
Coeficiente de adsorción de Freundlich |
-1/n (cm3) 1/n g—1 |
|
l/n |
Exponente de Freundlich |
|
|
|
Desorción porcentual al tiempo ti |
% |
|
|
Desorción porcentual en el intervalo de tiempo Δti |
% |
|
Kdes |
Coeficiente de desorción aparente |
cm3 |
|
|
Coeficiente de desorción de Freundlich |
µg -11/n (cm3) 1/ng-1 |
|
|
Masa de la sustancia problema desorbida del suelo al tiempo ti |
μg |
|
|
Masa de la sustancia problema desorbida del suelo durante el intervalo de tiempo Δti |
μg |
|
|
Masa de la sustancia determinada analíticamente en la fase acuosa en el equilibrio de desorción |
μg |
|
|
Masa total de la sustancia problema desorbida en el equilibrio de desorción |
μg |
|
|
Masa de la sustancia que permanece adsorbida en el suelo después del intervalo de tiempo Δti |
μg |
|
|
Masa de la sustancia que queda en solución después de alcanzado el equilibrio de adsorción debido a la sustitución incompleta del volumen |
μg |
|
|
Contenido de la sustancia problema que permanece adsorbida en el suelo en el equilibrio de desorción |
μg g-1 |
|
|
Concentración en masa de la sustancia problema en la fase acuosa en el equilibrio de desorción |
μg cm-3 |
|
vT |
Volumen total de la fase acuosa en contacto con el suelo durante el experimento de cinética de desorción llevado a cabo con el método en serie |
cm3 |
|
vR |
Volumen del sobrenadante retirado del tubo después de alcanzar el equilibrio de adsorción y sustituido por el mismo volumen de solución de CaCl20,01 M |
cm3 |
|
|
Volumen de la alícuota tomada para el análisis a partir del tiempo (i), durante el experimento de cinética de desorción realizado con el método en serie |
cm3 |
|
|
Volumen de la solución tomada del tubo (i) para la medida de la sustancia problema, en el experimento de cinética de desorción (método paralelo) |
cm3 |
|
|
Volumen de la solución tomada del tubo para la medida de la sustancia problema, en el equilibrio de desorción |
cm3 |
|
MB |
Balance de masa |
% |
|
mE |
Masa total de la sustancia problema extraída del suelo y de las paredes del recipiente del ensayo en dos pasos |
μg |
|
V rec |
Volumen del sobrenadante recuperado después del equilibrio de adsorción |
cm3 |
|
pow |
Coeficiente de reparto octanol/agua |
|
|
pKa |
Constante de disociación |
|
|
Sw |
Hidrosolubilidad |
gl-1 |
1.4. PRINCIPIO DEL MÉTODO DE ENSAYO
A las muestras de suelo de peso seco conocido que se han preequilibrado en CaCl20,01 M se añaden volúmenes conocidos de soluciones de concentración también conocida de la sustancia problema, no marcada ni radiomarcada, en CaCl20,01 M. La mezcla se agita durante un tiempo adecuado. Las suspensiones de suelo se separan entonces por centrifugación y, si se desea, por filtración, y se analiza la fase acuosa. La cantidad de sustancia problema adsorbida en la muestra de suelo se calcula como la diferencia entre la cantidad de sustancia problema inicialmente presente en la solución y la cantidad que permanece al final del experimento (método indirecto).
Como opción, la cantidad de sustancia problema adsorbida puede determinarse también directamente por análisis del suelo (método directo). Este procedimiento, que implica someter el suelo a extracción gradual con el solvente apropiado, se recomienda en caso de que no pueda determinarse exactamente la diferencia de concentración de la sustancia en la solución. Los siguientes son ejemplos de tales casos: adsorción de la sustancia problema en la superficie de los recipientes del ensayo, inestabilidad de la sustancia problema en la escala de tiempo del experimento, adsorción débil que dé solamente un pequeño cambio de concentración en la solu-ción, y adsorción fuerte que deje una concentración tan baja que no pueda determinarse exactamente. Si se utiliza una sustancia radiomarcada, la extracción del suelo puede evitarse mediante análisis de la fase edáfica por combustión y recuento de centelleo líquido. Sin embargo, el recuento de centelleo líquido es una técnica inespecífica que no puede distinguir entre productos parentales y productos de transformación; por lo tanto, debe utilizarse solamente si la sustancia problema es estable durante la duración del estudio.
1.5. INFORMACIÓN SOBRE LA SUSTANCIA PROBLEMA
Los reactivos químicos deben ser de grado analítico. Se recomienda el uso de sustancias problema no marcadas de composición conocida y pureza preferiblemente del 95 %, al menos, o de sustancias problema radio-marcadas de composición y radiopureza conocidas. En caso de marcadores de semivida breve, deben aplicarse correcciones para tener en cuenta su desintegración.
Antes de llevar a cabo una prueba de adsorción/desorción, debe disponerse de la siguiente información sobre la sustancia problema:
|
a) |
hidrosolubilidad (A.6); |
|
b) |
presión de vapor (A.4) o constante de la ley de Henry; |
|
c) |
degradación abiótica: hidrólisis en función del pH (C.7); |
|
d) |
coeficiente de reparto (A.8); |
|
e) |
biodegradabilidad fácil (C.4) o transformación aerobia y anaerobia en el suelo; |
|
f) |
pKa de las sustancias ionizables; |
|
g) |
fotólisis directa en agua (es decir, espectro de absorción ultravioleta-visible en el agua, rendimiento cuántico) y fotodegradación en el suelo. |
1.6. APLICABILIDAD DE LA PRUEBA
La prueba es aplicable a las sustancias químicas respecto de las cuales se dispone de un método analítico con la suficiente exactitud. Un parámetro importante que puede influir en la fiabilidad de los resultados, especialmente cuando se sigue el método indirecto, es la estabilidad de la sustancia problema en la escala de tiempo de la prueba. Así pues, es necesario comprobar la estabilidad en un estudio preliminar; si se observa alguna transformación en la escala de tiempo de la prueba, se recomienda que el estudio principal se realice analizando tanto la fase edáfica como la acuosa.
Pueden surgir dificultades en la realización de esta prueba con sustancias problema cuya hidrosolubilidad sea baja (Sw < 10 -4 g l-1), así como con sustancias muy cargadas, debido al hecho de que la concentración en la fase acuosa no puede medirse analíticamente con la suficiente exactitud. En estos casos hay que tomar medidas adicionales. En las secciones pertinentes del presente método se dan indicaciones sobre cómo tratar estos problemas.
Al probar sustancias volátiles, debe tenerse cuidado para evitar pérdidas durante el estudio.
1.7. DESCRIPCIÓN DEL MÉTODO
1.7.1. Instrumentos y reactivos químicos
Equipo normal de laboratorio, especialmente el siguiente:
|
a) |
tubos o recipientes para llevar a cabo los experimentos. Es importante que estos tubos o recipientes:
|
|
b) |
dispositivo de agitación: agitador rotatorio o equipo equivalente: el dispositivo de agitación debe mantener el suelo en suspensión durante la agitación; |
|
c) |
centrifugadora: preferiblemente de alta velocidad con, por ejemplo, fuerzas de centrifugación > 3 000 g, con temperatura controlada, capaces de retirar de la solución acuosa partículas con un diámetro mayor de 0,2 μm. Los recipientes deben estar cerrados durante la agitación y la centrifugación para evitar fenómenos de volatilidad y pérdidas de agua; para minimizar la adsorción sobre ellos, deben utilizarse tapones desactivados, tales como tapones de rosca con revestimiento de teflón; |
|
d) |
opcional: dispositivo de filtración: filtros de 0,2 μm de porosidad, estériles, de un solo uso. Debe tenerse especial cuidado en la selección del material del filtro, a fin de evitar cualquier pérdida de la sustancia problema sobre él; para las sustancias problema poco solubles no se recomienda usar material orgánico; |
|
e) |
instrumentación analítica, adecuada para medir la concentración de la sustancia problema; |
|
f) |
estufa de laboratorio, capaz de mantener una temperatura de 103 a 110 oC. |
1.7.2. Caracterización y selección de los suelos
Los suelos deben caracterizarse por tres parámetros considerados en gran parte responsables de la capacidad de adsorción: carbono orgánico, contenido de arcilla y textura de suelo, y pH. Como ya se ha mencionado (véase el punto 1.2) la adsorción/desorción de ciertas sustancias puede verse afectada por otras propiedades fisicoquímicas del suelo, que deben considerarse en tales casos.
Los métodos utilizados para la caracterización de suelo son muy importantes y pueden tener una influencia significativa en los resultados. Por lo tanto, se recomienda que se mida el pH del suelo en una solución de CaCl20,01 M (que es la solución utilizada en la prueba de adsorción/desorción) según el método correspondiente de la ISO (por ejemplo, ISO-10390-1). También se recomienda que las otras propiedades pertinentes del suelo se determinen según métodos normalizados [por ejemplo, el Manual de análisis de suelo (Handbook of Soil Analysis)] de la ISO: esto permite que el análisis de los datos de sorción se base en parámetros del suelo normalizados en conjunto. En el punto 3, «Referencias» (50-52), se da información sobre métodos normalizados existentes de análisis y caracterización del suelo. Para el calibrado de los métodos de prueba del suelo, se recomienda el uso de suelos de referencia.
En el cuadro 1 se da información sobre la selección de suelos para los experimentos de adsorción/desorción. Los 7 suelos seleccionados cubren los tipos de suelo de las zonas geográficas templadas. En caso de sustancias problema ionizables, los suelos seleccionados deben cubrir una gama amplia de pH, para poder evaluar la adsorción de la sustancia en sus formas ionizada y no ionizada. En el punto 1.9, «Realización de la prueba», figuran orientaciones sobre cuántos suelos distintos han de utilizarse en las diversas fases de la prueba.
Si se prefieren otros tipos de suelo, deben caracterizarse por los mismos parámetros y ser similares a los descritos en el cuadro 1 en cuanto a la variación de sus propiedades, incluso aunque no se ajusten exactamente a los criterios.
Cuadro 1
Información sobre la selección de muestras de suelo para los estudios de adsorción/desorción
|
Tipo de suelo |
Gama de pH (en CaCl20,01 M) |
Contenido de carbono orgánico ( %) |
Contenido de arcilla ( %) |
Textura del suelo (23) |
|
1 |
4,5-5,5 |
1,0-2,0 |
65-80 |
Arcilla |
|
2 |
> 7,5 |
3,5-5,0 |
20-40 |
Franco-arcilloso |
|
3 |
5,5-7,0 |
1,5-3,0 |
15-25 |
Franco-limoso |
|
4 |
4,0-5,5 |
3,0-4,0 |
15-30 |
Franco |
|
5 |
< 4,0-6,0 (24) |
< 0,5-1,5 (24) (25) |
< 10-15 (24) |
Franco-arenoso |
|
6 |
> 7,0 |
< 0,5-1,0 (24) (25) |
40-65 |
Franco-arcilloso/ arcilla |
|
7 |
< 4,5 |
> 10 |
< 10 |
Arena/franco-arenoso |
1.7.3. Recogida y conservación de muestras de suelo
1.7.3.1. Recogida
No se recomienda ninguna técnica o herramienta de muestreo específica; la técnica de muestreo depende del propósito del estudio (53) (54) (55) (56) (57) (58).
Deben considerarse los elementos siguientes:
|
a) |
es necesario tener información detallada sobre las circunstancias del yacimiento, incluyendo la situación, cubierta vegetal, tratamientos con plaguicidas y fertilizantes, adiciones biológicas o contaminación accidental. Deben seguirse las recomendaciones de la norma de la ISO sobre muestreo del suelo (ISO 10381-6) en cuanto a la descripción del sitio de muestreo; |
|
b) |
hay que definir el sitio de muestreo por sus coordenadas UTM (proyección universal transversal de Merca-tor/cero geodésico horizontal europeo) o geográficas; así se puede volver a recoger en el futuro un suelo particular o definir el suelo bajo diversos sistemas de clasificación utilizados en países diferentes. Solamente debe recogerse el horizonte A hasta una profundidad máxima de 20 cm. Especialmente en el caso del suelo de tipo 7, si hay un horizonte Oh, presente como parte del suelo, debe incluirse en el muestreo. |
Las muestras de suelo deben transportarse utilizando recipientes y bajo condiciones de temperatura que garanticen que no se alteran perceptiblemente las propiedades iniciales del suelo.
1.7.3.2. Conservción
Se prefiere el uso de suelos tomados recientemente del yacimiento. Solamente si esto no es posible, el suelo puede conservarse a temperatura ambiente y secado al aire. No se recomienda ningún límite de tiempo de conservación, pero los suelos almacenados durante más de 3 años deben volver a analizarse en cuanto a su contenido de carbono orgánico, pH y capacidad de intercambio catiónico (CEC), antes de que se utilicen.
1.7.3.3. Manipulación y preparación de las muestras de suelo para la prueba
Los suelos se secan al aire a temperatura ambiente (preferiblemente entre 20 y 25 oC). La desagregación debe llevarse a cabo con la fuerza mínima, de modo que la textura original del suelo cambie lo menos posible. Los suelos se tamizan a un tamaño de partícula ≤ 2 mm; deben seguirse las recomendaciones de la norma de la ISO sobre muestreo de suelo (ISO 10381-6) en cuanto al proceso de tamizado. Se recomienda una homoge-neización cuidadosa, ya que aumenta la reproducibilidad de los resultados. El índice de humedad de cada suelo se determina en tres alícuotas mediante calentamiento a 105 oC hasta que no haya ningún cambio significativo en el peso (aproximadamente 12 horas). Para todos los cálculos, la masa del suelo hace referencia a la masa seca en estufa, es decir, el peso del suelo corregido para descontar el índice de humedad.
1.7.4. Preparación de la sustancia problema para su aplicación al suelo
La sustancia problema se disuelve en una solución de CaCl20,01 M en agua destilada o desionizada; la solución de CaCl2 se utiliza como fase solvente acuosa para mejorar la centrifugación y minimizar el intercambio catiónico. La concentración de la solución madre debe ser preferiblemente 3 órdenes de magnitud más alta que el límite de detección del método analítico utilizado. Este umbral garantiza la exactitud de las medidas en cuanto a la metodología seguida en este método; además, la concentración de la solución madre debe estar por debajo de la hidrosolubilidad de la sustancia problema.
La solución madre debe prepararse de preferencia justo antes de la aplicación a las muestras de suelo y debe guardarse cerrada en la oscuridad a 4 oC. El tiempo de conservación depende de la estabilidad de la sustancia problema y de su concentración en la solución.
Solamente en el caso de las sustancias poco solubles (Sw < 10-4 g l-1), puede ser necesario añadir un agente de solubilización apropiado cuando sea difícil disolver la sustancia problema. Este agente de solubilización: a) debe ser miscible con agua, como el metanol o el acetonitrilo; b) su concentración no debe exceder del 1 % del volumen total de la solución madre y debe constituir menos de ese límite en la solución de la sustancia problema que se vaya a poner en contacto con el suelo (preferentemente menos del 0,1 %), y c) no debe ser un agente tensoactivo ni experimentar reacciones solvolíticas con la sustancia problema. El uso de un agente de solubilización debe constar y justificarse en el informe sobre los datos.
Otra opción para sustancias poco solubles es añadir al sistema de prueba sustancia problema ya disuelta: esta sustancia se disuelve en un solvente orgánico, del cual se añade una alícuota al sistema de suelo y solución de CaCl20,01 M en agua destilada o desionizada. El contenido del solvente orgánico en la fase acuosa debe mantenerse lo más bajo posible, normalmente sin exceder del 0,1 %. La utilización de un solvente orgánico puede adolecer de irreproducibilidad del volumen. Así pues, puede introducirse un error adicional ya que la concentración de sustancia problema y de cosolvente no será la misma en todas las pruebas.
1.8. REQUISITOS PREVIOS PARA LLEVAR A CABO LA PRUEBA DE ADSORCIÓN/DESORCIÓN
1.8.1. Método analítico
Entre los parámetros clave que pueden influir en la exactitud de las medidas de sorción se incluyen la exactitud del método analítico en el análisis de las fases tanto de solución como adsorbida, la estabilidad y la pureza de la sustancia problema, el logro del equilibrio de sorción, la magnitud del cambio de concentración de la solución, la proporción suelo/solución y los cambios en la estructura del suelo durante el proceso de equilibrado (35) (59) (60) (61) (62). En el apéndice 2 figuran algunos ejemplos donde se tratan estos problemas de exactitud.
Debe comprobarse la fiabilidad del método analítico utilizado en la gama de concentración que pueda darse durante la prueba. El experimentador debe ser libre para elaborar un método apropiado con la exactitud, la precisión, la reproducibilidad, los límites de detección y la recuperación apropiados. A continuación se dan orientaciones sobre cómo llevar a cabo tal prueba.
Un volumen apropiado de CaCl20,01 M (por ejemplo, 100 cm3) se agita durante 4 horas con un peso determinado de suelo (por ejemplo, 20 g) de alta capacidad de adsorción, es decir, con un alto contenido de carbono orgánico y de arcilla; estos pesos y volúmenes pueden variar dependiendo de las necesidades analíticas, pero una proporción suelo/solución de 1:5 es un punto de partida conveniente. Se centrifuga la mezcla y puede filtrarse la fase acuosa. Se añade a esta cierto volumen de la solución madre de sustancia problema para alcanzar una concentración nominal dentro de la gama de concentración que pueda darse durante la prueba. Este volumen no debe exceder del 10 % del volumen final de la fase acuosa, para cambiar lo menos posible la naturaleza de la solución de preequilibrado. Se analiza la solución.
Debe realizarse una prueba en blanco con el sistema suelo + solución de CaCl2 (sin la sustancia problema), para comprobar la presencia de artefactos en el método analítico y de efectos de matriz causados por el suelo.
Entre los métodos analíticos que pueden utilizarse para las medidas de sorción se incluyen la cromatografía gas-líquido (GLC), la cromatografía líquida de alta resolución (HPLC), la espectrometría (por ejemplo, cromatografía gaseosa/espectrometría de masas, HPLC/espectrometría de masas) y el recuento de centelleo líquido (para sustancias radiomarcadas). Sea cual sea el método analítico utilizado, se considera conveniente si la recuperación está entre el 90 y el 110 % del valor nominal. Para permitir la detección y la evaluación después de que haya tenido lugar el reparto, los límites de detección del método analítico deben estar al menos 2 órdenes de magnitud por debajo de la concentración nominal.
Las características y los límites de detección del método analítico disponible para llevar a cabo los estudios de adsorción desempeñan un papel importante en la definición de las condiciones de prueba y de la realización de todo el experimento. Este método sigue una vía experimental general y proporciona recomendaciones y orientaciones sobre soluciones alternativas cuando el método analítico y las instalaciones de laboratorio impongan limitaciones.
1.8.2. Selección de las proporciones óptimas suelo/solución
La selección de proporciones apropiadas suelo/solución para los estudios de sorción depende del coeficiente de distribución Kd y del grado relativo de adsorción deseado. El cambio de concentración de la sustancia en la solución determina la exactitud estadística de la medida basada en la forma de la ecuación de adsorción y el límite de la metodología analítica, respecto a la detección de la concentración de la sustancia química en la solución. Por lo tanto, en la práctica general es útil partir de algunas proporciones fijadas, para las cuales el porcentaje adsorbido sea de más del 20 %, y preferiblemente > 50 % (62), mientras que debe tenerse cuidado para mantener bastante alta la concentración de sustancia problema en la fase acuosa a fin de poder medirla con exactitud. Esto es particularmente importante en caso de altos porcentajes de adsorción.
Un planteamiento conveniente de la selección de las proporciones adecuadas suelo/agua se basa en una estimación del valor de Kd por estudios preliminares o por técnicas aceptadas de estimación (apéndice 3). La selección de una proporción adecuada puede entonces basarse en la gráfica de la proporción suelo/solución frente a Kd con porcentajes fijos de adsorción (figura 1). En esta gráfica se asume que la ecuación de adsorción es lineal (26). La relación aplicable se obtiene reordenando la ecuación (4) de Kd en la forma de la ecuación (1):
|
|
(1) |
o en su forma logarítmica suponiendo que R = msoil/V0 y Aeq %/100 = 
|
|
(2) |

La figura 1 muestra las proporciones suelo/solución necesarias en función de Kd para diversos niveles de adsorción. Por ejemplo, con una proporción suelo/solución de 1:5 y un Kd de 20, habrá aproximadamente un 80 % de adsorción. Para obtener una adsorción del 50 % con el mismo Kd, debe utilizarse una proporción de 1:25. Este planteamiento de la selección de las proporciones suelo/solución adecuadas da al investigador la flexibilidad suficiente para satisfacer sus necesidades experimentales.
Los casos que son más difíciles de tratar son aquellos en que la sustancia química se adsorbe mucho o muy poco. En los casos en que se dé adsorción baja, se recomienda una proporción suelo/solución de 1:1, aunque con algunos tipos de suelo muy orgánico pueden ser necesarias proporciones más pequeñas para obtener una mezcla fluida. Debe tenerse cuidado con la metodología analítica para medir pequeños cambios en la concentración de la solución; si no, la medida de la adsorción será inexacta. Por otra parte, en caso de coeficientes de distribución Kd muy altos, puede llegarse hasta una proporción suelo/solución de 1:100 para dejar una cantidad significativa de sustancia química en la solución. Sin embargo, debe tenerse cuidado para asegurar una buena mezcla, y dejarse tiempo suficiente para que el sistema se equilibre. Un planteamiento alternativo es predecir el valor de Kd aplicando técnicas de estimación basadas, por ejemplo, en los valores de Pow (apéndice 3). Esto podría ser útil especialmente para las sustancias químicas poco adsorbidas/polares con Pow < 20 y para las sustancias lipofílicas/de alta sorción con Pow > 104.
1.9. REALIZACIÓN DE LA PRUEBA
1.9.1. Condiciones de la prueba
Todos los experimentos se hacen a temperatura ambiente y, si es posible, a una temperatura constante situada entre 20 y 25 oC.
Las condiciones de la centrifugación permitirán eliminar de la solución las partículas de más de 0,2 μm. Este valor corresponde a la partícula más pequeña que se considera partícula sólida, y es el límite entre partículas sólidas y coloidales. En el apéndice 4 figuran orientaciones sobre cómo determinar las condiciones de la centrifugación.
Si el equipo de centrifugación no puede garantizar la eliminación de las partículas de más de 0,2 μm, puede utilizarse una combinación de centrifugación y de filtración con filtros de 0,2 μm. Estos filtros deben hacerse de un material inerte conveniente para evitar cualquier pérdida de la sustancia problema en ellos. En todo caso, debe demostrarse que no se produce durante la filtración ninguna pérdida de sustancia problema.
1.9.2. Etapa 1: Estudio preliminar
La finalidad de llevar a cabo un estudio preliminar figura ya en el punto 1.2 «Objeto». Con el experimento sugerido más adelante se dan orientaciones sobre la realización de tal prueba.
1.9.2.1. Selección de las proporciones óptimas suelo/solución
Se utilizan 2 tipos de suelo y 3 proporciones suelo/solución (6 experimentos). Un tipo de suelo tendrá alto contenido de carbono orgánico y bajo contenido de arcilla; el otro, bajo contenido de carbono orgánico y alto contenido de arcilla. Se sugieren las proporciones siguientes:
|
— |
50 g de suelo y 50 cm3 de solución acuosa de la sustancia problema (proporción 1:1), |
|
— |
10 g de suelo y 50 cm3 de solución acuosa de la sustancia problema (proporción 1:5), |
|
— |
2 g de suelo y 50 cm3 de solución acuosa de la sustancia problema (proporción 1:25). |
La cantidad mínima de suelo con la cual puede llevarse a cabo el experimento depende de las instalaciones del laboratorio y de las características de los métodos analíticos utilizados. Sin embargo, se recomienda utilizar por lo menos 1 g, y preferiblemente 2 g, para obtener resultados fiables en la prueba.
Una muestra de control con solamente la sustancia problema en solución de CaCl20,01 M (sin suelo) se somete precisamente al mismo tratamiento que los sistemas de prueba, para comprobar la estabilidad de la sustancia problema en solución de CaCl2 y su posible adsorción a las superficies de los recipientes del ensayo.
Una muestra en blanco por suelo con la misma cantidad de suelo y el volumen total de 50 cm3 de solución de CaCl20,01 M (sin sustancia problema) se somete al mismo procedimiento de prueba, con el fin de servir de contraste durante el análisis para detectar sustancias de interferencia o suelos contaminados.
Todos los experimentos, incluidos los controles y los blancos, deben llevarse a cabo al menos por duplicado. El número total de muestras que deben prepararse para el estudio puede calcularse en función de la metodología seguida.
Los métodos para el estudio preliminar y el estudio principal son generalmente los mismos; se mencionan las excepciones en su caso.
Las muestras de suelo secadas al aire se equilibran agitándolas con un volumen mínimo de 45 cm3 de CaCl20,01 M durante una noche (12 horas) antes del día del experimento. Después se añade cierto volumen de la solución madre de sustancia problema para ajustar el volumen final a 50 cm3. Este volumen añadido de la solución madre: a) no debe exceder del 10 % del volumen final de 50 cm3 de la fase acuosa para cambiar lo menos posible la naturaleza de la solución de preequilibrado, y b) debe suponer preferiblemente una concentración inicial de la sustancia problema en contacto con el suelo (Co) por lo menos dos órdenes de magnitud más alta que el límite de detección del método analítico; este umbral garantiza la posibilidad de llevar a cabo medidas exactas incluso en caso de adsorción fuerte (> 90 %) y de determinar más tarde las isotermas de adsorción. También se recomienda que, a ser posible, la concentración inicial de sustancia (Co) no supere la mitad de su límite de solubilidad.
Se da más abajo un ejemplo de cómo calcular la concentración de la solución madre (Cst). Se supone un límite de detección de 0,01 μg cm-3 y una adsorción del 90 %; así pues, la concentración inicial de la sustancia problema en contacto con el suelo debe ser preferiblemente 1 μg cm-3 (dos órdenes de magnitud más alta que el límite de detección). Suponiendo que se añade el volumen recomendado máximo de la solución madre (es decir, 5 cm3 a 45 cm3 de solución de equilibrado de CaCl20,01 M (= el 10 % de la solución madre hasta un volumen total de 50 cm3 de fase acuosa), la concentración de la solución madre debe ser 10 μg cm-3, que es tres órdenes de magnitud más alta que el límite de detección del método analítico.
El pH de la fase acuosa debe medirse antes y después del contacto con el suelo, puesto que desempeña un papel importante en todo el proceso de adsorción, especialmente en caso de sustancias ionizables.
Se agita la mezcla hasta que se alcance el equilibrio de adsorción. El tiempo de equilibrado de los suelos es muy variable, dependiendo de la sustancia química y del suelo; generalmente es suficiente un período de 24 horas (77). En el estudio preliminar pueden recogerse muestras secuencialmente durante un período de 48 horas desde la mezcla (por ejemplo, a las 4, 8, 24, 48 horas). Sin embargo, los tiempos de análisis deben considerarse con flexibilidad para tener en cuenta el horario de trabajo del laboratorio.
Hay dos opciones para el análisis de la sustancia problema en la solución acuosa: a) el método paralelo, y b) el método en serie. Debe insistirse en que, aunque el método paralelo sea experimentalmente más pesado, el tratamiento matemático de los resultados es más simple (apéndice 5). Sin embargo, la elección de la metodología seguida corresponde al experimentador, que habrá de considerar las instalaciones y los recursos disponibles del laboratorio.
|
a) |
Método paralelo: se preparan tantas muestras con la misma proporción de suelo/solución como intervalos de tiempo a que se desee estudiar la cinética de adsorción. Después de la centrifugación y, en su caso, filtración, la fase acuosa del primer tubo se recupera lo más completamente posible y se analiza después de, por ejemplo, 4 horas, la del segundo tubo después de 8 horas, la del tercero después de 24, etc. |
|
b) |
Método en serie: se prepara solamente una muestra por duplicado de cada proporción suelo/solución. A los intervalos de tiempo definidos se centrifuga la mezcla para separar las fases. Se determina inmediatamente la sustancia problema en una pequeña alícuota de la fase acuosa: el experimento continúa con la mezcla original. Si se aplica la filtración después de la centrifugación, el laboratorio debe tener instalaciones para realizar la filtración de pequeñas alícuotas acuosas. Se recomienda que el volumen total de las alícuotas tomadas no exceda del 1 % del volumen total de la solución, para no cambiar mucho la proporción suelo/solución ni disminuir la masa de soluto disponible para la adsorción durante la prueba. |
La adsorción porcentual se calcula a cada tiempo ![]() en función de la concentración inicial nominal y de la concentración medida a ese tiempo de muestreo (ti), corregido para tener en cuenta el valor del blanco. Se realizan las gráficas de
en función de la concentración inicial nominal y de la concentración medida a ese tiempo de muestreo (ti), corregido para tener en cuenta el valor del blanco. Se realizan las gráficas de ![]() frente al tiempo (figura 1 del apéndice 5) para estimar la llegada a la meseta de equilibrio (27). También se calcula el valor de Kd en el equilibrio. Tomando como base este valor de Kd, a partir de la figura 1 se seleccionan proporciones adecuadas suelo/solución, de modo que la adsorción porcentual alcance más del 20 % y, preferiblemente, > 50 % (61). Todas las ecuaciones y principios de gráficas aplicables figuran en el punto 2, de «Datos e informes», y en el apéndice 5.
frente al tiempo (figura 1 del apéndice 5) para estimar la llegada a la meseta de equilibrio (27). También se calcula el valor de Kd en el equilibrio. Tomando como base este valor de Kd, a partir de la figura 1 se seleccionan proporciones adecuadas suelo/solución, de modo que la adsorción porcentual alcance más del 20 % y, preferiblemente, > 50 % (61). Todas las ecuaciones y principios de gráficas aplicables figuran en el punto 2, de «Datos e informes», y en el apéndice 5.
1.9.2.2. Determinación del tiempo de equilibrado de adsorción y de la cantidad de sustancia problema adsorbida en el equilibrio
Como ya se ha mencionado, las gráficas de ![]() o
o ![]() frente al tiempo permiten la estimación del logro del equilibrio de adsorción y de la cantidad de sustancia problema adsorbida en el equilibrio. Las figuras 1 y 2 del apéndice 5 muestran ejemplos de tales gráficas. El tiempo de equilibrado es el que necesita el sistema para alcanzar una meseta.
frente al tiempo permiten la estimación del logro del equilibrio de adsorción y de la cantidad de sustancia problema adsorbida en el equilibrio. Las figuras 1 y 2 del apéndice 5 muestran ejemplos de tales gráficas. El tiempo de equilibrado es el que necesita el sistema para alcanzar una meseta.
Si, con un suelo determinado, no se llega a ninguna meseta sino que hay un aumento constante, puede deberse a la complicación por factores tales como la biodegradación o la difusión lenta. La biodegradación puede comprobarse repitiendo el experimento con una muestra esterilizada del suelo. Si no se logra ninguna meseta incluso en este caso, el experimentador debe realizar estudios específicos para buscar otros fenómenos que pudieran darse: puede hacerse con modificaciones apropiadas de las condiciones del experimento (temperatura, tiempos de agitación, proporciones suelo/solución). Corresponde al experimentador decidir si continúa con el procedimiento de prueba a pesar de que posiblemente no se llegue a lograr el equilibrio.
1.9.2.3. Adsorción en la superficie del recipiente del ensayo y estabilidad de la sustancia problema
Analizando las muestras de control puede obtenerse información sobre la adsorción de la sustancia problema en la superficie de los recipientes del ensayo, así como sobre su estabilidad. Si se observa una disminución superior al error típico del método analítico, puede haber fenómenos de degradación abiótica o adsorción en la superficie del recipiente del ensayo. Puede conseguirse distinguir entre estos dos fenómenos lavando a fondo las paredes del recipiente con un volumen conocido de un solvente apropiado y determinando la sustancia problema en la solución de lavado. Si no se observa ninguna adsorción en la superficie de los recipientes del ensayo, la disminución confirma la inestabilidad abiótica de la sustancia problema. Si se encuentra adsorción, es necesario cambiar el material de los recipientes del ensayo. Sin embargo, los datos sobre la adsorción en la superficie de los recipientes del ensayo obtenidos de este experimento no pueden extrapolarse directamente al experimento con suelo/solución. La presencia de suelo afecta a esta adsorción.
Puede obtenerse información adicional sobre la estabilidad de la sustancia problema mediante la determinación del balance de masa parental a lo largo del tiempo. Esto significa que se determina la sustancia problema en la fase acuosa, en los extractos de suelo y en las paredes del recipiente del ensayo. La diferencia entre la masa de la sustancia problema añadida y la suma de las masas de la sustancia problema en la fase acuosa, extractos de suelo y paredes de los recipientes del ensayo es igual a la masa degradada, volatilizada o no extraída. Para llevar a cabo una determinación del balance de masa, debe haberse alcanzado el equilibrio de adsorción en el tiempo que dure el experimento.
El balance de masa se lleva a cabo en ambos suelos y con una proporción suelo/solución por suelo que dé una disminución por encima del 20 % (preferiblemente > 50 %) en el equilibrio. Cuando se termine el experimento de selección de la proporción con el análisis de la última muestra de la fase acuosa después de 48 horas, las fases se separan por centrifugación y, si se desea, filtración. Se recupera lo más posible de la fase acuosa, y se añade al suelo un solvente de extracción conveniente (coeficiente de extracción de por lo menos el 95 %) para extraer la sustancia problema. Se recomienda hacer por lo menos dos extracciones sucesivas. Se determina la cantidad de sustancia problema presente en los extractos de recipientes del ensayo y del suelo, y se calcula el balance de masa (ecuación 10 en el punto 2.1.2). Si es inferior al 90 %, se considera que la sustancia problema es inestable en la escala de tiempo de la prueba. Sin embargo, los estudios podían aún continuar, teniendo en cuenta la inestabilidad de la sustancia problema; en este caso se recomienda analizar ambas fases en el estudio principal.
1.9.3. Etapa 2: Cinética de adsorción con una concentración de la sustancia problema
Se utilizan 5 suelos, seleccionados a partir del cuadro 1. Es conveniente la inclusión entre estos 5 suelos de algunos o de todos los suelos utilizados en el estudio preliminar, si procede. En tal caso, la etapa 2 no tiene que repetirse con los suelos utilizados en el estudio preliminar.
El tiempo de equilibrado, la proporción suelo/solución, el peso de la muestra de suelo, el volumen de la fase acuosa en contacto con el suelo y la concentración de la sustancia problema en la solución se seleccionan basándose en los resultados del estudio preliminar. Es mejor hacer el análisis aproximadamente después de un tiempo del contacto de 2, 4, 6, 8 (quizás también 10) y 24 horas; el tiempo de agitación puede ampliarse a un máximo de 48 horas en caso de que una sustancia requiera un tiempo más largo de equilibrado según los resultados de la selección de la proporción. Sin embargo, los tiempos de análisis pueden considerarse con flexibilidad.
Se hace cada experimento (un suelo y una solución) al menos por duplicado para poder estimar la varianza de los resultados. En cada experimento se lleva a cabo una prueba en blanco, con suelo y solución de CaCl20,01 M, sin sustancia problema, y con un peso y un volumen, respectivamente, idénticos a los del experimento. Se somete al mismo procedimiento de prueba una muestra de control con solamente la sustancia problema en solución de CaCl20,01 M (sin suelo) como precaución frente a fenómenos inesperados.
La adsorción porcentual se calcula a cada tiempo ![]() o intervalo de tiempo
o intervalo de tiempo ![]() (según sea necesario) y se representa gráficamente frente al tiempo. También se calcula el coeficiente de distribución Kd en el equilibrio, así como el coeficiente de adsorción normalizado para tener en cuenta el carbono orgánico Koc (con sustancias orgánicas no polares).
(según sea necesario) y se representa gráficamente frente al tiempo. También se calcula el coeficiente de distribución Kd en el equilibrio, así como el coeficiente de adsorción normalizado para tener en cuenta el carbono orgánico Koc (con sustancias orgánicas no polares).
Resultados de la prueba de cinética de adsorción
El valor lineal de Kd es generalmente exacto para describir el comportamiento respecto a la sorción en el suelo (35) (78) y refleja la movilidad inherente de las sustancias químicas en el suelo. Por ejemplo, en general, las sustancias con Kd < 1 cm3 g-1 se consideran cualitativamente móviles. Del mismo modo, MacCall et al. (16) han elaborado un sistema de clasificación de la movilidad basado en valores de Koc (16). Además, hay sistemas de clasificación de la lixiviación basados en la relación entre Koc y DT-50 (28) (32) (79).
También, según estudios de análisis de error (61), los valores de Kd inferiores a 0,3 cm3 g-1 no pueden estimarse exactamente a partir de una disminución de la concentración en la fase acuosa, incluso cuando se aplica la proporción más favorable suelo/solución (desde el punto de vista de la exactitud), es decir, 1:1. En este caso se recomienda el análisis de ambas fases, suelo y solución.
En cuanto a las observaciones mencionadas, se recomienda que el estudio del comportamiento de la adsorción de una sustancia química en el suelo y de su movilidad potencial continúe mediante la determinación de las isotermas de adsorción de Freundlich para los sistemas de los cuales sea posible una determinación exacta de Kd con el protocolo experimental seguido en este método de ensayo. La determinación exacta es posible si el valor que resulta de multiplicar Kd por la proporción suelo/solución es >, cuando las medidas se basan en la disminución de la concentración de la fase acuosa (método indirecto), o bien >, cuando se analizan ambas fases (método directo) (61).
1.9.4. Etapa 3: Isotermas de adsorción y cinética de desorción/isotermas de desorción
1.9.4.1. Isotermas de adsorción
Se utilizan 5 concentraciones de sustancia problema que cubran preferiblemente dos órdenes de magnitud; en la selección de estas concentraciones deben tenerse en cuenta la hidrosolubilidad y las concentraciones acuosas resultantes en el equilibrio. Debe mantenerse la misma proporción suelo/solución por suelo a lo largo del estudio. La prueba de adsorción se lleva a cabo según lo descrito anteriormente, con la única diferencia de que la fase acuosa se analiza solamente una vez, al tiempo necesario para alcanzar el equilibrio según lo determinado antes en la etapa 2. Se determinan las concentraciones de equilibrio en la solución y se calcula la cantidad adsorbida a partir de la disminución de la sustancia problema en la solución o con el método directo. La masa adsorbida por unidad de masa del suelo se representa gráficamente en función de la concentración de la sustancia problema en el equilibrio (véase el punto 2, «Datos e informes»).
Resultados del experimento de isotermas de adsorción
Entre los modelos matemáticos de adsorción propuestos hasta ahora, el de las isotermas de Freundlich es el utilizado más frecuentemente para describir procesos de adsorción. En las referencias (41) (45) (80) (81) (82) se proporciona información más detallada sobre la interpretación e importancia de los modelos de adsorción.
Nota: Debe indicarse que es posible comparar los valores de KF (coeficiente de adsorción de Freundlich) de diferentes sustancias solamente si estos valores de KF se expresan en las mismas unidades (83).
1.9.4.2. Cinética de desorción
El propósito de este experimento es investigar si una sustancia química se adsorbe reversible o irreversiblemente a un suelo. Esta información es importante, puesto que el proceso de desorción también desempeña un papel destacado en el comportamiento de las sustancias en el suelo de campo. Por otra parte, los datos de desorción son útiles para la modelización por ordenador de la lixiviación y la simulación del arrastre de sustancias disueltas. Si se desea hacer un estudio de desorción, se recomienda que el estudio descrito a continuación se lleve a cabo con cada sistema para el cual haya sido posible una determinación exacta de Kd en el experimento anterior de cinética de adsorción.
Análogamente al estudio de cinética de adsorción, hay dos opciones para proceder con el experimento de cinética de desorción: a) el método paralelo, y b) el método en serie. La selección de la metodología seguida corresponde al experimentador, que tendrá en cuenta las instalaciones y los recursos del laboratorio.
|
a) |
Método paralelo: de cada suelo seleccionado para realizar el estudio de desorción se preparan tantas muestras con la misma proporción suelo/solución como intervalos de tiempo a que se desee estudiar la cinética de desorción. Es preferible utilizar los mismos intervalos de tiempo que en el experimento de cinética de adsorción: sin embargo, el tiempo total puede ampliarse según sea necesario para que el sistema alcance el equilibrio de desorción. En cada experimento (un suelo, una solución) se lleva a cabo una prueba en blanco, con suelo y solución de CaCl20,01 M, sin la sustancia problema, y con un peso y un volumen, respectivamente, idénticos a los del experimento. Como muestra de control, la sustancia problema en solución de CaCl20,01 M (sin suelo) se somete al mismo procedimiento de prueba. Todas las mezclas del suelo con la solución se agitan basta que se alcance el equilibrio de adsorción (según lo determinado antes en la etapa 2). Entonces, las fases se separan por centrifugación y las fases acuosas se retiran en la mayor proporción posible. El volumen de solución retirado se sustituye con un volumen igual de CaCl20,01 M sin sustancia problema y las nuevas mezclas se agitan otra vez. La fase acuosa del primer tubo se recupera lo más completamente posible y se analiza después de, por ejemplo, 2 horas, la del segundo tubo después de 4 horas, la del tercero después de 6 horas, etc., hasta que se alcance el equilibrio de desorción. |
|
b) |
Método en serie: después del experimento de cinética de adsorción, se centrifuga la mezcla y se retira lo más posible la fase acuosa. El volumen de solución retirado se sustituye con un volumen igual de CaCl20,01 M sin sustancia problema. La nueva mezcla se agita hasta que se alcance el equilibrio de desorción. Durante este período, a intervalos de tiempo definidos, se centrifuga la mezcla para separar las fases. En una pequeña alícuota de la fase acuosa se determina inmediatamente la sustancia problema; el experimento continúa después con la mezcla original. El volumen de cada alícuota debe ser menos del 1 % del volumen total. Se añade a la mezcla la misma cantidad de solución de CaCl20,01 M para mantener la proporción suelo/solución, y continúa la agitación hasta el intervalo siguiente. |
La desorción porcentual se calcula a cada tiempo ![]() o intervalo de tiempo
o intervalo de tiempo ![]() (según las necesidades del estudio) y se representa frente al tiempo. También se calcula el coeficiente de desorción Kdes en el equilibrio. Todas las ecuaciones aplicables figuran en el punto 2, «Datos e informes», y en el apéndice 5.
(según las necesidades del estudio) y se representa frente al tiempo. También se calcula el coeficiente de desorción Kdes en el equilibrio. Todas las ecuaciones aplicables figuran en el punto 2, «Datos e informes», y en el apéndice 5.
Resultados del experimento de cinética de desorción
Las gráficas comunes de la desorción ![]() y la adsorción
y la adsorción ![]() porcentuales frente al tiempo permiten valorar la reversibilidad del proceso de adsorción. Si el equilibrio de desorción se logra incluso dentro del doble del tiempo de equilibrio de adsorción, y la desorción total es de más del 75 % de la cantidad adsorbida, se considera que la adsorción es reversible.
porcentuales frente al tiempo permiten valorar la reversibilidad del proceso de adsorción. Si el equilibrio de desorción se logra incluso dentro del doble del tiempo de equilibrio de adsorción, y la desorción total es de más del 75 % de la cantidad adsorbida, se considera que la adsorción es reversible.
1.9.4.3. Isotermas de desorción
Las isotermas de desorción de Freundlich se determinan con los suelos utilizados en el experimento de las isotermas de adsorción. La prueba de desorción se lleva a cabo según lo descrito en el punto 1.9.2.5.2, «Cinética de desorción», con la única diferencia de que la fase acuosa se analiza solamente una vez, en el equilibrio de desorción. Se calcula la cantidad de sustancia problema desorbida. El contenido de sustancia problema que permanece adsorbida al suelo en el equilibrio de desorción se representa en función de la concentración de equilibrio de la sustancia problema en la solución (véase a continuación «Datos e informes» y el apéndice 5).
2. DATOS E INFORMES
Los datos analíticos se presentan en forma de cuadro (véase el apéndice 6). Se dan las medidas y las medias calculadas. Se proporcionan las representaciones gráficas de las isotermas de adsorción. Se hacen los cálculos según lo descrito más adelante.
Para la prueba, se considera que el peso de 1 cm3 de solución acuosa es 1 g. La proporción suelo/solución puede expresarse en unidades de peso/peso o de peso/volumen con la misma cifra.
2.1. ADSORCIÓN
La adsorción ![]() se define como el porcentaje de sustancia adsorbida en el suelo en relación con la cantidad presente al principio de la prueba, en las condiciones de prueba. Si la sustancia problema es estable y no se adsorbe significativamente a la pared del recipiente,
se define como el porcentaje de sustancia adsorbida en el suelo en relación con la cantidad presente al principio de la prueba, en las condiciones de prueba. Si la sustancia problema es estable y no se adsorbe significativamente a la pared del recipiente, ![]() se calcula a cada tiempo ti, según la ecuación:
se calcula a cada tiempo ti, según la ecuación:
|
|
(3) |
donde:
|
|
= |
adsorción porcentual al tiempo ti ( %), |
|
|
= |
masa de la sustancia problema adsorbida en el suelo al tiempo ti (μg), |
|
m0 |
= |
masa de la sustancia problema en el tubo de ensayo, al principio de la prueba (μg). |
En el apéndice 5 figura información detallada sobre el cálculo de la adsorción porcentual ![]() con los métodos paralelo y en serie.
con los métodos paralelo y en serie.
El coeficiente de distribución Kd es la proporción entre el contenido de sustancia en la fase edáfica y la concentración en masa de la sustancia en la solución acuosa, en las condiciones de prueba, cuando se alcanza el equilibrio de adsorción:
|
(4) |
donde:
|
|
= |
contenido de la sustancia adsorbida en el suelo en el equilibrio de adsorción (μg g-1), |
|
|
= |
concentración en masa de la sustancia en la fase acuosa en el equilibrio de adsorción (μg cm-3); esta concentración se determina analíticamente teniendo en cuenta los valores obtenidos en las pruebas en blanco, |
|
|
= |
masa de la sustancia adsorbida en el suelo en el equilibrio de adsorción (μg), |
|
|
= |
masa de la sustancia en la solución en el equilibrio de adsorción (μg), |
|
msoil |
= |
cantidad de la fase edáfica, expresada en masa seca de suelo (g), |
|
Vo |
= |
volumen inicial de la fase acuosa en contacto con el suelo (cm3). |
La relación entre Aeq y Kd se da en la ecuación siguiente:
|
(5) |
donde:
|
Aeq |
= |
adsorción porcentual en el equilibrio de adsorción ( %). |
El coeficiente de adsorción normalizado para tener en cuenta el carbono orgánico Koc relaciona el coeficiente de distribución Kd con el contenido de carbono orgánico de la muestra de suelo:
|
(6) |
donde:
|
% OC |
= |
porcentaje de carbono orgánico en la muestra de suelo (g g-1). |
El coeficiente Koc representa un solo valor que caracteriza el reparto principalmente de las sustancias orgánicas no polares entre el carbono orgánico del suelo o sedimento y el agua. La adsorción de estas sustancias está correlacionada con el contenido orgánico del sólido de sorción (7): así pues, los valores de Koc dependen de las características específicas de las fracciones húmicas que difieren considerablemente en su capacidad de sorción, debido a diferencias de origen, génesis, etc.
2.1.1. Isotermas de adsorción
La ecuación de las isotermas de adsorción de Freundlich relaciona la cantidad de sustancia problema adsorbida con la concentración de sustancia problema en la solución en el equilibrio (ecuación 8).
Los datos se tratan como en el punto 2.1, «Adsorción», y, de cada tubo de ensayo, se calcula el contenido de sustancia problema adsorbida en el suelo después de la prueba de adsorción [ , en otras partes expresado como x/m]. Se acepta que se ha logrado el equilibrio y que
, en otras partes expresado como x/m]. Se acepta que se ha logrado el equilibrio y que  representa el valor de equilibrio:
representa el valor de equilibrio:
|
(7) |
La ecuación de adsorción de Freundlich es la siguiente (8):
|
(8) |
o, en forma lineal:
|
|
(9) |
donde:
|
|
= |
coeficiente de adsorción de Freundlich: su dimensión es cm3 g-1 solo si l/n = 1; en los demás casos, se introduce la pendiente 1/n en la dimensión ce |
|
n |
= |
constante de regresión: l/n generalmente oscila entre 0,7 y 1,0, indicando que los datos de sorción suelen ser ligeramente no lineales. |
Se representan gráficamente las ecuaciones (8) y (9) y se calculan los valores de ![]() y 1/n por análisis de regresión utilizando la ecuación 9. También se calcula el coeficiente de correlación r2 de la ecuación logarítmica. En la figura 2 se da un ejemplo de tales representaciones:
y 1/n por análisis de regresión utilizando la ecuación 9. También se calcula el coeficiente de correlación r2 de la ecuación logarítmica. En la figura 2 se da un ejemplo de tales representaciones:
2.1.2. Balance de masa
El balance de masa (MB) se define como el porcentaje de sustancia que puede recuperarse analíticamente después de una prueba de adsorción respecto a la cantidad nominal de sustancia presente al principio de la prueba.
El tratamiento de los datos será diferente si el solvente es completamente miscible con agua. En caso de un solvente miscible con agua, el tratamiento de los datos descrito en el punto 2.2, «Desorción» puede aplicarse para determinar la cantidad de sustancia recuperada por la extracción con el solvente. Si el solvente es menos miscible con agua, hay que hacer la determinación de la cantidad recuperada.
El balance de masa MB de la adsorción se calcula del siguiente modo; se supone que el término mE corresponde a la suma de las masas de la sustancia problema extraídas del suelo y de la superficie del recipiente del ensayo con un solvente orgánico:
|
|
(10) |
donde:
|
MB |
= |
balance de masa ( %) |
|
mE |
= |
masa total de la sustancia problema extraída del suelo y de las paredes del recipiente del ensayo en dos fases (μg), |
|
Co |
= |
concentración inicial en masa de la solución problema en contacto con el suelo (μg cm-3), |
|
Vrec |
= |
volumen de sobrenadante recuperado después del equilibrio de adsorción (cm-3). |
2.2. DESORCIÓN
La desorción (D) se define como el porcentaje de sustancia problema que se desorbe, en relación con la cantidad de sustancia adsorbida previamente, en las condiciones de prueba:
|
|
(11) |
donde:
|
|
= |
desorción porcentual a un tiempo ti ( %), |
|
|
= |
masa de la sustancia problema desorbida del suelo a un tiempo ti (μg), |
|
|
= |
masa de la sustancia problema adsorbida en el suelo en el equilibrio de adsorción (μg). |
En el apéndice 5 figura información detallada sobre cómo calcular la desorción porcentual ![]() con los métodos paralelo y en serie.
con los métodos paralelo y en serie.
El coeficiente de desorción aparente (Kdes) es, en las condiciones de prueba, la proporción entre el contenido de sustancia que permanece en la fase edáfica y la concentración en masa de la sustancia desorbida en la solución acuosa, cuando se alcanza el equilibrio de desorción:
|
(12) |
donde:
|
Kdes |
= |
coeficiente de desorción (cm3 g-1), |
|
|
= |
masa total de la sustancia problema desorbida del suelo en el equilibrio de desorción (μg), |
|
VT |
= |
volumen total de la fase acuosa en contacto con el suelo durante la prueba de cinética de desorción (cm3). |
En el apéndice 5, en la sección «Desorción», figuran directrices para calculan 
Observación
Si la prueba de adsorción precedente se ha llevado a cabo con el método paralelo, se considera que el volumen VT de la ecuación 12 es igual a Vo.
2.2.1. Isotermas de desorción
La ecuación de isotermas de desorción de Freundlich relaciona el contenido de la sustancia problema que permanece adsorbida en el suelo con la concentración de la sustancia problema en la solución en el equilibrio de desorción (ecuación 16).
Para cada tubo de prueba, el contenido de sustancia que permanece adsorbida en el suelo en el equilibrio de desorción se calcula del modo siguiente:
|
(13) |
 se define como:
se define como:
|
(14) |
donde:
|
|
= |
contenido de la sustancia problema que permanece adsorbida en el suelo en el equilibrio de desorción (μg g-1), |
|
|
= |
masa de la sustancia determinada analíticamente en la fase acuosa en el equilibrio de desorción (μg), |
|
|
= |
masa de la sustancia problema que queda en solución después de alcanzado el equilibrio de adsorción debido a la sustitución incompleta del volumen (μg), |
|
|
= |
masa de la sustancia en la solución en el equilibrio de adsorción (μg); |
|
|
(15) |
|
|
= |
volumen de solución tomada del tubo para la medida de la sustancia problema, en el equilibrio de desorción (cm3), |
|
VR |
= |
volumen del sobrenadante retirado del tubo después de alcanzar el equilibrio de adsorción y sustituido por el mismo volumen de solución de CaCl20,01 M (cm3). |
A continuación se muestra la ecuación de desorción de Freundlich:
|
(16) |
o, en forma lineal:
|
|
(17) |
donde:
|
|
= |
coeficiente de desorción de Freundlich, |
|
n |
= |
constante de regresión, |
|
|
= |
concentración en masa en la sustancia en la fase acuosa en el equilibrio de desorción (μg cm-3). |
Las ecuaciones 16 y 17 pueden representarse gráficamente y el valor de ![]() y el de 1/n se calculan por análisis de regresión utilizando la ecuación 17.
y el de 1/n se calculan por análisis de regresión utilizando la ecuación 17.
Observación
Si el exponente l/n de adsorción o desorción de Freundlich es igual a 1, la constante de enlace de adsorción o desorción de Freundlich (![]() y
y ![]() ) será respectivamente igual a la constante de equilibrio de adsorción o desorción (Kd y Kdes), y las gráficas de Cs frente a Caq serán lineales. Si los exponentes no son iguales a 1, las gráficas de Cs frente a Caq no serán lineales y las constantes de adsorción y desorción variarán a lo largo de las isotermas.
) será respectivamente igual a la constante de equilibrio de adsorción o desorción (Kd y Kdes), y las gráficas de Cs frente a Caq serán lineales. Si los exponentes no son iguales a 1, las gráficas de Cs frente a Caq no serán lineales y las constantes de adsorción y desorción variarán a lo largo de las isotermas.
2.2.2. Informe de la prueba
El informe de la prueba debe incluir la información siguiente:
|
— |
identificación completa de las muestras de suelo utilizadas, incluyendo: |
|
— |
referencia geográfica del sitio (latitud, longitud), |
|
— |
fecha del muestreo, |
|
— |
tipo de uso (por ejemplo, suelo agrícola, bosque, etc.), |
|
— |
profundidad del muestreo, |
|
— |
contenido de arena/limo/arcilla, |
|
— |
valores de pH (en CaCl20,01 M), |
|
— |
contenido de carbono orgánico, |
|
— |
contenido de materia orgánica, |
|
— |
contenido de nitrógeno, |
|
— |
proporción C/N, |
|
— |
capacidad de intercambio canónico (mmol/kg), |
|
— |
toda la información relativa a la recogida y conservación de las muestras de suelo, |
|
— |
en su caso, toda información pertinente para la interpretación de la adsorción/desorción de la sustancia problema, |
|
— |
referencia de los métodos utilizados para la determinación de cada parámetro; |
|
— |
información sobre la sustancia problema según el caso; |
|
— |
temperatura de los experimentos; |
|
— |
condiciones de centrifugación; |
|
— |
procedimiento analítico utilizado para analizar la sustancia problema; |
|
— |
justificación del uso eventual de agentes de solubilización para la preparación de la solución madre de la sustancia problema; |
|
— |
explicación de las correcciones hechas en los cálculos, en su caso; |
|
— |
datos según las fichas (apéndice 6) y representaciones gráficas; |
|
— |
toda la información y observaciones útiles para la interpretación de los resultados de la prueba. |
3. REFERENCIAS
|
(1) |
Kukowski H. and Brummer G., (1987). Investigations on the Adsorption and Desorption of Selected Chemicals in Soils. UBA Report 106 02045, Part II. |
|
(2) |
Fränzle O., Kuhnt G. and Vetter L, (1987). Selection of Representative Soils in the EC-Territory. UBA Report 106 02045, Part I. |
|
(3) |
Kuhnt G. and Muntau H. (Eds.) EURO-Soils: Identification, Collection, Treatment, Characterisation. Special Publication no. 1.94.60, Joint Research Centre. European Commission, ISPRA, December 1994. |
|
(4) |
OECD Test Guidelines Programme, Final Report of the OECD Workshop on Selection of Soils/Sediments, Belgirate, Italy, 18-20 January 1995 (June 1995). |
|
(5) |
US-Environment Protection Agency: Pesticide Assessment Guidelines, Subdivision N., Chemistry; Environmental Fate, Series 163-1, Leaching and Adsorption/Desorption Studies, Addendum 6 on Data Reporting. 540/09-SS-096, Date: 1/1988. |
|
(6) |
US-Environment Protection Agency: Prevention, Pesticides and Toxic Substances. OPPTS Harmonized Test Guidelines. Series 835-Fate, Transpon and Transformation Test Guidelines, OPPTS No: 835.1220 Sediment and Soil Adsorption/Desorption Isotherm. EPA No: 71 2-C-96-048. April 1996. |
|
(7) |
ASTM Standards, E 1195-85, Standard Test Method for Determining a Sorption Constant (Koc) for an Organic Chemical in Soil and Sediments. |
|
(8) |
Agriculture Canada: Environmental Chemistry and Fate. Guidelines for registration of pesticides in Canada, 15 July 1987. |
|
(9) |
Netherlands Commission Registration Pesticides (1995): Application for registration of a pesticide. Section G. Behaviour of the product and its metabolites in soil, water and air. |
|
(10) |
Danish National Agency of Environmental Protection (October 1988): Criteria for registration of pesticides as especially dangerous to health or especially harmful to the environment. |
|
(11) |
BBA (1990), Guidelines for the Official Testing of Plant Protection Products, Biological Research Centre for Agriculture and Forestry, Braunschweig, Germany. |
|
(12) |
Calvet R., (1989), 'Evaluation of adsorption coefficients and the prediction of the mobilities of pesticidas in soils', in Methodological Aspects of the Study of Pesticide Behaviour in Soil (ed. P. Jamet), INRA, París, (Review). |
|
(13) |
Calvet R., (1980) 'Adsorption-Desorption Phenomena' in Interactions between herbicides and the soil. (R.J. Hance ed.), Academic Press, London, pp. 83-122. |
|
(14) |
Hasset J.J., and Banwart W.L. (1989), The sorption of nonpolar organics by soils and sediments' in Reactions and Movement of Organic Chemicals in Soils. Soil Science Society of America (S.S.S.A), Special Publication no. 22, pp 31-44. |
|
(15) |
van Genuchten M. Th., Davidson J.M., and Wierenga P.J., (1974), 'An evaluation of kinetic and equilibrium equations for the prediction of pesticide movement through porous media'. Soil Sci. Soc. Am. Proc, Vol. 38(1), 29-35. |
|
(16) |
McCall P.J., Laskowski D.A. Swann R.I., and Dishburger H.J., (1981), 'Measurement of sorption coefficients of organic chemicals and their use, in environmental fate analysis', in Test Protocols for Environmental Fate and Movement of Toxicants. Proceedings of AOAC Symposium, AOAC, Washington DC. |
|
(17) |
Lambert S.M., Porter P.E., and Schieferrstein R.H., (1965), 'Movement and sorption of chemicals applied to the soil'. Weeds, 13, 185-190. |
|
(18) |
Rhodes R.C., Belasco I.J., and Pease H.L., (1970) 'Determination of mobility and adsorption of agrochemicals in soils'. J.Agric.Food Chem., 18, 524-528. |
|
(19) |
Russell M.H., (1995), 'Recommended approaches to assess pesticide mobility in soil' in Environmental Behavior of Agrochemicals (ed. T.R. Roberts and P.C. Kearney). John Wiley & Sons Ltd. |
|
(20) |
Esser H.O., Hemingway R.J., Klein W., Sharp D.R., Vonk J.W. and Holland P.T., (1988), 'Recommended approach to the evaluation of the environmental behavior of pesticides', IUPAC Reports on Pesticidas (24). Pure Appl. Chem., 60, 901-932. |
|
(21) |
Guth J.A., Burkhard N., and D.O. Eberle, (1976), 'Experimental models for studying the persistence of pesticides in soils'. Proc. BCPC Symposium: Persistence of Insecticides and Herbicides, pp 137-157, BCPC, Surrey, UK. |
|
(22) |
Furminge C.G.L., and Osgerby J.M., (1967), 'Persistence of herbicides in soil'. J. Sci. Fd Agric, 18, 269-273. |
|
(23) |
Burkhard N., and Guth J.A., (1981). 'Chemical hydrolysis of 2-Chloro-4,6-bis(aikylamino)-l,3,5-triazine herbicides and their breakdown in soil under the influence of adsorption'. Pestic. Sci. 12, 45-52. |
|
(24) |
Guth J.A., Gerber H.R., and Schlaepfer T., (1977). 'Effect of adsorption, movement and persistence on the biological availability of soil-applied pesticides'. Proc. Br. Crop Prot. Conf., 3, 961-971. |
|
(25) |
Osgerby J.M., (1973), 'Process affecting herbicide action in soil'. Pestic. Sci., 4, 247-258. |
|
(26) |
Guth J.A., (1972), 'Adsorptions- und Einwascheverhalten von Pflanzenschutzmitteln in Böden'. Schr. Reihe Ver. Wass.-Boden-Lufthyg. Berlm-Dahlem, Heft 37, 143-154. |
|
(27) |
Hamaker J.W., 1975), The interpretation of soil leaching experiments', in Environmental Dynamics of Pesticides (eds R. Flaque and V.H. freed). pp. 1 3 5-172, Plenum Press, NY. |
|
(28) |
Helling C.S., (1971). 'Pesticide mobility in soils'. Soil Sci. Soc. Amer. Proc. 35, 732-210. |
|
(29) |
Hamaker J.W., (1972), 'Diffusion and volatilization' in Organic chemicals in the soil environment (C.A.I. Goring and J.W. Hamaker eds), Vol. I, 49-143. |
|
(30) |
Burkhard N. and Guth J.A., (1981), 'Rate of volatilisation of pesticides from soil surfaces; Comparison of calculated results with those determined in a laboratory model system'. Pestic. Sci. 12, 37-44. |
|
(31) |
Cohen S.Z., Creeger S.M., Carsel R.F., and Enfield C.G., (1984), 'Potential pesticide contamination of groundwater from agricultural uses', in Treatment and Disposal of Pesticide Wastes, pp. 297-325, Acs Symp. Ser. 259, American Chemical Society, Washington, DC. |
|
(32) |
Gustafson D.I., (1989), 'Groundwater ubiquity score: a simple method for assessing pesticide leachability'. J. Environ. Toxic. Chem., 8(4). 339-357. |
|
(33) |
Leistra M., and Dekkers W.A., (1976). 'Computed effects of adsorption kinetics on pesticide movement in soils'. J. of Soil Sci., 28, 340-350. |
|
(34) |
Bromilov R.H., and Leistra M. (1980), 'Measured and simulated behavior of aldicarb and its oxydation producís in fallow soils'. Pest. Sci., 11, 389-395. |
|
(35) |
Green R.E., and Karickoff S.W., (1990), 'Sorption estimates for modeling', in Pesticides in the Soil Environment: Process, Impacts and Modeling (ed. H.H. Cheng). Soil Sci. Soc. Am., Book Series no. 2, pp. 80-101. |
|
(36) |
Lambert S.M., (1967), 'Functional relationship between sorption in soil and chemical structure'. J. Agri. Food Chem., 15, 572-576. |
|
(37) |
Hance R.J., (1969), 'An empirical relationship between chemical structure and the sorption of some herbicides by soils'. J. Agri. Food Chem., 17, 667-668. |
|
(38) |
Briggs G.G. (1969), 'Molecular structure of herbicides and their sorption by soils'. Nature, 223, 1288. |
|
(39) |
Briggs G.G. (1981). 'Theoretical and experimental relationships between soil adsorption, octanol-water partition coeffícients, water solubilities, bioconcentration factors, and the parachor'. J. Agric. Food Chem., 29, 1050-1059. |
|
(40) |
Sabljic A., (1984), 'Predictions of the nature and strength of soil sorption of organic polutance by molecular topology'. J. Agrie. Food Chem., 32, 243-246. |
|
(41) |
Bailey G.W., and White J.L., (1970), 'Factors influencing the adsorption, desorption, and movement of pesticides in soil'. Residue Rev., 32, 29-92. |
|
(42) |
Bailey G.W., J.L. White and Y. Rothberg. (1968), 'Adsorption of organic herbicides by montomorillonite: Role of pH and chemical character of adsorbate'. Soil Sci. Soc. Amer. Proc. 32:222-234. |
|
(43) |
Karickhoff S.W., (1981) 'Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils'. Chemosphere 10, 83 3-846. |
|
(44) |
Paya-Pérez A., Riaz M. and Larsen B., (1989), 'Soil Sorption of 6 Chlorobenzenes and 20 PCB Congeners'. Environ. Toxicol. Safety 21, 1-17. |
|
(45) |
Hamaker J.W., and Thompson J.M, (1972). 'Adsorption in organic cbemicals' in Organic Chemicals ir. the Soil Environment (Goring C.A.I. and Hamaker J.W., eds), Vol I and II, Marcel Dekker, Inc., New York. NY, 1972, pp. 49-143. |
|
(46) |
Deli J., and Warren G.F., 1971, 'Adsorption, desorption and leaching of diphenamid in soils'. Weed Sci. 19:67-69. |
|
(47) |
Chu-Huang Wu, Buehring N., Davinson J.M, and Santelmann, (1975), 'Napropamide Adsorption, desorption and Movement in soils'. Weed Science, Vol. 23, 454-457. |
|
(48) |
Haues M.H.B., Stacey M., and Thompson J.M., (1968) 'Adsorption of s-triazine herbicides by soil organic preparations' in Isotopes and Radiation in Soil Organic Studies, p. 75, International. Atomic Energy Agency, Vienna. |
|
(49) |
Pionke H.B., and Deangelis R.J. (1980), 'Methods for distributing pesticide loss in field run-off between the solution and adsorbed phase', CREAMS. in A Field Scale Model for Chemicals, Run-off and Erosión from Agricultural Management Systems, Chapter 19. Vol. III: Supporting Documentation, USDA Conservation Research report. |
|
(50) |
ISO Standard Compendium Environment: Soil Quality — General aspeets; chemical and physical methods of analysis; biological methods of analysis. First Edition (1994). |
|
(51) |
Scheffer F. and Schachtschabel, Lehrbuch der Bodenkunde, F., Enke Verlag. Stuttgart (1982), 11 th edition. |
|
(52) |
Black, Evans D.D., White J.L., Ensminger L.E., and Clark F.E., eds. «Methods of Soil Analysis», Vol 1 and 2, American Society of Agronomy, Madison, WI, 1982. |
|
(53) |
ISO/DIS 10381-1 Soil Quality — Sampling — Part 1: Guidance on the design of sampling programmes. |
|
(54) |
ISO/DIS 10381-2 Soil Quality — Sampling — Part 2: Guidance on sampling techniques. |
|
(55) |
ISO/DIS 10381-3 Soil Quality — Sampling — Part 3: Guidance on safety of sampling. |
|
(56) |
ISO/DIS 10381-4 Soil Quality — Sampling — Part 4: Guidance on the investigation of natural and cultivated soils. |
|
(57) |
ISO/DIS 10381-5 Soil Quality — Sampling — Part 5: Guidance on the investigation of soil contamination of urban and industrial sites. |
|
(58) |
ISO 10381-6, 1993: Soil Quality — Sampling — Part 6: Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory. |
|
(59) |
Green R.E., and Yamane V.K., (1970) «Precision in pesticide adsorption measurements». Soil Sci. Am. Proc, 34, 353-354. |
|
(60) |
Grover R., and Hance R.J. (1970), «Effect of ratio of soil to water on adsorption of linuron and atrazine». Soil Sci., 109-138. |
|
(61) |
Boesten, J.J.T.I, 'Influence of soil/liquid ratio on the experimental error of sorption coefficients in pesticide/soil system'. Pest. Sci. 1990, 30, 31-41. |
|
(62) |
Boesten, J.J.T.I. «Influence of soil/liquid ratio on the experimental error of sorption coefficients in relation to OECD guideline 106» Proceedings of 5th international workshop on environmental behaviour of pesticides and regulatory aspects, Bmssels, 26-29 April 1994. |
|
(63) |
Bastide J., Cantier J.M., et Coste C, (1980), «Comportement de substances herbicides dans le sol en fonction de leur structure chimique». Weed Res. 21, 227-231. |
|
(64) |
Brown D.S., and Flagg E.W., (1981), «Empirical prediction of organic pollutants sorption in natural sediments»). Environ.Qual., 10(3), 382-386. |
|
(65) |
Chiou C.T., Porter P.E., and Schmedding D.W., (1983), «Partition equilibria of non-ionic organic compounds between soil organic matter and water». Environ. Sci. Technol., 17(4), 227-231. |
|
(66) |
Gerstl Z., and Mingelgrin U., (1984), 'Sorption of organic substances by soils and sediments'. J. Environm. Sci. Health, B19 (3), 297-312. |
|
(67) |
Vowles P.D., and Mantoura R.F.C., (1987), 'Sediment-water partition coefficient and HPLC retention factors of aromatic hydrocarbons'. Chemosphere, 16(1), 109-116. |
|
(68) |
Lyman W.J., Reehl W.F.and Rosenblatt D.H. (1990). Handbook of Chemical Property Estimation Methods. Environmental Behaviour of Organic Compounds. American Chemical Society, Washington DC. |
|
(69) |
Keniga E.E., and Goring, C.A.I. (1980). «Relationship between water solubility, soil sorption, octanol-water partitioning and concentration of chemicals in the biota» in Aquatic Toxicology (eds J.G. Eaton, et al.), pp.78-115, ASTM STP 707, Philadelphia. |
|
(70) |
Chiou C.T., Peters L.J., and Freed V.H., (1979), «A physical concept of soil-water equilibria for non-ionic organic compounds». Science, Vol. 206, 831-832. |
|
(71) |
Hassett J.J., Banwart W.I., Wood S.G., and Means J.C., (1981). 'Sorption of/-Naphtol: implications concerning the limits of hydrophobic sorption'. Soil Sci. Soc. Am. J. 45, 38-42. |
|
(72) |
Karickhoff S.W., (1981), «Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils». Chemosphere, Vol. 10(8), 8 3 3-846. |
|
(73) |
Moreale A., van Bladel R.. (1981), 'Adsorption de 13 herbicides et insecticides par le sol. Relation solubilité — reactivité. Revue de l'Agric. 34 (4), 319-322'. |
|
(74) |
Müller M., Kördel W. (1996), «Comparison of screening methods for the determination/estimation of adsorption coefficients on soil». Chemosphere. 32(12), 2493-2504. |
|
(75) |
Kördel \V., Kotthoff G.. Müller M. (1995), 'HPLC — screening method for the determination of the adsorption coefficient on soil — results of a ring test'. Chemosphere 30 (7), 1373-1 3S4. |
|
(76) |
Kördel W., Stutte J., Kotthoff G. (1993), 'HPLC — screening method for the determination of the adsorption coefficient on soil — comparison of different stationary phases'. Chemosphere 27 (12), pp. 2341-2352. |
|
(77) |
Hance, R.J., (1967), 'The Speed of Attainment of Sorption Equilibria in Some Systems Involving Herbicides'. Weed Research, Vol. 7, pp. 29-36. |
|
(78) |
Koskinen W.C., and Harper S S., (1990), 'The retention processes: mechanisms' in Pesticides in the Soil Environment: Processes, Impacts and Modelling (ed. H.H. Cheng). Soil Sci. Soc. Am. Book Series, No. 2, Madison, Wisconsin. |
|
(79) |
Cohen S.Z., Creeger S.M., Carsel R.F., and Enfield C.G. (1984), 'Potential pesticide contamination of groundwater from agricultural uses', in Treatment and Disposal of Pesticide Wastes, pp.297-325, ACS Symp. Ser. 259, American Chemical Society, Washington, DC. |
|
(80) |
Giles C.H., (1970), 'Interpretation and use of sorption isotherms' in Sorption and Transport Processes in Soils. S.C.I. Monograph No. 37, pp. 14-32. |
|
(81) |
Giles, C.H.; McEwan J.H.; Nakhwa, S.N. and Smith, D, (1960), 'Studies in adsorption: XI, A system of classification of solution adsorption isotherms and its use in the diagnosis of adsorption mechanisms and in measurements of pesticides surface areas of soils'. J. Chem. Soc, pp. 3973-93. |
|
(82) |
Calvet R., Tercé M, and Arvien J.C., (1980), 'Adsorption des pesticides par les sols et leurs constituants: 3. Caractéristiques générales de l'adsorption'. Ann. Agron. 31: pp. 239-251. |
|
(83) |
Bedbur E., (1996), 'Anomalies in the Freundlich equation', Proc. COST 66 Workshop, Pesticides in soil and the environment, 13-15 May 1996, Stratford-upon-Avon, UK. |
|
(84) |
Guth, J.A., (1985), 'Adsorption/desorption', in Joint International Symposium, Physicochemical Properties and their Role in Environmental Hazard Assessment, July 1-3, Canterbury, UK. |
|
(85) |
Soil Texture Classification (US and FAO systems): Weed Science, 3 3, Suppl. 1 (1985) and Soil Sci. Soc. Amer. Proc. 26:305 (1962). |
Apéndice 1
Esquema de la prueba

Apéndice 2
INFLUENCIA DE LA EXACTITUD DEL MÉTODO ANALÍTICO Y DEL CAMBIO DE CONCENTRACIÓN SOBRE LA EXACTITUD DE LOS RESULTADOS DE LA ADSORCIÓN
A partir del cuadro siguiente (84), se ve que, cuando la diferencia entre la masa inicial (m0 = 110 μg) y la masa en el equilibrio [ = 100 μg] de la sustancia problema en la solución es muy pequeña, un error del 5 % en la medida de la concentración en el equilibrio origina un error del 50 % en el cálculo de la masa de la sustancia adsorbida en el suelo [
= 100 μg] de la sustancia problema en la solución es muy pequeña, un error del 5 % en la medida de la concentración en el equilibrio origina un error del 50 % en el cálculo de la masa de la sustancia adsorbida en el suelo [ ] y del 52,4 % en el cálculo de Kd.
] y del 52,4 % en el cálculo de Kd.
|
Cantidad de suelo |
msoil |
= 10 g |
|
Volumen de solución |
Vo |
= 100 cm3 |
|
|
FOR-L_2008142ES.01044401.notes.0187.xml.jpg
(μg) |
FOR-L_2008142ES.01044401.notes.0188.xml.jpg
(μg cm-3) |
R |
FOR-L_2008142ES.01044401.notes.0189.xml.jpg
(μg) |
FOR-L_2008142ES.01044401.notes.0190.xml.jpg
(μg g-1) |
Rt |
Kd* |
Rt |
|
|
PARA A = 9 % |
|||||||
|
m0 = 110 μg o Co= 1,100 μg/cm3 |
100 |
1,000 |
Valor verd. |
10 |
1,00 |
Valor verd. |
1 |
|
|
101 |
1,010 |
1 % |
9 |
0,90 |
10 % |
0,891 |
10,9 % |
|
|
105 |
1,050 |
5 % |
5 |
0,50 |
50 % |
0,476 |
52,4 % |
|
|
109 |
1,090 |
9 % |
1 |
0,10 |
90 % |
0,092 |
90,8 % |
|
|
|
PARA A = 55 % |
|||||||
|
m0 = 110 μg o Co = 1,100 μg/cm3 |
50,0 |
0,500 |
Valor verd. |
60,0 |
6,00 |
Valor verd. |
12,00 |
|
|
50,5 |
0,505 |
1 % |
59,5 |
5,95 |
0,8 % |
11,78 |
1,8 % |
|
|
52,5 |
0,525 |
5 % |
57,5 |
5,75 |
4,0 % |
10,95 |
8,8 % |
|
|
55,0 |
0,550 |
10 % |
55,0 |
5,50 |
8,3 % |
10,00 |
16,7 % |
|
|
|
PARA A = 99 % |
|||||||
|
m0 = 110 μg o Cn = 1,100 μg/cm3 |
1,100 |
0,011 |
Valor verd. |
108,9 |
10,89 |
Valor verd. |
990 |
|
|
1,111 |
0,01111 |
1 % |
108,889 |
10,8889 |
0,01 % |
980 |
1,0 % |
|
|
1,155 |
0,01155 |
5 % |
108,845 |
10,8845 |
0,05 % |
942 |
4,8 % |
|
|
1,21 |
0,0121 |
10 % |
108,790 |
10,8790 |
0,10 % |
899 |
9,2 % |
donde:
|
|
= |
|
|
|
= |
masa de la sustancia problema en la fase edáfica en el equilibrio (μg), |
|
|
= |
masa de la sustancia problema en la fase acuosa en el equilibrio (μg), |
|
|
= |
contenido de la sustancia problema en la fase edáfica en el equilibrio (μg g-1), |
|
|
= |
concentración en masa de la sustancia problema en la fase acuosa en el equilibrio (μg cm-3), |
|
R |
= |
error analítico en la determinación de |
|
R: |
= |
error calculado debido al error analítico R. |
Apéndice 3
TÉCNICAS DE ESTIMACIÓN DE Kd
|
1. |
Las técnicas de estimación permiten dar un valor de Kd a partir de correlaciones observadas con, por ejemplo, valores de POW (12) (39) (63) (64) (65) (66) (67) (68), datos de hidrosolubilidad (12) (19) (21) (39) (68) (69) (70) (71) (72) (73), o datos de polaridad obtenidos por aplicación de HPLC en fase inversa (74) (75) (76). Como se muestra en los cuadros 1 y 2, a partir de estas ecuaciones se calcula Koc o Kom y después, indirectamente, se obtiene Kd con las ecuaciones:
|
|
2. |
Estas correlaciones se basan en dos suposiciones: 1) la materia orgánica del suelo es el factor que más influye en la adsorción de una sustancia, y 2) las interacciones implicadas son principalmente no polares. En consecuencia, estas correlaciones: 1) no son aplicables a las sustancias polares (o solo lo son de forma limitada), y 2) no son aplicables a los casos en que el contenido de materia orgánica del suelo es muy pequeño (12). Por otra parte, aunque se han encontrado correlaciones satisfactorias entre Pow y adsorción (19), no puede decirse lo mismo de la relación entre hidrosolubilidad y grado de adsorción (19) (21): en este sentido, los estudios son muy contradictorios. |
|
3. |
En los cuadros 1 y 2 se dan ejemplos de correlaciones del coeficiente de adsorción con el coeficiente de reparto octanol-agua y con la hidrosolubilidad, respectivamente. Cuadro 1 Ejemplos de correlaciones entre el coeficiente de distribución de la adsorción y el coeficiente de reparto octanol-agua; pueden verse más ejemplos en (12) (68)
Cuadro 2 Ejemplos de correlaciones entre el coeficiente de distribución de la adsorción y la hidrosolubilidad; pueden verse más ejemplos en (68) (69)
|
Apéndice 4
CÁLCULOS PARA DEFINIR LAS CONDICIONES DE LA CENTRIFUGACIÓN
|
1. |
El tiempo de centrifugación viene dado por la siguiente fórmula, suponiendo partículas esféricas:
Con fines de simplificación, todos los parámetros se dan en unidades ajenas al SI (g, cm), donde:
En la práctica general, se utiliza un tiempo doble del calculado para conseguir una separación completa. |
|
2. |
La ecuación (1) puede simplificarse más si consideramos que la viscosidad (η) y la densidad (ρaq) de la solución son iguales a la viscosidad y la densidad del agua a 25 oC: por tanto, η = 8,95 × 10-3 g s-1 cm-1 and ρaq = 1,0 g, cm-3. Entonces, el tiempo de centrifugación se obtiene con la ecuación 2):
|
|
3. |
En la ecuación 2 se ve que hay dos parámetros importantes para definir las condiciones de la centrifugación, es decir, el tiempo (t) y la velocidad (rpm) necesarios para conseguir la separación de las partículas de un tamaño determinado (en nuestro caso, de 0,1 μm de radio): 1) la densidad del suelo, y 2) la altura de la mezcla en el tubo de centrífuga (Rb-Rt), es decir, la distancia que recorre una partícula de suelo desde el nivel superior de la solución hasta el fondo del tubo: evidentemente, con un volumen determinado, la altura de la mezcla en el tubo dependerá del cuadrado del radio del tubo. |
|
4. |
La figura 1 presenta las variaciones del tiempo de centrifugación (1) frente a la velocidad de centrifugación (rpm) con diferentes densidades de suelo (ρs) (figura 1a) y diferentes alturas de la mezcla en los tubos de centrífuga (figura 2a). En la figgura 1a queda de manifiesto la influencia de la densidad del suelo: por ejemplo, con una centrifugación clásica de 3 000 rpm el tiempo de centrifugación es de aproximadamente 240 minutos para una densidad de suelo de 1,2 g cm-3, mientras que es de solo 50 minutos para 2,0 g cm-3. Análogamente, según la figura 1b, con una centrifugación clásica de 3 000 rpm el tiempo de centrifugación es de aproximadamente 50 minutos para una altura de la mezcla de 10 cm y de solo 7 minutos para una altura de 1 cm. No obstante, es importante encontrar la relación óptima entre la centrifugación que requiera la menor altura posible y una manipulación fácil para el experimentador al separar las fases tras la centrifugación. |
|
5. |
Por otra parte, al definir las condiciones experimentales para la separación de las fases suelo/solución, es importante considerar la posible existencia de una tercera «pseudofase», los coloides. Estas partículas, de tamaño inferior a 0,2 μm, pueden afectar considerablemente a todo el mecanismo de la adsorción de una sustancia en una suspensión de suelo. Cuando se realiza la centrifugación según se describe anteriormente, los coloides se quedan en la fase acuosa y se someten a análisis junto con la fase acuosa, con lo que se pierde la información sobre su influencia. Si el laboratorio donde se realiza la prueba tiene equipos de ultracentrifugación o ultrafiltración, es posible estudiar con mayor profundidad la adsorción/desorción de una sustancia en el suelo, con información sobre la adsorción de la sustancia en los coloides. En este caso, para separar las 3 fases (suelo, coloides, solución) debe realizarse una ultracentrifugación a 60 000 rpm/minuto o una ultrafiltración con una porosidad de 100 000 Dalton. También hay que modificar en consonancia el protocolo del ensayo, a fin de determinar la sustancia en las 3 fases. |
Figura 1a.

Figura 1b.

Apéndice 5
CÁLCULO DE LA ADSORCIÓN A (%) Y DE LA DESORCIÓN D (%)
El esquema temporal del procedimiento es el siguiente:

A efectos de los cálculos, se supone que la sustancia problema es estable y no se adsorbe de forma importante a las paredes del recipiente.
ADSORCIÓN A (A%)
a) Método paralelo
La adsorción porcentual se calcula con cada tubo de ensayo (i) a cada tiempo (ti), según la ecuación:
|
(1) |
Los términos de esta ecuación pueden calcularse de la forma siguiente:
|
m0 = C0 · V0 (μg) |
(2) |
|
(3) |
donde:
|
|
= |
adsorción porcentual ( %) al tiempo ti, |
|
|
= |
masa de la sustancia problema en el suelo al tiempo ti en que se realiza el análisis (μg), |
|
m0 |
= |
masa de la sustancia problema en el tubo de ensayo, al inicio de la prueba (μg), |
|
C0 |
= |
concentración inicial en masa de la solución problema en contacto con el suelo (µg cm-3), |
|
|
= |
concentración en masa de la sustancia en la fase acuosa al tiempo ti en que se realiza el análisis (μg cm-3); esta concentración se determina analíticamente teniendo en cuenta los valores obtenidos en la prueba en blanco, |
|
V0 |
= |
volumen inicial de la solución problema en contacto con el suelo (cm3). |
Los valores de la adsorción porcentual ![]() o
o  se representan gráficamente frente al tiempo y se determina el tiempo al que se alcanza el equilibrio de sorción. En las figuras 1 y 2 se recogen ejemplos de tales gráficas.
se representan gráficamente frente al tiempo y se determina el tiempo al que se alcanza el equilibrio de sorción. En las figuras 1 y 2 se recogen ejemplos de tales gráficas.

b) Método en serie
Las siguientes ecuaciones tienen en cuenta que el procedimiento de adsorción se sigue con mediciones de la sustancia problema en pequeñas alícuotas de la fase acuosa a intervalos de tiempo especificados.
|
— |
Durante cada intervalo de tiempo, la cantidad de sustancia adsorbida en el suelo se calcula de la manera siguiente:
|
|
— |
El porcentaje de adsorción a cada intervalo de tiempo;
mientras que el porcentaje de adsorción
Los valores de la adsorción |
|
— |
Al tiempo de equilibrado teq:
|
Los parámetros usados en estas ecuaciones se definen de la forma siguiente:
|
|
= |
masa de la sustancia adsorbida en el suelo durante los intervalos de tiempo Δt1, Δt2,..., Δtn respectivamente (μg); |
|
|
= |
masa de la sustancia medida en una alícuota v |
|
|
= |
masa de la sustancia adsorbida en el suelo en el equilibrio de adsorción (μg); |
|
|
= |
masa de la sustancia en la solución en el equilibrio de adsorción (μg); |
|
|
= |
volumen de la alícuota donde se mide la sustancia problema (cm3); |
|
|
= |
adsorción porcentual correspondiente al intervalo de tiempo Δti (%); |
|
|
= |
adsorción porcentual en el equilibrio de adsorción ( %) |
DESORCIÓN D (%)
El tiempo t0 al que se inicia el experimento de cinética de desorción se considera que es el momento en que el volumen recuperado máximo de la solución de sustancia problema (después de alcanzarse el equilibrio de adsorción) es sustituido por un volumen igual de solución de CaCl20,01 M.
a) Método paralelo
A un tiempo ti, la masa de la sustancia problema se mide en la fase acuosa tomada del tubo i (![]() ), y la masa desor-bida se calcula con arreglo a la ecuación siguiente:
), y la masa desor-bida se calcula con arreglo a la ecuación siguiente:
|
|
(13) |
En el equilibrio de desorción ti = teq por lo que 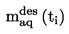 =
= 
La masa de la sustancia problema desorbida durante un intervalo de tiempo (Ati) se da en la ecuación:
|
|
(14) |
La desorción porcentual se calcula:
— a un tiempo ti a partir de la ecuación:
|
|
(15) |
— y durante un intervalo de tiempo (Δti) a partir de la ecuación:
|
|
(16) |
donde:
|
|
= |
desorción porcentual al tiempo ti ( %), |
|
|
= |
desorción porcentual correspondiente al intervalo de tiempo Δti ( %), |
|
|
= |
masa de la sustancia problema desorbida al tiempo ti (μg), |
|
|
= |
masa de la sustancia problema desorbida durante el intervalo de tiempo Δti (μg), |
|
|
= |
masa de la sustancia problema medida analíticamente al tiempo ti en un volumen de solución |
|
|
= |
masa de la sustancia problema que queda en solución una vez alcanzado el equilibrio de adsorción debido a la sustitución incompleta del volumen (μg). |
|
|
(17) |
|
|
= |
masa de la sustancia problema en la solución en el equilibrio de adsorción (μg), |
|
VR |
= |
volumen de sobrenadante retirado del tubo después de que se haya alcanzado el equilibrio de adsorción y sustituido por el mismo volumen de una solución de CaCl20,01 M (cm |
|
|
= |
volumen de solución tomado del tubo (i) para la medida de la sustancia problema, en el experimento de cinética de desorción (cm3). |
Los valores de desorción ![]() o
o ![]() (según las necesidades del estudio) se representan frente al tiempo y se determina el tiempo al que se alcanza el equilibrio de desorción.
(según las necesidades del estudio) se representan frente al tiempo y se determina el tiempo al que se alcanza el equilibrio de desorción.
b) Método en serie
Las siguientes ecuaciones tienen en cuenta que el procedimiento de adsorción presentado se ha seguido con mediciones de la sustancia problema en pequeñas alícuotas (![]() ) de la fase acuosa (método en serie en el punto 1.9, «Realización de la prueba»). Se supone que: a) el volumen de sobrenadante retirado del tubo tras el experimento de cinética de adsorción se ha sustituido con el mismo volumen de solución de CaCl20,01 M (VR), y b) el volumen total de fase acuosa en contacto con el suelo (VT) durante el experimento de cinética de desorción se mantiene constante y viene dado por la ecuación:
) de la fase acuosa (método en serie en el punto 1.9, «Realización de la prueba»). Se supone que: a) el volumen de sobrenadante retirado del tubo tras el experimento de cinética de adsorción se ha sustituido con el mismo volumen de solución de CaCl20,01 M (VR), y b) el volumen total de fase acuosa en contacto con el suelo (VT) durante el experimento de cinética de desorción se mantiene constante y viene dado por la ecuación:
|
|
(18) |
Al tiempo ti:
|
— |
La masa de la sustancia problema se mide en una pequeña alícuota
|
|
— |
Al equilibrio de desorción ti = teq, por lo que |
|
— |
La desorción porcentual
|
Al intervalo de tiempo (Δti):
Durante cada intervalo de tiempo, la cantidad de sustancia desorbida se calcula de la forma siguiente:
|
— |
para el primer intervalo de tiempo Δt1 = t1 —t0
|
|
— |
para el segundo intervalo de tiempo Δt2 = t2-t1
|
|
— |
para el enésimo intervalo de tiempo Δtn = tn-tn-1
|
Finalmente, la desorción porcentual a cada intervalo de tiempo, ![]() , se calcula con la ecuación siguiente:
, se calcula con la ecuación siguiente:
|
|
(24) |
mientras que la desorción porcentual ![]() al tiempo ti viene dada por la ecuación:
al tiempo ti viene dada por la ecuación:
|
|
(25) |
definiéndose los parámetros utilizados de la forma siguiente:
|
= |
masa de la sustancia que permanece adsorbida en el suelo tras los intervalos de tiempo Δt1, Δt2,..., Δtn respectivamente (μg), |
||
|
= |
masa de la sustancia que permanece adsorbida en el suelo tras los intervalos de tiempo Δt1, Δt2,..., Δtn respectivamente (μg), |
||
|
= |
masa de la sustancia medida en una alicuota |
||
|
VT |
= |
volumen total de la fase acuosa en contacto con el suelo durante et el experimento de cinética de desorcióh realizado con el método en serie (cm3), |
||
|
|
= |
masa de la sustancia problema que queda en solución una vez alcanzado el equilibr de adsorcióh debido a la sustitución incompleta del volumen (µg):
|
||
|
VR |
= |
volumen de sobrenadante retirado del tubo después de alcanzar el equilibrio de adsorción y sustituido por el mismo volumen de solución de CaCl20,01 M (cm3), |
||
|
|
= |
volumen de la alícuota tomada del tubo para el análisis (i), durante el experimento de cinética de desorción realizado con el método en serie (cm3):
|
Apéndice 6
ADSORCIÓN-DESORCIÓN EN SUELOS: FICHAS DE COMUNICACIÓN DE DATOS
Sustancia estudiada:
Suelo estudiado:
Contenido en masa seca del suelo (105 oC, 12 horas): … %
Temperatura: … oC
Adecuación del método analítico
|
Suelo pesado |
g |
|
|
Suelo: masa seca |
g |
|
|
Volumen de la solución de CaCl2 |
cm3 |
|
|
Concentración final nominal sol de la solución |
μg cm-3 |
|
|
Concentración final analítica sol de la solución |
μg cm-3 |
|
Principio del método analítico utilizado:
Calibración del método analítico:
Sustancia estudiada:
Suelo estudiado:
Contenido en masa seca del suelo (105 oC, 12 horas): … %
Temperatura: … oC
|
Metodología analítica seguida: |
Indirecta |
|
Paralela |
|
En serie |
|
|
|
Directa |
|
|
|
|
|
Prueba de adsorción: muestras de ensayo
|
|
Simbolo |
Unidades |
Tiempo equilibrado |
Tiempo de equilibrado |
Tiempo de equilibrado |
Tiempo de equilibrado |
||||
|
No tubo |
|
|
|
|
|
|
|
|
|
|
|
Suelo pesado |
— |
g |
|
|
|
|
|
|
|
|
|
Suelo: masa seca |
msoil |
g |
|
|
|
|
|
|
|
|
|
Volumen de agua en el suelo pesado (calculado) |
Vws |
cm3 |
|
|
|
|
|
|
|
|
|
Volumen de solución CaCl,0,01 M para equilibrar el suelo |
|
cm3 |
|
|
|
|
|
|
|
|
|
Volumen de solución madre |
|
cm3 |
|
|
|
|
|
|
|
|
|
Volumen total tic fase acuosa en contacto con el suelo |
Vo |
cm3 |
|
|
|
|
|
|
|
|
|
Concentración inicial de la solución problema |
Co |
μg cm-3 |
|
|
|
|
|
|
|
|
|
Masa de la sustancia problema ni inicio de la prueba |
m 0 |
μg |
|
|
|
|
|
|
|
|
|
Iras agitación y centrifugación |
||||||||||
|
MÉTODO INDIRECTO |
||||||||||
|
Método paralelo |
||||||||||
|
Concentración de la sustancia problema lase acuosa, incluida corrección por blanco |
|
μg cm-3 |
|
|
|
|
|
|
|
|
|
Método en serie |
||||||||||
|
Musa de la sustancia problema medida en la alícuota Va |
|
μg |
|
|
|
|
|
|
|
|
|
MÉTODO DIRECTO |
||||||||||
|
Masa de la sustancia problema adsorbida en el suelo |
|
μg |
|
|
|
|
|
|
|
|
|
Calculo de la adsorción |
||||||||||
|
Adsorción |
|
% |
|
|
|
|
|
|
|
|
|
|
|
% |
|
|
|
|
|
|
|
|
|
Me-Jias |
|
|
|
|
|
|
||||
|
Coeficiente |
Ke |
cm3 g-1 |
|
|
|
|
|
|
|
|
|
Medias |
|
|
|
|
|
|
||||
|
Coefidence |
Koc |
cm3 g-1 |
|
|
|
|
|
|
|
|
|
Medias |
|
|
|
|
|
|
Sustancia estudiada:
Suelo estudiado:
Contenido en masa seca del suelo (105 oC, 12 horas): … %
Temperatura: … oC
Prueba de adsorción: blancos y control
|
|
Símbolo |
Unidad |
Blanco |
Blanco |
Control |
|||
|
No tubo |
|
|
|
|
|
|
|
|
|
Suelo pesado |
|
g |
|
|
|
|
0 |
0 |
|
Cantidad de agua en el suelo pesado (calculada) |
|
cm3 |
|
|
|
|
— |
— |
|
Volumen añadido de solución CaCl20,01 M |
|
cm3 |
|
|
|
|
|
|
|
Volumen añadido de la solución madre de le sustancia problema |
|
cm3 |
0 |
0 |
|
|
|
|
|
Volumen total de fase acuosa (calculado) |
|
cm3 |
|
|
|
|
— |
— |
|
Concentración inicial de la sustancia problema en la fase acuosa |
|
μg cm-3 |
|
|
|
|
|
|
|
Tras agitación y centrifugación |
||||||||
|
Concentración en la fase acuosa |
|
μg cm-3 |
|
|
|
|
|
|
Nota: Añádanse columnas en caso necesario.
Sustancia estudiada:
Suelo estudiado:
Contenido en masa seca del suelo (105 oC, 12 horas): … %
Temperatura: … oC
Balance de masa
|
|
Símbolo |
Unidad |
|
|
|
|
|
No tubo |
|
|
|
|
|
|
|
Suelo pesado |
— |
g |
|
|
|
|
|
Suelo: masa seca |
msoil |
g |
|
|
|
|
|
Volumen de agua en suelo pesado (calculado) |
Vws |
ml |
|
|
|
|
|
Volumen sol de la solución de CaCl20,01 M para equilibrar el suelo |
|
ml |
|
|
|
|
|
Volumen de solución madre |
|
cm3 |
|
|
|
|
|
Volumen total de fase acuosa en contacto con el suelo |
V0 |
cm3 |
|
|
|
|
|
Concentración inicial de la solución problema |
C0 |
μg cm-3 |
|
|
|
|
|
Tiempo de equilibrado |
— |
h |
|
|
|
|
|
Tras agitación y centrifugación |
||||||
|
Concentración de la sustancia problema en fase acuosa en equilibrio adsorción, incluida corrección por blanco |
|
μg cm-3 |
|
|
|
|
|
Tiempo de equilibrado |
teq |
h |
|
|
|
|
|
1a dilución con solvente |
||||||
|
Volumen retirado fase acuosa |
Vrec |
cm3 |
|
|
|
|
|
Volumen añadido solvente |
ΔV |
cm3 |
|
|
|
|
|
1a extracción con solvente |
||||||
|
Señal analizada en fase solvente |
SE1 |
var. |
|
|
|
|
|
Concentración de la sustancia problema en solvente |
CE1 |
μg cm-3 |
|
|
|
|
|
Masa sustancia extraída del suelo y paredes recipiente |
mE1 |
μg |
|
|
|
|
|
2a dilución con solvente |
||||||
|
Volumen retirado solvente |
ΔV5 |
cm3 |
|
|
|
|
|
Volumen añadido solvente |
ΔV |
cm3 |
|
|
|
|
|
2a extracción con solvente |
||||||
|
Señal analizada en fase solvente |
SE2 |
var |
|
|
|
|
|
Concentración de la sustancia problema en sol-vente |
CE2 |
μg cm-3 |
|
|
|
|
|
Masa sustancia extraída del suelo y paredes recipiente |
mE2 |
μg |
|
|
|
|
|
Total masa sustancia problema extraída en dos pasos |
mE |
μg |
|
|
|
|
|
Balance de masa |
MB |
% |
|
|
|
|
Sustancia estudiada:
Suelo estudiado:
Contenido en masa seca del suelo (105 oC, 12 horas): … %
Temperatura: … oC
Isotermas de adsorción
|
|
Símbol |
Unidad |
|
|
|
|
|
|
|
|
|
No tubo |
|
|
|
|
|
|
|
|
|
|
|
Suelo pesado |
— |
g |
|
|
|
|
|
|
|
|
|
Suelo: masa seca |
E |
g |
|
|
|
|
|
|
|
|
|
Volumen de agua en suelo pesado (calculado) |
Vws |
cm3 |
|
|
|
|
|
|
|
|
|
Volumen de la solución de CaCl20,01 M para equilibrar el suelo |
|
cm3 |
|
|
|
|
|
|
|
|
|
Volumen de solución madre añadido |
|
cm3 |
|
|
|
|
|
|
|
|
|
Volumen total de fase acuosa en contacto con suelo (calculado) |
V0 |
cm3 |
|
|
|
|
|
|
|
|
|
Concentración solución |
C0 |
Μg cm-3 |
|
|
|
|
|
|
|
|
|
Tiempo de equilibrado |
— |
h |
|
|
|
|
|
|
|
|
|
Tras agitación y centrifugación |
||||||||||
|
Concentración de la sustan-cia en fase acuosa, incluida corrección por blanco |
|
μg cm-3 |
|
|
|
|
|
|
|
|
|
Temperatura |
|
oC |
|
|
|
|
|
|
|
|
|
Masa adsorbida por unidad suelo |
|
μg g-1 |
|
|
|
|
|
|
|
|
Análisis de regresión:
valor de KF ads:
valor de 1/n:
coeficiente de regresión r2:
Sustancia estudiada:
Suelo estudiado:
Contenido en masa seca del suelo (105 oC, 12 horas): … %
Temperatura: … oC
|
Metodología analítica seguida: |
Indirecta |
|
Paralela |
|
En serie |
|
Prueba de desorción
|
|
Símbolo |
Unidades |
Intervalo de tiempo |
Intervalo de tiempo |
Intervalo de tiempo |
Intervalo de tiempo |
|
|
No tubo de la fase de adsorción |
|
|
|
|
|
|
|
|
Masa de sustancia adsorbida en el suelo en el equilibrio de adsorción |
|
μg |
|
|
|
|
|
|
Volumen retirado de fase acuosa, sustituido por CaCl20,01 M |
VR |
cm3 |
|
|
|
|
|
|
Volumen total de fase acuosa en contacto con suelo |
PM SM |
V0 |
cm3 |
|
|
|
|
|
SM |
VT |
cm3 |
|
|
|
|
|
|
Masa de la sustancia problema que queda en la solución una vez alcanzado el equilibrio de adsorción debido a sustitución incompleta del volumen |
|
μg |
|
|
|
|
|
|
Cinética de desorción |
|||||||
|
Masa medida de sustancia desorbida del suelo al tiempo ti |
|
μg |
|
|
|
|
|
|
Volumen de solución tomado del tubo (i) para medir la sustancia problema |
PM |
V1 r |
cm3 |
|
|
|
|
|
SM |
|
cm3 |
|
|
|
|
|
|
Masa de sustancia desorbida del suelo al tiempo ti (calculada) |
|
μg |
|
|
|
|
|
|
Masa de sustancia desorbida del suelo durante el intervalo de tiempo Δti (calculada) |
|
μg |
|
|
|
|
|
|
Desorción porcentual |
|||||||
|
Desorción al tiempo ti |
|
% |
|
|
|
|
|
|
Desorción al intervalo de tiempo Δti |
|
% |
|
|
|
|
|
|
Coeficiente de desorción aparente |
|
|
|
|
|
|
PM: Método paralelo.
SM: Método en serie.
C.19. CALCULO DEL COEFICIENTE DE ADSORCIÓN (KOC ) EN SUELOS Y EN LODOS DE AGUAS RESIDUALES MEDIANTE CROMATOGRAFÍA LÍQUIDA DE ALTA RESOLUCIÓN (HPLC)
1. MÉTODO
El presente método de ensayo reproduce las directrices del documento TG 121 de la OCDE (2000).
1.1. INTRODUCCIÓN
El comportamiento que presentan los compuestos en cuanto a su adsorción en suelos o en lodos de agua; residuales puede describirse mediante ciertos parámetros, para cuya determinación experimental se puede aplicar el método de ensayo C.18. Un parámetro importante es el coeficiente de adsorción, definido como la relación entre la concentración del compuesto en la muestra de suelo o lodo y la concentración del mismo en la fase acuosa, cuando la adsorción llega al equilibrio. El coeficiente de adsorción normalizado con respecto a contenido de carbono orgánico presente en el suelo, Koc, constituye un útil indicador de la capacidad que presenta un producto químico para unirse a la materia orgánica de los suelos o los lodos de aguas residuales; este valor permite establecer comparaciones entre los diversos productos químicos. Se puede calcular este parámetro a través de correlaciones con la hidrosolubilidad y con el coeficiente de reparto n-octanol/agua (1) (2) (3) (4) (5) (6) (7).
El método experimental descrito en este ensayo hace uso de una técnica de HPLC para calcular el coeficiente de adsorción, Koc, en suelos y lodos de aguas residuales (8). La fiabilidad de los valores así obtenidos es superior a la de los cálculos efectuados por QSAR (9). Como método de cálculo, no puede sustituir completamente a los experimentos de equilibrio de lotes contemplados en el método de ensayo C.18. A pesar de ello, el Koc calculado puede ser útil para escoger los parámetros de ensayo adecuados en los estudios de adsorción/desorción que siguen el método de ensayo C.18 calculando los valores Kd (coeficiente de distribución) o Kf (coeficiente de adsorción de Freundlich) según la ecuación 3 (véase el punto 1.2 a continuación).
1.2. DEFINICIONES
Kd : coeficiente de distribución, que se define como la relación existente entre las concentraciones en el equilibrio (C) de una sustancia problema disuelta en un sistema de dos fases: un adsorbente (suelo o lodo) más una fase acuosa; es un valor adimensional, cuando las concentraciones en ambas fases se expresan como peso/peso. Si la concentración del compuesto en la fase acuosa viene expresada como peso/volumen, las unidades de Kd son ml g-1. Kd puede variar en función de las propiedades del adsorbente.
|
|
(1) |
donde:
|
Csoil |
= |
concentración de la sustancia problema en el suelo (soil), una vez alcanzado el equilibrio (μg . g-1) |
|
Csludge |
= |
concentración de la sustancia problema en el lodo (sludge), una vez alcanzado el equilibrio (μg . g-1) |
|
Caq |
= |
concentración de la sustancia problema en la fase acuosa, una vez alcanzado el equilibrio (μg . g-1) μg . ml-1). |
Kf: coeficiente de adsorción de Freundlich, que se define como la concentración de la sustancia problema en el suelo o en el lodo de aguas residuales (x/m) cuando la concentración en la fase acuosa, llegado el equilibrioaq, toma el valor 1; unidades: μg . g-1 de adsorbente. El valor puede variar en función de las propiedades del adsorbente.
|
|
(2) |
donde:
|
x/m |
= |
cantidad de sustancia problema x (μg g-1) adsorbida por masa de adsorbente m (g), en el equilibrio, |
|
1/n |
= |
pendiente de la isoterma de adsorción de Freundlich, |
|
Caq |
= |
concentración de la sustancia problema en la fase acuosa, en el equilibrio (μg ml-1). |
At
Cuando
Koc: coeficiente de distribución (Kd) o coeficiente de adsorción de Freundlich (Kf) normalizado con respecto al contenido de carbono orgánico (foc) de un adsorbente; en el caso particular de productos químicos no ionizados, resulta un indicador aproximado para conocer la magnitud de la adsorción de un compuesto y permite establecer comparaciones entre diversos productos químicos. Según sean las dimensiones de Kd y Kf, Koc puede ser adimensional o bien venir expresado en las unidades siguientes: ml . g-1 o μg . g-1 de materia orgánica.
|
|
(3) |
La relación entre los coeficientes Koc y Kd no siempre es lineal; así pues, la posible diferencia de los valores de Koc entre uno y otro suelo es muy reducida, en comparación con la de los valores Kd o Kf.
A partir del coeficiente de distribución másica (k'), empleando un gráfico de calibración de log k' frente a log Koc (registrado con los compuestos de referencia elegidos), se deduce el coeficiente de adsorción (Koc).
|
|
(4) |
donde:
|
tR |
= |
tiempo de retención (en el cromatograma de HPLC) del compuesto problema o del compuesto de referencia (minutos), |
|
t0 |
= |
tiempo de retención de la fase móvil en la columna de HPLC (minutos) (véase el punto 1.8.2). |
Pow coeficiente de reparto octanol/agua, que se define como el cociente entre la concentración de la sustancia disuelta en n-octanol y la concentración de la misma disuelta en la fase acuosa; se trata de un valor adimensional.
|
|
(5) |
1.3. COMPUESTOS DE REFERENCIA
Es necesario conocer, antes de aplicar el método, cuál es la fórmula estructural del compuesto, así como su pureza y su constante de disociación (en caso pertinente). Resulta útil contar con datos adicionales tales como la solubilidad en agua y, en disolventes orgánicos, el coeficiente de reparto octanol/agua y las características de hidrólisis.
A fin de establecer una correlación entre los datos de retención obtenidos mediante HPLC, relativos a la sustancia problema, y el coeficiente de adsorción de la misma, Koc, se debe trazar un gráfico de calibración (log Koc frente a log k'). Se empleará un mínimo de 6 puntos de referencia, al menos uno de ellos por encima y otro por debajo del valor previsto para la sustancia problema. La exactitud del método mejorará significativamente si se emplean compuestos de referencia que guarden una relación estructural con la sustancia problema. Si el analista no cuenta con este tipo de datos, deberá ser él quien elija los compuestos adecuados para la calibración. En tal caso, se optará por una serie más general de compuestos heterogéneos desde el punto de vista estructural. En el apéndice figura una lista de compuestos y de valores de Koc aplicables para los análisis de lodos de aguas residuales (cuadro 1) o de suelos (cuadro 3). La elección de otros compuestos para la calibración deberá ser justificada.
1.4. PRINCIPIO DEL MÉTODO DE ENSAYO
Se lleva a cabo una HPLC en columnas analíticas rellenas de una fase sólida de tipo cianopropilo, de las disponibles en el mercado, que contiene tanto grupos lipófilos como grupos polares. Se utiliza una fase estacionaria moderadamente polar basada en una matriz de sílice:
|
— O — Si |
— CH2 — CH2 — CH2 |
— CN |
|
silice |
grupo apolar separador |
grupo polar |
El principio del método de ensayo es similar al del método de ensayo A.8 (coeficiente de reparto, método de HPLC). Al ir atravesando la columna, junto con la fase móvil, la sustancia problema interacciona con la fase estacionaria. Como resultado del reparto entre la fase móvil y la fase estacionaria, la sustancia problema experimenta una retención. La composición ambifílica de la fase estacionaria (partes polares y partes apolares) permite una interacción de los diversos grupos polares o apolares de cada molécula similar a la que tiene lugar en el caso de la materia orgánica en matrices tales como lodos de aguas residuales o suelos. Este hecho posibilita establecer una relación entre el tiempo de retención en la columna y el coeficiente de adsorción en la materia orgánica.
El pH ejerce una significativa influencia sobre la adsorción, particularmente en el caso de los compuestos polares. En el caso de suelos de cultivo o tanques de plantas de tratamiento de aguas residuales, el pH suele estar comprendido entre 5,5 y 7,5. Cuando se desea analizar compuestos ionizables, se debe llevar a cabo un ensayo con la forma ionizada y otro con la no ionizada, empleando las soluciones tampón adecuadas, aunque solo en aquellos casos en que al menos el 10 % del compuesto analizado se disocie a un pH de entre 5,5 y 7,5.
Para la evaluación se utiliza exclusivamente la relación entre la retención en la columna de HPLC y el coeficiente de adsorción, de modo que no es preciso aplicar un método de análisis cuantitativo: tan solo es necesario determinar el tiempo de retención. Si se dispone de una serie adecuada de compuestos de referencia y es posible aplicar las condiciones experimentales normales, el método constituye un modo rápido y eficaz para calcular el coeficiente de adsorción, Koc.
1.5. APLICABILIDAD DEL ENSAYO
El método de HPLC es aplicable a aquellos productos químicos (marcados o no) para los cuales se disponga de un sistema de detección apropiado (por ejemplo, un espectrofotómetro, un detector de radiactividad) y que sean suficientemente estables durante todo el experimento. Puede resultar especialmente útil para analizar productos químicos cuyo estudio sea difícil mediante otros sistemas experimentales (compuestos volátiles, compuestos cuya solubilidad en agua no alcance una concentración que pueda determinarse desde el punto de vista analítico, compuestos con una elevada afinidad por la superficie de los sistemas de incubación). Se puede utilizar este método para analizar mezclas que originen al eluir bandas sin resolución. En este caso, se debe definir un margen de límites superior e inferior para los valores de log Koc correspondientes a los compuestos de la mezcla problema.
La presencia de impurezas puede dificultar a veces la interpretación de los resultados de la HPLC; no obstante, su importancia será de índole menor en la medida en que se cuente con análisis que permitan identificar con claridad la sustancia problema y distinguirla de las impurezas.
Se ha validado el uso del método para analizar las sustancias citadas en el cuadro 1 del apéndice; igualmente, el método ha sido aplicado al análisis de muchos otros productos químicos comprendidos en las familias químicas siguientes:
|
— |
aminas aromáticas (por ejemplo, trifluralina, 4-cloroanilina, 3,5-dinitroanilina, 4-metilanilina, N-metilani-lina, l-naftilamina), |
|
— |
ésteres de ácidos carboxílicos aromáticos (por ejemplo, benzoato de metilo, 3,5-dinitrobenzoato de etilo), |
|
— |
hidrocarburos aromáticos (por ejemplo, tolueno, xileno, etilbenceno, nitrobenceno), |
|
— |
ésteres de ácidos ariloxifenoxipropiónicos (por ejemplo, metil diclofop, etil fenoxaprop, P-etil fenoxaprop), |
|
— |
fungicidas con estructura de tipo imidazol o benzimidazol (por ejemplo, carbendazima, fuberidazol, tria-zóxido), |
|
— |
amidas de ácidos carboxílicos (por ejemplo, 2-clorobenzamida, N,N-dimetilbenzamida, 3,5-dinitrobenza-mida, N-metilbenzamida, 2-nitrobenzamida, 3-nitrobenzamida), |
|
— |
hidrocarburos clorados (por ejemplo, endosulfán, DDT, hexaclorobenceno, quintoceno, 1,2,3-tricloroben-ceno), |
|
— |
insecticidas organofosforados (por ejemplo, metil azinfos. disulfotón, fenamifos, isofenfos, pirazofos, sul-profos, triazofos), |
|
— |
fenoles (por ejemplo, fenol, 2-nitrofenol, 4-nitrofenol, pentaclorofenol, 2.4,6-triclorofenol, 1-naftol), |
|
— |
derivados de la fenilurea (por ejemplo, isoproturón, monolinurón, pencicurón), |
|
— |
colorantes (por ejemplo, Acid Yellow 219, Basic Blue 41, Direct Red 81), |
|
— |
hidrocarburos poliaromáticos (por ejemplo, acenafteno, naftaleno), |
|
— |
herbicidas con estructura de 1,3,5-triazina (por ejemplo, prometrina, propazina, simazina, terbutrina), |
|
— |
derivados de tipo triazol (por ejemplo, tebuconazol, triadimefon, tradimenol, triapentenol). |
No es aplicable el método a aquellas sustancias que den lugar a una reacción con el eluyente o con la fase estacionaria. Tampoco a aquellas sustancias que interaccionen con componentes inorgánicos de modo específico (por ejemplo, las que formen complejos en racimo con los minerales de las arcillas). Puede ocurrir que el método no sirva en el caso de tensioactivos, compuestos inorgánicos y ácidos y bases orgánicos moderados o fuertes. Es posible determinar valores de log Koc comprendidos entre 1,5 y 5,0. Para analizar compuestos ioni-zables, se debe emplear una fase móvil tamponada; no obstante, se tendrá cuidado de evitar que los componentes del tampón o la sustancia problema precipiten.
1.6. CRITERIOS DE CALIDAD
1.6.1. Exactitud
Normalmente, se puede calcular que el coeficiente de adsorción de una sustancia problema estará comprendido en el intervalo de ±0,5 unidades logarítmicas del valor determinado por el método de equilibrio de lotes (véase el cuadro 1 del apéndice). Es posible lograr una mayor exactitud si los compuestos de referencia empleados guardan una relación estructural con la sustancia problema.
1.6.2. Repetibilidad
Se debe efectuar al menos un duplicado de cada determinación. Los valores de log Koc obtenidos a partir de cada medición deben encontrarse en un intervalo de 0,25 unidades logarítmicas.
1.6.3. Reproducibilidad
La experiencia hasta ahora adquirida en la aplicación del método confirma la validez del mismo. Un estudio del método de HPLC, en el que se utilizaron 48 sustancias (plaguicidas en su mayoría) para las que se disponía de datos fiables respecto al Koc en suelos, dio como resultado un coeficiente de correlación R = 0,95 (10) (11).
Con objeto de mejorar y validar el método, se llevó a cabo un ensayo comparativo en el que participaron 11 laboratorios (12). Los resultados están recogidos en el cuadro 2 del apéndice.
1.7. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
1.7.1. Estimación preliminar del coeficiente de adsorción
Tanto el coeficiente de reparto octanol/agua Pow (= Kow) como, en cierta medida, la hidrosolubilidad pueden servir de indicadores del grado de adsorción, particularmente en el caso de compuestos no ionizados, de modo que pueden ser utilizados para prever la gama de valores. Existen numerosas correlaciones útiles publicadas, relativas a varios grupos de productos químicos (1) (2) (3) (4) (5) (6) (7).
1.7.2. Aparato
Se debe disponer de un cromatógrafo de líquidos equipado con una bomba continua y un detector adecuado. Es conveniente utilizar una válvula de inyección junto con un bucle al efecto. Se emplearán resinas comerciales con grupos cianopropílicos enlazados covalentemente a un sustrato de sílice (por ejemplo, Hypersil y Zor-bax CN). Se puede intercalar una precolumna del mismo material entre el sistema de inyección y la columna en la que se realiza el análisis. Puede existir una considerable variabilidad entre las columnas que procedan de diferentes proveedores, manifestada en su eficacia para lograr la separación. Se deberán lograr los siguientes coeficientes de distribución másica, k', expuestos aquí como valores orientativos: log k' > 0,0 cuando log Koc = 3,0, y log k' > -0,4 for cuando log Koc = 2,0, al emplear como fase móvil una mezcla de metanol/agua (55 %/45 %).
1.7.3. Fases móviles
Se han estudiado varias fases móviles: se recomienda utilizar las dos siguientes:
|
— |
metanol/agua (55 %/45 % v/v), |
|
— |
metanol/tampón de citrato 0,01M (pH = 6,0), (55 %/45 % v/v). |
Para preparar el disolvente de elución, se emplea una mezcla de metanol para HPLC y agua destilada o tampón de citrato. La mezcla se desgasifica antes de ser utilizada. Se efectuará una elución isocrática. En el caso de que las mezclas de metanol/agua no sean apropiadas, es posible investigar mezclas de otros disolventes con agua, por ejemplo etanol/agua o bien acetonitrilo/agua. En el caso de compuestos ionizables, se recomienda emplear una solución tampón a fin de estabilizar el pH. Se debe procurar evitar la precipitación de sales y el deterioro de la columna, situaciones posibles al utilizar algunas mezclas de fase orgánica + tampón.
No es posible incluir aditivos tales como reactivos tipo par iónico, ya que pueden alterar las características de adsorción de la fase estacionaria. Tales alteraciones de la fase estacionaria pueden ser irreversibles. Así pues, los experimentos en los que intervengan aditivos deben ser realizados en columnas independientes.
1.7.4. Solutos
Se utilizará la fase móvil para disolver tanto la sustancia problema como los compuestos de referencia.
1.8. REALIZACIÓN DEL ENSAYO
1.8.1. Condiciones de ensayo
Se registrará la temperatura a la que se realizan las mediciones. Es muy conveniente servirse de un compartimento termostatizado para la columna, para garantizar la constancia de las condiciones en que transcurre la calibración y las cromatografías para los cálculos o las mediciones de la sustancia problema.
1.8.2. Determinación del tiempo de retención de la fase móvil, t0
Para determinar el tiempo de retención de la fase móvil, t0, se pueden emplear dos métodos distintos (véase también el punto 1.2).
1.8.2.1. Determinación del tiempo de retención de la fase móvil, t0, mediante una serie homóloga
Se ha demostrado que este procedimiento da lugar a valores de t0 fiables y normalizados. Véase información detallada en el método de ensayo A.8: coeficiente de reparto (n-octanol/agua), método de HPLC.
1.8.2.2. Determinación del tiempo de retención de la fase móvil, t0, utilizando compuestos inertes no retenidos por la columna
Esta técnica consiste en inyectar soluciones de formamida, urea o nitrato de sodio. Se debe efectuar al menos un duplicado de cada medición.
1.8.3. Determinación de los tiempos de retención, tR
Se elegirán compuestos de referencia conforme a lo descrito en el punto 1.3. Se puede efectuar una inyección de los mismos a modo de patrón mixto para determinar sus tiempos de retención, siempre que se haya confirmado que no existen interferencias recíprocas que alteren estos valores. Periódicamente, al menos 2 veces al día, se llevará a cabo una calibración, para tener en cuenta las potenciales variaciones imprevistas en cuanto a la eficacia de la columna. Una práctica muy adecuada consiste en efectuar las inyecciones de calibración tanto antes como después de inyectar la sustancia problema, al objeto de confirmar que no hayan variado los tiempos de retención. Se inyecta por separado cada sustancia problema, en cantidades tan reducidas como sea posible (para evitar una sobrecarga de la columna) y se procede a determinar sus tiempos de retención.
Para lograr una mayor fiabilidad de las mediciones, se realizará al menos un duplicado de cada determinación. Los valores de log Koc obtenidos a partir de cada medición deben encontrarse en un intervalo de 0,25 unidades logarítmicas.
1.8.4. Evaluación
A partir del tiempo de retención de la fase móvil, t0, y de los tiempos de retención de los compuestos de referencia elegidos, tR, se calculan los coeficientes de distribución másica, k' aplicando la ecuación 4 (véase el punto 1.2). A continuación, se traza un gráfico en el que se representan los datos relativos al valor log k' de los compuestos de referencia frente a los respectivos valores log Koc procedentes de los experimentos de equilibrio de lotes, mostrados en los cuadros 1 y 3 del apéndice. Interpolando en este gráfico el valor log k' correspondiente a cada sustancia problema, se obtiene entonces el respectivo valor log Koc. Si los resultados reales indican que el log Koc de la sustancia problema se sale del intervalo de calibración, se debe repetir el ensayo, empleando en este caso otros compuestos de referencia más apropiados.
2. DATOS E INFORME
El informe debe incluir la información siguiente:
|
— |
identidad y pureza de la sustancia problema y de los compuestos de referencia, así como valores de pKa en caso de que sea relevante. |
|
— |
descripción del equipo y de las condiciones de trabajo, por ejemplo: tipo y dimensiones de la columna en la que se efectúa el análisis (y de la precolumna), medios de detección, fase móvil (proporción de los distintos componentes, pH), intervalo de temperaturas durante las mediciones, |
|
— |
tiempo de retención de la fase móvil y método empleado para determinarlo, |
|
— |
cantidades de la sustancia problema y de los compuestos de referencia introducidas en la columna, |
|
— |
tiempos de retención de los compuestos de referencia utilizados para la calibración, |
|
— |
representación gráfica y detalles relativos a la recta de regresión ajustada (log k' frente a log Koc), |
|
— |
datos sobre la retención media y valor log Koc calculado para el compuesto problema, |
|
— |
cromatogramas. |
3. REFERENCIAS
|
(1) |
W. J. Lyman, W. F. Reehl, D. H. Rosenblatt (ed). (1990). Handbook of chemical property estimation methods, Chap. 4, McGraw-Hill, New York. |
|
(2) |
J. Hodson, N. A. Williams (1988). The estimation of the adsorption coefficient (Koc) for soils by HPLC. Chemosphere, 17, 167. |
|
(3) |
G. G. Briggs (1981). Theoretical and experimental relationships between soil adsorption, octanol-water partition coefficients, water solubilities, bioconcentration factors, and the parachor. J. Agric. Food Chem., 29, pp. 1050-1059. |
|
(4) |
C. T. Chiou, P. E. Porter, D.W. Schmedding (1983). Partition equilibria of nonionic organic compounds between soil organic matter and water. Environ. Sci. Technol, 17, pp. 227-231. |
|
(5) |
Z. Gerstl, U. Mingelgrin (1984). Sorption of organic substances by soils and sediment. J. Environm. Sci. Health, B19, pp. 297-312. |
|
(6) |
C. T. Chiou, L. J. Peters, V. H. Freed (1979). A physical concept of soil water equilibria for nonionic organic compounds, Science, 106, pp. 831-832. |
|
(7) |
S. W. Karickhoff (1981). Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils. Chemosphere, 10, pp. 83 3-846. |
|
(8) |
W. Kördel, D. Hennecke, M. Herrmann (1997). Application of the HPLC-screening method for the determination of the adsorption coefficient on sewage sludges. Chemosphere, 35(1/2), pp. 121-128. |
|
(9) |
M. Mueller, W. Kördel (1996). Comparison of screening methods for the estimation of adsorption coefficients on soil. Chemosphere, 32(12), pp. 2493-2504. |
|
(10) |
W. Kördel,). Stutte, G. Kotthoff (1993). HPLC-screening method for the determination of the adsorption coefficient in soil-comparison of different stationary phases, Chemosphere, 27(12), pp. 2341-2352. |
|
(11) |
B. von Oepen, W. Kördel, W. Klein (1991). Sorption of nonpolar and polar compounds to soils: Processes, measurements and experience with the applicability of the modified OECD Guideline 106, Chemosphere, 22, pp. 285-304. |
|
(12) |
W. Kördel, G. Kotthoff, J. Müller (1995). HPLC-screening method for the determination of the adsorption coefficient on soil-results of a ring test. Chemosphere, 30(7), pp. 1373-1384. |
Apéndice
Cuadro 1
Comparación entre los valores de Koc observados en suelos y en lodos de aguas residuales y los datos calculados aplicando el método de HPLC (29) (30)
|
Compuesto |
No CAS |
log Koc (lodos) |
log Koc (HPLC) |
Δ |
log KOC (suelos) |
Log KOC (HPLC) |
Δ |
|
Atrazina |
1912-24-9 |
1,66 |
2,14 |
0,48 |
1,81 |
2,20 |
0,39 |
|
Linurón |
330-55-2 |
2,43 |
2,96 |
0,53 |
2,59 |
2,89 |
0,30 |
|
Fentión |
55-38-9 |
3,75 |
3,58 |
0,17 |
3,31 |
3,40 |
0,09 |
|
Monurón |
150-68-5 |
1,46 |
2,21 |
0,75 |
1,99 |
2,26 |
0,27 |
|
Fenantreno |
85-01-8 |
4,35 |
3,72 |
0,63 |
4,09 |
3,52 |
0,57 |
|
Benzoato de fenilo |
93-99-2 |
3,26 |
3,03 |
0,23 |
2,87 |
2,94 |
0,07 |
|
Benzamida |
55-21-0 |
1,60 |
1,00 |
0,60 |
1,26 |
1,25 |
0,01 |
|
4-Nitrobenzamida |
619-80-7 |
1,52 |
1,49 |
0,03 |
1,93 |
1,66 |
0,27 |
|
Acetanilida |
103-84-4 |
1,52 |
1,53 |
0,01 |
1,26 |
1,69 |
0,08 |
|
Anilina |
62-53-3 |
1,74 |
1,47 |
0,27 |
2,07 |
1,64 |
0,43 |
|
2,5-Dichloroanilina |
95-82-9 |
2,45 |
2,59 |
0,14 |
2,55 |
2,58 |
0,03 |
Cuadro 2
Resultados de un estudio comparativo, realizado en diferentes laboratorios para mejorar y validar el método de HPLC (31) (11 laboratorios participantes)
|
Compuesto |
No CAS |
log Koc |
Koc |
log Koc |
|
(OCDE 106) |
(método de HPLC) |
|||
|
Atrazina |
1912-24-9 |
1,81 |
78 ± 16 |
1,89 |
|
Monuron |
150-68-5 |
1,99 |
100 ± 8 |
2,00 |
|
Triapentenol |
77608-88-3 |
2,37 |
292 ± 58 |
2,47 |
|
Linuron |
330-55-2 |
2,59 |
465 ± 62 |
2,67 |
|
Fention |
55-38-9 |
3,31 |
2062 ± 648 |
3,31 |
Cuadro 3
Compuestos de referencia recomendados para aplicar el método de HPLC conforme a los datos de adsorción a los suelos
|
Compuesto de referencia |
No CAS |
log KOC (valores promedio), equilibrio de lotes |
No de datos (KOC) |
log de la desviación estándar |
Fuente |
|
Acetanilida |
103-84-4 |
1,25 |
4 |
0,48 |
(32) |
|
Fenol |
108-95-2 |
1,32 |
4 |
0,70 |
(32) |
|
2-Nitrobenzamida |
610-15-1 |
1,45 |
3 |
0,90 |
(33) |
|
N, N-dimetilbenzamida |
611-74-5 |
1,52 |
2 |
0,45 |
(32) |
|
4-Metilbenzamida |
619-55-6 |
1,78 |
3 |
1,76 |
(32) |
|
Benzoato de metilo |
93-58-3 |
1,80 |
4 |
1,08 |
(32) |
|
Atrazina |
1912-24-9 |
1,81 |
3 |
1,08 |
(34) |
|
Isoproturón |
34123-59-6 |
1,86 |
5 |
1,53 |
(34) |
|
3-Nitrobenzamida |
645-09-0 |
1,95 |
3 |
1,31 |
(33) |
|
Anilina |
62-53-3 |
2,07 |
4 |
1,73 |
(33) |
|
3,5-Dinitrobenzamida |
121-81-3 |
2,31 |
3 |
1,27 |
(33) |
|
Carbendazima |
10605-21-7 |
2,35 |
3 |
1,37 |
(34) |
|
Triadimenol |
55219-65-3 |
2,40 |
3 |
1,85 |
(34) |
|
Triazóxido |
72459-58-6 |
2,44 |
3 |
1,66 |
(34) |
|
Triazofos |
24017-47-8 |
2,55 |
3 |
1,78 |
(34) |
|
Linurón |
330-55-2 |
2,59 |
3 |
1,97 |
(34) |
|
Naftaleno |
91-20-3 |
2,75 |
4 |
2,20 |
(32) |
|
Endosulfán-diol |
2157-19-9 |
3,02 |
5 |
2,29 |
(34) |
|
Metiocarb |
2032-65-7 |
3,10 |
4 |
2,39 |
(34) |
|
Acid Yellow 219 |
63405-8 5-6 |
3,16 |
4 |
2,83 |
(32) |
|
1,2,3-Triclorobenceno |
87-61-6 |
3,16 |
4 |
1,40 |
(32) |
|
γ-HCH |
58-89-9 |
3,23 |
5 |
2,94 |
(32) |
|
Fention |
55-38-9 |
3,31 |
3 |
2,49 |
(34) |
|
Direct Red 81 |
2610-11-9 |
3,43 |
4 |
2,68 |
(32) |
|
Pirazofos |
13457-18-6 |
3,65 |
3 |
2,70 |
(34) |
|
α-Endosulfán |
959-98-8 |
4,09 |
5 |
3,74 |
(34) |
|
Metil diclofop |
51338-27-3 |
4,20 |
3 |
3,77 |
(34) |
|
Fenantreno |
85-01-8 |
4,09 |
4 |
3,83 |
(32) |
|
Basic Blue 41 (mezcla) |
26850-47-5 122,0-13-2 |
4,89 |
4 |
4,46 |
(32) |
|
DDT |
50-29-3 |
5,63 |
1 |
— |
(33) |
C.20. ENSAYO DE REPRODUCCIÓN EN DAPHNIA MAGNA
1. MÉTODO
El presente ensayo de toxicidad para la reproducción es copia de las directrices TG 211 de la OCDE (1998).
1.1. INTRODUCCIÓN
La principal finalidad de este ensayo es evaluar el efecto de los productos químicos sobre el resultado reproductor de Daphnia magna.
1.2. DEFINICIÓN Y UNIDADES
Parentales: hembras de Dafnia presentes al principio del ensayo y cuyo resultado reproductor constituye el objeto del presente estudio.
Descendientes: ejemplares de Dafnia jóvenes producidos por los parentales en el curso del ensayo.
Concentración mínima con efecto observado (LOEC): concentración de ensayo mínima a la cual se ha observado que la sustancia ejerce un efecto significativo sobre la reproducción y la mortalidad de los parentales (para p < 0,05) en comparación con el control y dentro del período de exposición establecido. Además, todas las concentraciones de ensayo superiores a la LOEC deben ejercer un efecto nocivo igual o mayor que el observado a dicha concentración. Si no se cumplen estas dos condiciones, es preciso dar una explicación completa del modo en que se ha elegido la LOEC (y, por tanto, la NOEC).
Concentración sin efecto observado (NOEC): concentración de ensayo inmediatamente inferior a la LOEC que, en comparación con el control, no ejerce ningún efecto estadísticamente significativo (p < 0,05) dentro del período de exposición establecido.
EC X : concentración de la sustancia de ensayo disuelta en agua que produce una reducción del x % en la reproducción de Daphnia magna dentro del período de exposición establecido.
Tasa intrínseca de aumento: medida del crecimiento de la población que integra el resultado reproductor y la mortalidad específica de la edad (20) (21) (22). En poblaciones en equilibrio estacionario es cero. En poblaciones en crecimiento es positiva, y negativa en las que disminuyen. Obviamente, toda tasa negativa es insostenible y conduce a la extinción.
Límite de detección: concentración mínima que puede detectarse, aunque no cuantificarse.
Límite de determinación: concentración mínima que puede medirse cuantitativamente.
Mortalidad: un animal se considera muerto cuando está inmóvil, es decir, cuando es incapaz de nadar o cuando no mueve los apéndices ni la región posterior del abdomen 15 segundos después de la agitación suave del recipiente de ensayo (si se usa otra definición, debe documentarse junto con la referencia correspondiente).
1.3. PRINCIPIO DEL MÉTODO DE ENSAYO
Las hembras jóvenes de Dafnia (los parentales), con una edad inferior a 24 horas al principio del ensayo, se exponen a la sustancia de ensayo añadida al agua a una gama de concentraciones. La duración del ensayo es de 21 días. Al final de este se evalúa el número total de descendientes vivos producidos por cada parental vivo. Esto significa que se excluyen de los cálculos los organismos inmaduros producidos por adultos que han muerto durante el ensayo. El resultado reproductor de los parentales se puede expresar de otras formas (por ejemplo, como número de descendientes vivos producidos por animal y por día a partir del primero en que se observa descendencia), pero estas deben documentarse además del número total de inmaduros producidos por parental vivo al final del ensayo. El resultado reproductor de los animales expuestos a la sustancia de ensayo se compara con el de los controles para determinar la concentración mínima con efecto observado (LOEC) y, por tanto, la concentración sin efecto observado (NOEC). Además, y en la medida de lo posible, los datos se analizan con ayuda de un modelo de regresión para estimar la concentración que provocaría una reducción del x % en el resultado reproductor (EC50, EC20 o EC10).
También hay que documentar la supervivencia de los parentales y el momento de la producción de la primera camada. Asimismo, pueden examinarse otros efectos vinculados con la sustancia sobre variables tales como el crecimiento (la longitud) y, posiblemente, la tasa intrínseca de aumento.
1.4. INFORMACIÓN SOBRE LA SUSTANCIA DE ENSAYO
Es preciso disponer de resultados de un ensayo de toxicidad aguda (véase la parte I del método C.2) realizado con Daphnia magna. El resultado puede ser útil para seleccionar un intervalo adecuado de concentraciones en los ensayos de reproducción. Deben conocerse la solubilidad en agua y la presión de vapor de la sustancia de ensayo, así como contar con un método analítico fiable para cuantificar la sustancia en las soluciones de ensayo con la eficiencia de recuperación y el límite de determinación establecidos.
Otros datos de la sustancia de ensayo potencialmente útiles para establecer las condiciones de ensayo son la fórmula estructural, la pureza de la sustancia, la estabilidad a la luz, la estabilidad en las condiciones del ensayo, los valores de pKa y Pow y los resultados del ensayo de biodegradabilidad fácil (véase el método C.4).
1.5. VALIDEZ DEL ENSAYO
Para que el ensayo sea válido, los controles deben cumplir los criterios de comportamiento siguientes:
|
— |
la mortalidad de los parentales (hembras de Dafnia) no debe ser superior al 20 % al final del ensayo, |
|
— |
el número medio de descendientes vivos producidos por cada parental superviviente al final del ensayo ha de ser ≥ 60. |
1.6. DESCRIPCIÓN DEL MÉTODO
1.6.1. Equipo
Los recipientes de ensayo y demás instrumentos que hayan de entrar en contacto con las soluciones de ensayo serán íntegramente de vidrio o de otro material químicamente inerte. Los recipientes de ensayo serán normalmente matraces de vidrio.
Además harán falta algunos de los instrumentos siguientes o todos ellos:
|
— |
aparato para medir el oxígeno (con microelectrodo o cualquier otro equipo que sea conveniente para medir el oxígeno disuelto en muestras de pequeño volumen), |
|
— |
aparato adecuado para el control de la temperatura, |
|
— |
medidor de pH, |
|
— |
aparato para determinar la dureza del agua, |
|
— |
aparato para determinar la concentración total de carbono orgánico (TOC) del agua o la demanda química de oxígeno (COD), |
|
— |
aparato adecuado para controlar el régimen de iluminación y medir la intensidad luminosa. |
1.6.2. Organismo de ensayo
La especie usada en el ensayo es Daphnia magna Straus. También pueden usarse otras especies de Dafnia siempre que reúnan los criterios de validez apropiados (el criterio de validez relativo al resultado reproductor de los controles debe ser relevante para la especie elegida). Si se usan otras especies de Dafnia, es preciso identificarlas con claridad y justificar su uso.
De preferencia, el clon debe identificarse genotípicamente. Las investigaciones realizadas (1) han demostrado que el comportamiento reproductor del clon A (procedente del IRCHA en Francia) (3) cumple siempre el criterio de validez de una media de ≥ 60 60 descendientes por parental que sobrevive cuando se cultiva en las condiciones descritas en este método. No obstante, son aceptables otros clones siempre que se demuestre que el cultivo de Dafnia cumple los criterios de validez del ensayo.
Al principio del ensayo, los animales tendrán una edad inferior a 24 horas y no deben ser progenie de primera camada. Han de proceder de una estirpe saludable (es decir, sin signos de agresión, tales como mortalidad elevada, presencia de machos y efipios, retraso de la producción de la primera camada, animales decolorados, etc.). Los animales de la estirpe deben mantenerse en condiciones de cultivo (luz, temperatura, medio, nutrición y animales por unidad de volumen) similares a las usadas en el ensayo. Cuando el medio de cultivo que vaya a usarse en el ensayo sea distinto del usado en el cultivo sistemático de Dafnia, es recomendable prever un período de aclimatación anterior al ensayo, por lo general de 3 semanas de duración (es decir, una generación), para evitar el estrés en los parentales.
1.6.3. Medio de ensayo
Es recomendable usar en este ensayo un medio definido completamente. Con ello se evita el uso de aditivos (tales como algas marinas, extracto de suelo, etc.) difíciles de caracterizar y aumentan en consecuencia las oportunidades de normalización entre laboratorios. Se ha observado que los medios Elendt M4 (4) y M7 (véase el apéndice 1) son adecuados para este propósito. No obstante, son aceptables medios distintos [por ejemplo, (5) (6)], siempre que se demuestre que el comportamiento del cultivo de Dafnia cumple los criterios de validez del ensayo.
Si se usan medios con aditivos sin definir, estos deben especificarse con claridad, y en el informe del ensayo hay que aportar información sobre su composición, en particular el contenido en carbono, pues podría contribuir a la dieta suministrada. Es recomendable determinar en la preparación madre del aditivo orgánico el carbono orgánico total (TOC), la demanda química de oxígeno (TOC) o las dos cosas, así como hacer una estimación de la contribución resultante al TOC y a la COD en el medio de ensayo. Conviene que la concentración de TOC en el medio (antes de incorporar las algas) sea inferior a 2 mg/l (7).
Si se ensayan sustancias que contengan metales, es importante tener en cuenta que las propiedades del medio de ensayo (por ejemplo, dureza, capacidad quelante) pueden afectar a la toxicidad de dichas sustancias. Por ello es deseable usar un medio totalmente definido. Sin embargo, en este momento, los únicos medios completamente definidos adecuados para el cultivo prolongado de Daphnia magna que se conocen son los Elendt M4 y M7. Ambos contienen el agente quelante EDTA. La práctica ha demostrado (2) que la «toxicidad aparente» del cadmio es generalmente inferior cuando el ensayo de reproducción se hace en medios M4 o M7 que cuando se hace en medios exentos de EDTA. Por tanto, M4 y M7 no son recomendables para ensayar sustancias con metales, y deben evitarse asimismo en estos casos otros medios que contengan agentes quelan-tes conocidos. En el caso de sustancias que contengan metales, puede ser aconsejable usar otros medios como, por ejemplo, agua dulce dura reconstituida según ASTM (7), que no contiene EDTA, enriquecida con extracto de algas marinas (8). Esta combinación de agua dulce dura reconstituida según ASTM y extracto de algas marinas es también apropiada para el cultivo prolongado y los ensayos con Daphnia magna (2), aun cuando ejerce una ligera acción quelante debida al componente orgánico del extracto de algas marinas.
Al principio del ensayo y durante este, la concentración de oxígeno disuelto debe ser superior a 3 mg/l. El pH debe estar comprendido entre 6 y 9, y normalmente no debe oscilar en más de 1,5 unidades en ninguno de los ensayos. Se recomienda utilizar una dureza superior a 140 mg/l (expresada como CaCO3). Los ensayos realizados con valores iguales o superiores a este han demostrado un comportamiento reproductor compatible con los criterios de validez (9) (10).
1.6.4. Soluciones de ensayo
Las soluciones de ensayo de las concentraciones elegidas se preparan por dilución de una solución de reserva. A ser posible, estas soluciones madre deben prepararse disolviendo la sustancia en medio de ensayo.
En algunos casos puede ser necesario usar disolventes o dispersantes orgánicos para obtener una solución madre de concentración adecuada, aunque es preciso evitar en la medida de lo posible el uso de esta clase de materiales. Son ejemplos de disolventes adecuados la acetona, el etanol, el metanol, la dimetilformamida y el trietilenglicol. Son dispersantes adecuados el Cremophor RH40, la metilcelulosa al 0,01 % y el HCO-40. En cualquier caso, la cantidad de sustancia de ensayo contenida en las soluciones de ensayo no debe sobrepasar el límite de solubilidad en el medio de ensayo.
Los disolventes se usan para obtener una solución madre que pueda dosificarse con exactitud en agua. A la concentración de disolvente recomendada en el medio de ensayo final (< 0,1 ml/l), los disolventes indicados en el párrafo anterior no son tóxicos y no aumentan la solubilidad en agua de la sustancia.
Los dispersantes pueden facilitar la dosificación exacta y la dispersión. A la concentración recomendada en el medio de ensayo final (< 0,1 ml/l), los dispersantes indicados en el párrafo anterior no son tóxicos y no aumentan la solubilidad en agua de la sustancia.
1.7. DISEÑO DEL ENSAYO
Hay que asignar los tratamientos a los recipientes de ensayo y realizar todas las manipulaciones posteriores de dichos recipientes de forma aleatoria. De otro modo podría introducirse un sesgo susceptible de ser interpretado como efecto de una concentración. En particular, si las unidades experimentales se manipulan en orden de tratamiento o de concentración, determinados efectos vinculados con el tiempo, como la fatiga u otros errores del operador, podrían ejercer un efecto más acusado a concentraciones más elevadas. Además, si hay alguna probabilidad de que los resultados se vean afectados por una condición inicial o ambiental del ensayo, como la posición en el laboratorio, hay que pensar en la conveniencia de practicar agrupamientos.
1.8. PROCEDIMIENTO
1.8.1. Condiciones de exposición
1.8.1.1. Duración
La duración del ensayo es de 21 días.
1.8.1.2. Carga
Los parentales se mantienen separados, uno por recipiente de ensayo, con un volumen de 50 a 100 ml de medio en cada recipiente.
En ocasiones hay que emplear un volumen mayor para satisfacer los requisitos del método analítico usado para determinar la concentración de la sustancia de ensayo, aunque también se permite acumular los recipientes en paralelo para el análisis químico. Si se usan volúmenes superiores a 100 ml, puede ser necesario aumentar la ración administrada a Dafnia para garantizar una disponibilidad suficiente de nutrientes y para cumplir con los criterios de validez. En ensayos dinámicos pueden considerarse, por razones técnicas, otros diseños (por ejemplo, 4 grupos de 10 animales en un volumen de ensayo mayor); en cualquier caso, es preciso documentar cualquier modificación del diseño.
1.8.1.3. Número de animales
En ensayos semiestáticos, se mantienen por separado al menos 10 animales a cada concentración de ensayo y al menos 10 animales en la serie de control.
En ensayos dinámicos, se ha demostrado (1) que es adecuado repartir 40 animales en 4 grupos de 10 a cada concentración de ensayo. Puede usarse un número inferior de organismos de ensayo, y se recomienda un mínimo de 20 animales por concentración repartidos en 2 o más recipientes en paralelo de igual número de animales (por ejemplo, 4 recipientes en paralelo con 5 dáfnidos cada uno). Obsérvese que en los ensayos en los cuales se mantengan los animales en grupos, si los parentales mueren, no será posible expresar el resultado reproductor como número total de descendientes vivos producidos por parental vivo al final del ensayo. En tal caso, el resultado reproductor debe expresarse como «número total de descendientes vivos producidos por parental presente al principio del ensayo».
1.8.1.4. Alimentación
En ensayos semiestáticos, la alimentación debe ser preferentemente diaria; en cualquier caso, tendrá una frecuencia mínima de 3 veces por semana (es decir, en correspondencia con los cambios de medio). Deben documentarse las desviaciones con respecto a esta pauta (por ejemplo, ensayos dinámicos).
Durante el ensayo, la dieta de los parentales estará formada preferentemente por algas vivas de una o varias de las siguientes especies: Chlorella sp, Selenastrum capricornutum (ahora Pseudokirchneriella subcapitata (11) y Scenedesmus subspicatus. La dieta suministrada se basará en la cantidad de carbono orgánico (C) proporcionado a cada parental. Las investigaciones realizadas (12) han demostrado que, en el caso de Daphnia magna, son suficientes raciones comprendidas entre 0,1 y 0,2 mg C/Dafnia/día para alcanzar el número de descendientes necesarios para cumplir los criterios de validez. La ración puede administrarse a una tasa constante durante todo el período de ensayo o, si se prefiere, a una tasa inferior al principio que se va incrementando en función del crecimiento de los parentales. No obstante, en este caso, las raciones deben permanecer en todo momento dentro del intervalo recomendado de 0,1 a 0,2 mg C/Dafnia/día.
Si es preciso recurrir a parámetros indirectos, como el número de células de algas o la absorbancia luminosa, para determinar la ración adecuada (por razones de comodidad, pues medir el contenido de carbono requiere mucho tiempo), cada laboratorio debe crear su propio nomograma para la medición indirecta del contenido de carbono del cultivo de algas (véanse algunos consejos sobre construcción de nomogramas en el apéndice 2). Los nomogramas deben revisarse al menos una vez al año, y con mayor frecuencia si cambian las condiciones de los cultivos de algas. Se ha observado que la absorbancia luminosa proporciona una indicación del contenido de carbono mejor que el número de células (13).
Para reducir al mínimo el volumen de medio de cultivo de algas trasvasado a los recipientes de ensayo, la suspensión de algas administrada a Dafnia como alimento debe ser concentrada. Las algas pueden concentrarse mediante centrifugación seguida de resuspensión en agua destilada, agua desionizada o medio de cultivo de Dafnia.
1.8.1.5. Iluminación
16 horas de luz a una intensidad no superior a 15-20μE m-2 s-1.
1.8.1.6. Temperatura
La temperatura del medio de ensayo debe encontrarse en el intervalo 18-22 oC. No obstante, en un mismo ensayo, la temperatura no debe variar, a ser posible, más de 2 oC dentro de estos límites (por ejemplo, 18-20, 19-21 o 20-22 oC). Puede ser conveniente utilizar un recipiente de ensayo adicional con el fin de vigilar la temperatura.
1.8.1.7. Aireación
Los recipientes de ensayo no recibirán aireación durante el ensayo.
1.8.2. Concentraciones de ensayo
Normalmente se prepararán al menos 5 concentraciones de ensayo en una serie geométrica con un factor de separación preferentemente no superior a 3,2; también hay que usar un número apropiado de recipientes en paralelo con cada concentración de ensayo (véase el punto 1.8.1.3). Si se usan menos de 5 concentraciones, hay que justificarlo. Las sustancias no deben someterse a ensayo en concentraciones superiores a su límite de solubilidad en el medio de ensayo.
Al determinar la gama de concentraciones, debe tenerse en cuenta lo siguiente:
|
i) |
si el objetivo es determinar los valores LOEC y NOEC, la concentración de ensayo mínima ha de ser lo suficientemente baja para que la fecundidad a esa concentración no sea significativamente inferior a la del control. En caso contrario, será necesario repetir el ensayo a una concentración mínima menor; |
|
ii) |
si el objetivo es determinar los valores LOEC y NOEC, la concentración de ensayo máxima ha de ser lo suficientemente alta para que la fecundidad a esa concentración sea significativamente inferior a la del control. En caso contrario, será necesario repetir el ensayo a una concentración máxima mayor; |
|
iii) |
si se estima el valor de ECX para los efectos sobre la reproducción, es aconsejable usar concentraciones suficientes para definir dicho valor con un nivel de confianza adecuado. Si se estima la EC50 para los efectos sobre la reproducción, es aconsejable que la concentración de ensayo máxima sea superior a dicha EC50. De otro modo, aunque seguiría siendo posible estimar el EC50, el intervalo de confianza sería muy grande y no se podría evaluar satisfactoriamente la idoneidad del modelo ajustado; |
|
iv) |
A ser posible, la gama de concentraciones de ensayo no debe incluir ninguna concentración que ejerza un efecto estadísticamente significativo sobre la supervivencia de adultos, puesto que esto cambiaría la naturaleza del ensayo, que de simple ensayo de reproducción se transformaría en un ensayo de reproducción y mortalidad, que exige un análisis estadístico mucho más complejo. |
El conocimiento previo de la toxicidad de la sustancia de ensayo (derivado, por ejemplo, de estudios de toxicidad aguda y de determinación de gama) ayudará a elegir las concentraciones de ensayo adecuadas.
Si se usa un disolvente o un dispersante para facilitar la preparación de las soluciones de ensayo (véase el punto 1.6.4), su concentración final en los recipientes de ensayo no sobrepasará el valor de 0,1 ml/l, y será igual en todos los recipientes.
1.8.3. Controles
Además de la serie de ensayo, debe realizarse una serie de control con el medio de ensayo y, si procede, otra que contenga el disolvente o el dispersante. En este caso, la concentración de disolvente o dispersante ha de ser igual a la usada en los recipientes que contienen la sustancia de ensayo. Hay que emplear el número adecuado de recipientes en paralelo (véase el punto 1.8.1.3).
Por lo general, en un ensayo bien ejecutado, el coeficiente de variación con respecto al número medio de supervivientes vivos producido por cada parental de control debe ser ≤ 25 %; este valor debe documentarse para diseños de ensayo en los que se usen animales mantenidos por separado.
1.8.4. Renovación del medio de ensayo
La frecuencia de renovación del medio depende de la estabilidad de la sustancia de ensayo, pero debe ser al menos de 3 veces por semana. Si de los ensayos de estabilidad preliminares (véase el punto 1.4) se deduce que la concentración de la sustancia de ensayo no es estable (es decir, si está fuera del intervalo comprendido entre el 80 y el 120 % del valor nominal o si cae por debajo del 80 % de la concentración inicial medida) a lo largo del período de renovación máximo (3 días), hay que considerar la renovación más frecuente del medio o la realización de un ensayo dinámico.
Al renovar el medio en ensayos semidinámicos, se prepara una segunda serie de recipientes de ensayo y los parentales se transfieren a estos con ayuda, por ejemplo, de una pipeta de vidrio de diámetro adecuado. El volumen de medio transferido con los ejemplares de Dafnia debe reducirse al mínimo.
1.8.5. Observaciones
Los resultados de las observaciones hechas durante el ensayo deben consignarse en fichas de datos (véanse ejemplos en los apéndices 3 y 4). Si hacen falta otras mediciones (véanse los puntos 1.3 y 1.8.8), puede ser necesario realizar más observaciones.
1.8.6. Descendientes
Preferiblemente, los descendientes producidos por cada parental se retirarán y contarán a diario desde el nacimiento de la primera camada para evitar que consuman el alimento destinado al adulto. A los efectos de este método, solo es necesario contar el número de descendientes vivos, aunque debe registrarse la presencia de huevos abortados o de descendientes muertos.
1.8.7. Mortalidad
La mortalidad de los parentales se registrará preferiblemente a diario y al menos en las mismas ocasiones en que se cuenten los descendientes.
1.8.8. Otras variables
Aunque este método se ha diseñado principalmente para evaluar los efectos sobre la reproducción, quizá sea posible cuantificar otros efectos en medida suficiente para admitir el análisis estadístico. Las determinaciones relativas al crecimiento son muy interesantes, pues aportan información sobre posibles efectos subletales que pueden resultar más útiles que la simple determinación relativa a la reproducción; es recomendable proporcionar la determinación relativa a los parentales (la longitud del cuerpo sin contar la espina anal) al término del ensayo. Otras variables que podrían medirse o calcularse son el tiempo transcurrido hasta la producción de la primera camada (y de las siguientes), el número y el tamaño de las camadas por animal, el número de cama-das abortadas, la presencia de machos o efipios y la tasa intrínseca de aumento de la población.
1.8.9. Frecuencia de los análisis y mediciones
La concentración de oxígeno, la temperatura, la dureza y el pH deben medirse al menos una vez a la semana en los medios nuevo y antiguo, en los controles y a la concentración máxima de la sustancia de ensayo.
Durante el ensayo, las concentraciones de la sustancia analizada deben determinarse a intervalos regulares.
En ensayos semiestáticos en los que se espere que la concentración de la sustancia de ensayo se mantenga dentro del ± 20 % de la concentración nominal (es decir, en el intervalo del 80-120 %, véanse los puntos 1.4 y 1.8.4), se recomienda que las concentraciones de ensayo máxima y mínima se analicen recién preparadas y en el momento de su renovación al menos una vez durante la primera semana del ensayo (es decir, los análisis deben efectuarse en una muestra de la misma solución, recién preparada y en el momento de su renovación). A partir de este momento, las determinaciones han de repetirse a intervalos al menos semanales.
En ensayos en los que no se espere que la concentración de la sustancia de ensayo se mantenga dentro del ± 20 % de la concentración nominal, es preciso analizar todas las concentraciones de ensayo recién preparadas y en el momento de su renovación. No obstante, en los ensayos en los que la concentración inicial medida de la sustancia de ensayo no esté dentro del ± 20 % de la concentración nominal, pero en los que puedan aportarse pruebas suficientes de que las concentraciones iniciales son repetibles y estables (es decir, que están dentro del intervalo del 80-120 % de la concentración inicial), las determinaciones químicas podrían limitarse en las semanas 2 y 3 del ensayo a las concentraciones máxima y mínima. En todos los casos, la determinación de las concentraciones de la sustancia de ensayo antes de la renovación solo debe realizarse en un recipiente en paralelo de cada concentración de ensayo.
Para los ensayos dinámicos es apropiado un régimen de muestreo similar al descrito para los semiestáticos (si bien en este caso no cabe hacer determinaciones antes de renovar las soluciones). Sin embargo, puede ser aconsejable aumentar el número de ocasiones de muestreo durante la primera semana (tres series de mediciones, por ejemplo) para garantizar que las concentraciones de ensayo permanezcan estables. En esta clase de ensayos, los regímenes de flujo de diluyente y sustancia de ensayo deben verificarse a diario.
Si se demuestra que la concentración de la sustancia de ensayo se ha mantenido satisfactoriamente a lo largo de todo el ensayo dentro de un intervalo de ± 20 % de la concentración nominal o de la medida inicialmente, los resultados pueden basarse en los valores nominales o medidos inicialmente. Si la desviación con respecto a la concentración nominal o inicial medida es superior al ± 20 %, los resultados deben expresarse en términos de media ponderada en función del tiempo (véase el apéndice 5).
2. RESULTADOS E INFORME
2.1. TRATAMIENTO DE LOS RESULTADOS
El objeto de este ensayo es determinar el efecto de la sustancia de ensayo sobre el número total de descendientes vivos producidos por cada parental vivo al final del mismo. El número total de descendientes por parental debe calcularse para cada recipiente de ensayo (es decir, para cada recipiente en paralelo). Si en uno cualquiera de los recipientes en paralelo el parental muere durante el ensayo o resulta ser macho, dicho recipiente se excluye del análisis. Por tanto, este se basará en un número reducido de recipientes en paralelo.
Para estimar la LOEC y, por tanto, la NOEC, correspondiente a los efectos del producto químico sobre el resultado reproductor, es preciso calcular el resultado reproductor medio de los recipientes en paralelo para cada concentración y la desviación estándar residual acumulada, lo que puede hacerse mediante un análisis de la varianza (ANOVA). A continuación se compara la media obtenida a cada concentración con la del control aplicando un método de comparación múltiple apropiado. Las pruebas de Dunnett o Williams pueden ser útiles (14) (15) (16) (17). Hay que comprobar si se sostiene la hipótesis del ANOVA de homogeneidad de la varianza. Es recomendable realizar esta verificación gráficamente en lugar de mediante una prueba formal de significación (18); una alternativa adecuada es aplicar una prueba de Bartlett. Si la hipótesis no se sostiene, hay que considerar la transformación de los datos para homogeneizar las varianzas antes de aplicar el ANOVA, o bien la realización de un ANOVA ponderado. Hay que calcular y documentar la magnitud del efecto detectable con el ANOVA (es decir, la mínima diferencia significativa).
Para estimar la concentración que provocaría la reducción en un 50 % del resultado reproductor (es decir, la EC50), hay que ajustar a los datos una curva adecuada, tal como la curva logística, mediante un método estadístico como el de los mínimos cuadrados. La curva puede parametrarse de manera que la EC50 y su error estándar puedan estimarse directamente. Esto facilitará considerablemente el cálculo de los límites de confianza en torno a la EC50. Salvo que haya razones de peso para preferir otros, deben darse límites de confianza bilaterales del 95 %. El método de ajuste debe proporcionar preferentemente un medio para evaluar la significación de la falta de ajuste. Esto puede hacerse gráficamente o dividiendo la suma residual de cuadrados en «falta de ajuste» y «componentes de error puro» y efectuando una prueba de significación para la falta de ajuste. Como los tratamientos que inducen fecundidad elevada tienen más probabilidades de inducir una varianza más elevada del número de inmaduros producidos que aquellos que inducen fecundidad reducida, hay que considerar la conveniencia de ponderar los valores observados de manera que reflejen las distintas varianzas de los diferentes grupos de tratamiento [véase información de tipo general en la referencia (18)].
En el análisis de los datos del estudio final interlaboratorios (2) se ha ajustado una curva logística usando el modelo siguiente, aunque pueden usarse otros adecuados:
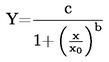
donde:
|
Y |
= |
número total de inmaduros por parental vivo al final del ensayo (calculado para cada recipiente) |
|
x |
= |
concentración de sustancia |
|
c |
= |
número esperado de inmaduros cuando x = 0 |
|
x0 |
= |
EC50 en la población |
|
b |
= |
parámetro «pendiente». |
Este modelo será probablemente adecuado en gran número de situaciones, pero habrá ensayos para los cuales no sea apropiado. Es preciso comprobar la validez del modelo tal como se ha sugerido en párrafos anteriores. En algunos casos puede estar indicado un modelo de hormesis, según el cual concentraciones bajas producen efectos amplificados (19).
También pueden estimarse otras concentraciones que provoquen un porcentaje de efecto determinado, como la EC10 o EC20, aunque quizá sea preferible parametrar el modelo de una forma distinta a la empleada para estimar la EC50.
2.2. INFORME DEL ENSAYO
El informe del ensayo debe incluir la información siguiente:
2.2.1. Sustancia de ensayo:
|
— |
naturaleza física y propiedades fisicoquímicas pertinentes, |
|
— |
identificación química, incluida la pureza. |
2.2.2. Especie sometida a ensayo:
|
— |
clon (si se ha tipificado genéticamente), proveedor u origen (si se conoce) y condiciones de cultivo aplicadas. Si se ha usado una especie distinta de Daphnia magna, debe consignarse y justificarse. |
2.2.3. Condiciones de ensayo:
|
— |
método de ensayo utilizado (por ejemplo, semiestático o dinámico, con indicación de volumen y carga expresada en número de individuos de Dafnia por litro), |
|
— |
fotoperíodo e intensidad luminosa, |
|
— |
diseño del ensayo (por ejemplo, número de recipientes en paralelo, número de parentales por recipiente en paralelo), |
|
— |
detalles del medio de cultivo usado, |
|
— |
si se usan, complementos de materia orgánica, con su composición, origen, método de preparación, TOC/COD de las preparaciones madre, estimación de los valores TOC/COD obtenidos en el medio de ensayo, |
|
— |
información detallada de la alimentación, con indicación de la cantidad (en mg C/Dafnia/día) y el programa (tipo de alimento, con indicación en su caso del nombre específico (especie) del alga y, si se conocen, la cepa y las condiciones de cultivo), |
|
— |
método de preparación de las soluciones madre y frecuencia de renovación (si se usan, hay que indicar el disolvente o dispersante y su concentración). |
2.2.4. Resultados:
|
— |
resultados de cualesquiera estudios preliminares de estabilidad de la sustancia de ensayo, |
|
— |
concentraciones de ensayo nominales y resultados de todos los análisis para determinar la concentración de la sustancia de ensayo en los recipientes de ensayo (véase un ejemplo de ficha de datos en el apéndice 4); también deben indicarse la eficiencia de recuperación del método y el límite de determinación, |
|
— |
calidad del agua en los recipientes de ensayo (pH, temperatura y concentración de oxígeno disuelto además de TOC y COD y dureza cuando proceda) (véase un ejemplo de ficha de datos en el apéndice 3), |
|
— |
registro completo de los descendientes vivos de cada parental (véase un ejemplo de ficha de datos en el apéndice 3), |
|
— |
número de muertes entre los parentales y día en que se hayan producido (véase un ejemplo de ficha de datos en el apéndice 3), |
|
— |
coeficiente de variación de la fecundidad de los controles (basada en el número total de descendientes vivos por parental vivo al final del ensayo), |
|
— |
representación gráfica del número total de descendientes vivos por parental (para cada recipiente en paralelo) vivo al final del ensayo frente a concentración de la sustancia de ensayo, |
|
— |
concentración mínima con efecto observado (LOEC) para la reproducción, acompañada de una descripción de los métodos estadísticos aplicados y de una indicación de la magnitud del efecto detectable; concentración (máxima) sin efecto observado (NOEC) para la reproducción; si procede, valores de LOEC y NOEC para la mortalidad de los parentales, |
|
— |
si procede, ECX para la reproducción e intervalos de confianza, así como una representación gráfica del modelo ajustado usado para su cálculo, la pendiente de la curva dosis-respuesta y su error estándar, |
|
— |
otros efectos biológicos observados o medidos; hay que documentar todos los demás efectos biológicos observados o medidos (crecimiento de los parentales, por ejemplo) y la correspondiente justificación, |
|
— |
explicación de cualquier desviación respecto al método de ensayo. |
3. REFERENCIAS
|
(1) |
OECD Test Guideline Programme, Report of the Workshop on the Daphnia magna Pilot Ring Test, Sheffield University, UK, 20-21 March 1993. |
|
(2) |
OECD Environmental Health and Safety Publications. Series on Testing and Assessment No. 6. Report of the Final Ring Test of the Daphnia magna Reproduction Test Paris. 1997. |
|
(3) |
Baird D. J., Barber J., Bradley M. C, Soares A. M. V. M. and Calow P. (1991). A comparative study of genotype sensitivity to acute toxic stress using clones of Daphnia magna Strauss. Ecotoxicology and Environmental Safety, 21, pp. 257-265. |
|
(4) |
Elendt B. P., (1990). Selenium deficiency in Crustacea; An ultrastructural approach to antennal damage in Daphnia magna Straus. Protoplasma, 154, pp. 25-33. |
|
(5) |
EPA (1993). Methods for Measuring the Acute Toxicity of Effluents and Receiving Waters to Freshwater and Marine Organisms. (Fourth ed.). EPA/600/4-90/027F. C. I. Weber (ed), USEPA, Cincinnati, Ohio. |
|
(6) |
Vigano L, (1991) Suitability of commercially available spring waters as standard medium for culturing Daphnia magna. Bull. Environ. Contam. Toxicol., 47, pp. 775-782. |
|
(7) |
ASTM (1988). Standard Guide for Conducting Acute Toxicity Tests with Fishes, Macroinvertebrates and Amphibians. E729-88a. American Society for Testing and Materials, Philadelphia P.A. 20 pp. |
|
(8) |
Baird D. J., Soares A. M. V. M., Girling A., Barber J., Bradley M. C. and Calow P. (1989). The long term maintenance of Daphnia magna Straus for use in ecotoxicological tests; problems and prospects. In: Proceedings of the 1st European Conference on Ecotoxicology. Copenhagen 1988 (H.Løkke, H. Tyle & F. Bro-Rasmussen. Eds.), pp. 144-148. |
|
(9) |
Parkhurst B. R., Forte J. L. and Wright G. P. (1981). Reproducibility of a life-cycle toxicity test with Daphnia magna. Bull. Environ. Contam. and Toxicol., 26, pp. 1-8. |
|
(10) |
Cowgill U. M. and Milazzo D. P. (1990) The sensitivity of two cladocerans to water quality variables: salinity and hardness. Arch. Hydrobiol., 120(2), pp. 185-196. |
|
(11) |
Korshikov (1990). Pseudokirchneriella subcapitata Hindak, F-1990. Biologice Prace, 36, 209. |
|
(12) |
Sims 1. R., Watson S. and Holmes D. (1993). Toward a standard Daphnia juvenile production test. Environmental Toxicology and Chemistry, 12, pp. 2053-2058. |
|
(13) |
Sims I. (1993). Measuring the growth of phytoplankton: the relationship between total organic carbon with three commonly used parameters of algal growth. Arch. Hydrobiol., 128, pp. 459-466. |
|
(14) |
Dunnett C. W., (1955). A multiple comparisons procedure for comparing several treatments with a control.]. Amer. Statist. Assoc., 50, pp. 1096-1121. |
|
(15) |
Dunnett C. W., (1964). New tables for multiple comparisons with a control. Biometrics, 20, pp. 482-491. |
|
(16) |
Williams D. A. (1971). A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27, pp. 103-117. |
|
(17) |
Williams D. A. (1972). The comparison of several dose levels with a zero dose control. Biometrics, 28, pp. 510-531. |
|
(18) |
Draper N. R. and Smith H. (1981). Applied Regression Analysis, second edition, Wiley, N.Y. |
|
(19) |
Brain P. and Cousens R. (1989). An equation to describe dose responses where there is stimulation of growth at low doses. Weed Research, 29, pp. 93-96. |
|
(20) |
Wilson E. O. and Bossert, W. H. (1971). A Primer of Population Biology. Sinauer Associates Inc. Publishers. |
|
(21) |
Poole R. W. (1974). An Introduction to quantitative Ecology. McGraw-Hill Series in Population Biology, New York, pp. 532. |
|
(22) |
Meyer J. S., Ingersoll C. G., McDonald L. L. and Boyce M. S. (1986). Estimating uncertainty in population growth rates: Jackknife vs bootstrap techniques. Ecology, 67, pp. 1156-1166. |
Apéndice 1
PREPARACIÓN DE MEDIOS ELENDT M7 Y M4 TOTALMENTE DEFINIDOS
Aclimatación a los medios Elendt M7 y M4
Algunos laboratorios han tenido dificultades para transferir directamente Dafnia a los medios M4 (1) y M7. Se han obtenido resultados bastante buenos con una aclimatación gradual, esto es, cambiando del medio propio a Elendt al 30 %, luego al 60 % y por último al 100 %. El período de aclimatación necesario puede ser bastante largo, incluso de un mes.
PREPARACIÓN
Oligoelementos
Se empieza por preparar soluciones madre distintas (I) de cada oligoelemento en agua de pureza apropiada (desionizada, destilada o tratada mediante ósmosis inversa). A partir de estas soluciones madre (I) se prepara una única solución madre (II) que contiene todos los oligoelementos (solución combinada):
|
Soluciones madre I (una sola sustancia) |
Cantidad añadida al agua (mg/l) |
Concentración (en relación con el medio M4) (veces) |
Para preparar la solución madre II combinada, se añade al agua la siguiente cantidad de solución madre I (ml/l) |
|
|
M 4 |
M 7 |
|||
|
H 3 BO 3 |
57 190 |
20 000 |
1,0 |
0,25 |
|
MnCl2 * 4 H2O |
7 210 |
20 000 |
1,0 |
0,25 |
|
LiCl |
6 120 |
20 000 |
1,0 |
0,25 |
|
RbCl |
1 420 |
20 000 |
1,0 |
0,25 |
|
SrCl2 * 6 H20 |
3 040 |
20 000 |
1,0 |
0,25 |
|
NaBr |
320 |
20 000 |
1,0 |
0,25 |
|
Na2MoO4 * 2 H2O |
1 260 |
20 000 |
1,0 |
0,25 |
|
CuCl2 * 2 H20 |
335 |
20 000 |
1,0 |
0,25 |
|
ZnCl2 |
260 |
20 000 |
1,0 |
1,0 |
|
CoCl2 * 6 H2O |
200 |
20 000 |
1,0 |
1,0 |
|
KI |
65 |
20 000 |
1,0 |
1,0 |
|
Na2SeO3 |
43,8 |
20 000 |
1,0 |
1,0 |
|
NH4VO3 |
11,5 |
20 000 |
1,0 |
1,0 |
|
Na2EDTA * 2 H20 |
5 000 |
2 000 |
— |
— |
|
FeSO4 * 7 H2O |
1 991 |
2 000 |
— |
— |
|
Las soluciones Na2EDTA v FeSO4 se preparan por separado, se vierten juntas y se esterilizan en autoclave inmediatamente. Se obtiene así: |
||||
|
Solución 21 Fe-EDTA |
|
1 000 |
20,0 |
5,0 |
Medios M4 y M7
Los medidos M4 y M7 se preparan a partir de la solución madre II con los macronutrientes y las vitaminas que siguen:
|
|
Cantidad añadida al agua (mg/l) |
Concentración (en relación con el medio M4) (vecces) |
Cantidad de solución madre añadida para preparar el medio (ml/l) |
|
|
M 4 |
M 7 |
|||
|
Solución madre II (combinación de oli-goelementos) II |
|
20 |
50 |
50 |
|
Soluciones madre de macronutrientes (una sustancia por solución) |
||||
|
CaCl2 * 2 H20 |
293 800 |
1 000 |
1,0 |
1,0 |
|
MgSO4 * 7 H2O |
246 600 |
2 000 |
0,5 |
0,5 |
|
KCl |
58 000 |
10 000 |
0,1 |
0,1 |
|
NaHCO3 |
64 800 |
1 000 |
1,0 |
1,0 |
|
Na2SiO3 * 9 H2O |
50 000 |
5 000 |
0,2 |
0,2 |
|
NaNO3 |
2 740 |
10 000 |
0,1 |
0,1 |
|
KH2PO4 |
1 430 |
10 000 |
0,1 |
0,1 |
|
K2HPO4 |
1 840 |
10 000 |
0,1 |
0,1 |
|
Solución madre de vitaminas combinadas |
— |
10 000 |
0,1 |
0,1 |
|
La solución madre de vitaminas combinadas se prepara añadiendo las 3 vitaminas a 1 litro de agua como se indica a continuación: |
||||
|
Clorhidrato de tiamina |
750 |
10 000 |
— |
— |
|
Cianocobalamina (B12) |
10 |
10 000 |
— |
— |
|
Biotina |
7,5 |
10 000 |
— |
— |
|
La solución madre de vitaminas combinadas se conserva congelada en pequeñas partes alícuotas. Las vitaminas se incorporan a los medios poco antes de usarlos.
|
Apéndice 2
ANÁLISIS DEL CARBONO ORGÁNICO TOTAL (TOC) Y ELABORACIÓN DE UN NOMOGRAMA DEL CONTENIDO EN COT DEL ALIMENTO DE ALGAS
Es sabido que el contenido en carbono del alimento de algas no suele medirse directamente, sino por medio de correlaciones (es decir, de nomogramas) con medidas indirectas, como el número de células de algas o la absorbancia de luz.
El TOC debe medirse por oxidación a alta temperatura y no con los métodos UV o del persulfato. (Véase: The Instrumental Determination of Total Organic Carbon, Total Oxygen Demand and Related Determinands 1979, HMSO 1980; 49 High Holborn, London WC1V 6HB).
Para elaborar el nomograma, las algas se separan del medio de crecimiento por centrifugación seguida de resuspensión en agua destilada. Se mide la variable indirecta y la concentración de TOC en cada una de las muestras por triplicado. Hay que analizar controles con agua destilada únicamente y deducir la concentración de TOC a partir de la observada en la muestra de algas.
El nomograma debe ser lineal dentro del intervalo necesario de concentraciones de carbono. A continuación se ilustran algunos ejemplos.
Nota: No deben usarse para hacer conversiones; es esencial que cada laboratorio prepare sus propios nomogramas.



Apéndice 3
EJEMPLO DE FICHA DE DATOS RELATIVA A LA RENOVACIÓN DEL MEDIO, LA VIGILANCIA FISICOQUÍMICA Y LA ALIMENTACIÓN, REPRODUCCIÓN Y MORTALIDAD DE ADULTOS DE DAFNIA
|
Experimento No: |
Fecha de inicio: |
Clon: |
Medio: |
Tipo de alimento: |
Sustancia de ensayo: |
Concentración nominal: |
||||||||||||||||||
|
Día |
0 |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
21 |
|
|
|
Renov. medio (marcar) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
pH |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
nuevo |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
antiguo |
|
|
|
O2 mg/l |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
neuevo |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
antiguo |
|
|
|
Temperatura ( oC) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
nuevo |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
antiguo |
|
|
|
Aporte alim. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
No descendientes vivos (35) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
Recipiente 1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
9 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
|
Mortalidad acumulada adul-tos (36) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Apéndice 4
EJEMPLO DE FICHA DE DATOS RELATIVA A LOS RESULTADOS DE LOS ANÁLISIS QUÍMICOS
(a) Concentraciones medidas
|
Concentración nominal |
Muestra semana 1 |
Muestra semana 2 |
Muestra semana 3 |
|||
|
Después de la renovación |
Antes de la renovación |
Después de la renovación |
Antes de la renovación |
Después de la renovación |
Antes de la renovación |
|
|
|
|
|
|
|
|
|
(b) Concentraciones medidas en porcentaje de la nominal
|
Concentración nominal |
Muestra semana 1 |
Muestra semana 2 |
Muestra semana 3 |
|||
|
Después de la renovación |
Antes de la renovación |
Después de la renovación |
Antes de la renovación |
Después de la renovación |
Antes de la renovación |
|
|
|
|
|
|
|
|
|
Apéndice 5
CÁLCULO DE UNA MEDIA PONDERADA EN FUNCIÓN DEL TIEMPO
Media ponderada en función del tiempo
Puesto que la concentración de la sustancia de ensayo puede disminuir a lo largo del tiempo comprendido entre las sucesivas renovaciones del medio, es preciso considerar cuál es la concentración que debe elegirse como representativa de la gama de concentraciones aplicadas a los parentales de Dafnia. La elección debe basarse en consideraciones biológicas y estadísticas. Si, por ejemplo, se piensa que la mayor influencia en la reproducción la ejerce la máxima concentración aplicada, debe usarse este valor máximo. En cambio, si se considera más importante el efecto acumulado o a plazo más largo, la concentración media es más relevante. En este caso, una media apropiada es la concentración media ponderada en función del tiempo, pues tiene en cuenta la variación de la concentración instantánea a lo largo del tiempo.
figura 1
Ejemplo de media ponderada en función del tiempo

La figura 1 ilustra un ejemplo de ensayo (simplificado) de 7 días de duración en el cual el medio se renueva los días 0, 2 y 4.
|
— |
La línea delgada en zigzag representa la concentración a lo largo del tiempo. Se supone que el descenso de la concentración es exponencial. |
|
— |
Los 6 puntos representados corresponden a las concentraciones medidas al principio y al final de cada período de renovación. |
|
— |
La línea gruesa indica la posición de la media ponderada en función del tiempo. |
La media ponderada en función del tiempo se calcula de manera que el área situada bajo ella sea igual al área situada bajo la curva de concentración. El cálculo correspondiente al ejemplo anterior se ilustra en el cuadro 1.
Cuadro 1
Cálculo de la media ponderada en función del tiempo
|
Renovación no |
Días |
Conc0 |
Conc1 |
Ln(Conc0) |
Ln(Conc1) |
Área |
|
1 |
2 |
10,000 |
4,493 |
2,303 |
1,503 |
13,767 |
|
2 |
2 |
11,000 |
6,037 |
2,398 |
1,798 |
16,544 |
|
3 |
3 |
10,000 |
4,066 |
2,303 |
1,403 |
19,781 |
|
Días totales: 7 |
Área total |
50,091 |
||||
|
Media PT |
7,156 |
|||||
|
Días: número de días del período de renovación. Conc0: concentración medida al principio de cada período de renovación. Conc1: concentración medida al final de cada período de renovación. Ln(Conc0): logaritmo natural de Conc0. Área: área comprendida bajo la curva exponencial para cada período de renovación. Se calcula como sigue: Ln(Conc1): logaritmo natural de Conc1.
|
La media ponderada en función del tiempo (Media PT) es igual al Área total dividida por los Días totales.
Naturalmente, en el caso de un ensayo de reproducción de Dafnia, el cuadro debería cubrir 21 días.
Es obvio que, si solo se hacen observaciones al principio y al final de cada período de renovación, es imposible confirmar que el proceso de disminución sea, en efecto, exponencial. Una curva distinta daría lugar a un cálculo de Área distinto. En cualquier caso, la disminución exponencial es una hipótesis plausible, y esta es probablemente la curva mejor a falta de más datos.
Es preciso hacer una advertencia para el caso de que el análisis químico no logre detectar ninguna sustancia al final del período de renovación. Salvo que se pueda estimar la velocidad con que la sustancia desaparece de la solución, será imposible obtener un área bajo la curva realista y, por tanto, obtener una media ponderada en función del tiempo que sea razonable.
C.21. MICROORGANISMOS DEL SUELO: ENSAYO DE TRANSFORMACIÓN DEL NITRÓGENO
1. MÉTODO
El presente método reproduce las directrices de ensayo de la OCDE TG 216 (2000).
1.1. INTRODUCCIÓN
Este método de ensayo es un método de laboratorio destinado a investigar los efectos a largo plazo de productos químicos, tras una exposición única, en la transformación del nitrógeno por los microorganismos del suelo. El ensayo se basa principalmente en las recomendaciones de la Organización Europea y Mediterránea para la Protección de las Plantas (1). Sin embargo, se han tenido en cuenta otras directrices, como las del Biologische Bundesanstalt (2) de Alemania, la Environmental Protection Agency de los Estados Unidos (3) la Sociedad de Química y Toxicología Medioambientales (Society of Environmental Toxicology and Chemistry, SETAC) (4) y la Organización Internacional de Normalización (ISO) (5). En un taller de la OECD sobre selección de suelos/sedimentos, celebrado en Belgirate, Italia, en 1995 (6) se acordó el número y el tipo de suelos que deben usarse en este ensayo. Las recomendaciones sobre recogida, manejo y almacenamiento de muestras de suelo se basan en un documento orientativo de la ISO (7) y en las recomendaciones del taller de Belgirate. En la valoración y evaluación de las características de las sustancias de ensayo, puede resultar necesaria la determinación de los efectos de la actividad microbiana en el suelo, por ejemplo, cuando se requieran datos sobre los posibles efectos secundarios de los productos fitosanitarios en la microflora del suelo o cuando sea previsible la exposición de los microorganismos del suelo a los productos químicos distintos de los fitosanitarios. El ensayo de transformación del nitrógeno se lleva a cabo para determinar los efectos de estos productos químicos en la microflora del suelo. Si se ensayan productos agroquímicos (por ejemplo, productos fitosanitarios, abonos y productos químicos para la silvicultura), se llevarán a cabo tanto ensayos de transformación del nitrógeno como del carbono. Si se ensayan productos no agroquímicos, es suficiente el ensayo de transformación del nitrógeno. Sin embargo, si los valores EC50 del ensayo de transformación del nitrógeno de estos productos químicos se sitúan dentro del intervalo encontrado para los inhibidores de la nitrificación disponibles en el comercio (por ejemplo, la nitrapirina), puede hacerse un ensayo de transformación del carbono para obtener más información.
Los suelos están formados por componentes vivos y no vivos que se dan en mezclas complejas y heterogéneas. Los microorganismos desempeñan un papel importante en la descomposición y transformación de la materia orgánica en los suelos fértiles. Existen muchas especies que contribuyen de diversas maneras a la fertilidad del suelo. Cualquier interferencia a largo plazo en estos procesos bioquímicos podría perturbar el ciclo de los nutrientes y alterar la fertilidad del suelo. La transformación del carbono y del nitrógeno se da en los suelos fértiles. Aunque las comunidades microbianas responsables de estos procesos difieren de unos suelos a otros, las vías de transformación son esencialmente las mismas.
Este método de ensayo está destinado a detectar los efectos negativos a largo plazo de una sustancia en el proceso de transformación del nitrógeno en los suelos aerobios de superficie. El método también permite calcular los efectos de una sustancia en la transformación del carbono por la microflora del suelo. La formación de nitratos se produce tras la degradación de los enlaces carbono-nitrógeno. Por tanto, si se encuentran índices iguales de producción de nitratos en suelos tratados y suelos de control, es muy probable que las principales rutas de degradación del carbono estén intactas y funcionales. El sustrato elegido para el ensayo (la harina de alfalfa) tiene una proporción carbono/nitrógeno favorable (normalmente entre 12/1 y 16/1). Por eso, la privación de carbono durante el ensayo es reducida y las comunidades microbianas, si resultan dañadas por una sustancia química, pueden recobrarse dentro de un plazo de 100 días.
Los ensayos a partir de los cuales se desarrolló este método estaban pensados principalmente para sustancias respecto a las cuales puede preverse la cantidad que llegará al suelo. Este es el caso, por ejemplo, de los productos fitosanitarios cuyo índice de aplicación al campo es conocido. Para los productos agroquímicos, es suficiente el ensayo de 2 dosis con arreglo al índice de aplicación estimado o previsto. Los productos agroquímicos pueden someterse a ensayo en forma de principios activos (p.a.) o de productos formulados. Sin embargo, el ensayo no se limita a los productos agroquímicos. Cambiando tanto las cantidades de la sustancia de ensayo aplicada al suelo como la manera en que se evalúan los datos, el ensayo puede usarse también para productos químicos cuando se desconozca la cantidad de estos que llegará al suelo. De este modo, en el caso de los productos químicos distintos de los agrícolas, se determinan los efectos de una serie de concentraciones en la transformación del nitrógeno. A continuación, se utilizan los datos de estos ensayos para trazar una curva dosis-respuesta y calcular los valores ECX , donde x es el porcentaje de efecto definido.
1.2. DEFINICIONES
Transformación del nitrógeno: es la degradación última por microorganismos de materia orgánica que contenga nitrógeno, mediante un proceso de amonificación y nitrificación, hasta llegar al nitrato inorgánico respectivo.
EC X (concentración efectiva): es la concentración de la sustancia de ensayo en el suelo que da lugar a un porcentaje x de inhibición de la transformación de nitrógeno en nitrato.
EC 50 (concentración efectiva mediana): es la concentración de la sustancia de ensayo en el suelo que da lugar a un 50 % de inhibición de la transformación de nitrógeno en nitrato.
1.3 COMPUESTOS DE REFERENCIA
Ninguno.
1.4 PRINCIPIO DEL MÉTODO DE ENSAYO
Al suelo tamizado se le añade harina vegetal y, a continuación, se trata una parte con la sustancia de ensayo y se deja la otra sin tratar (control). Cuando se ensayen productos agroquímicos, se recomienda un mínimo de 2 concentraciones de ensayo, que deben elegirse en relación con la concentración más alta prevista para el campo. Después de 0,7, 14 y 28 días de incubación, se extraen muestras de suelos tratados y de control con un disolvente apropiado y se determinan las cantidades de nitrato en los extractos. El índice de formación de nitratos en las muestras tratadas se compara con el de las de control y se calcula el porcentaje de desviación de las muestras tratadas respecto a las de control. Todos los ensayos tendrán una duración mínima de 28 días. Si, al 28o día, la diferencia entre los suelos tratados y los no tratados es igual o superior al 25 %, se continuarán las mediciones hasta un máximo de 100 días. Cuando se ensayen productos químicos no agrícolas, se añadirá a las muestras de suelo una serie de concentraciones de la sustancia de ensayo y se medirán al cabo de 28 días de incubación la cantidades de nitratos formadas en las muestras tratadas y de control. Los resultados de los ensayos con concentraciones múltiples se analizarán utilizando un modelo de regresión y se calcularán los valores ECX (es decir, EC50, EC25 y/o EC10). Véanse las definiciones.
1.5. VALIDEZ DEL ENSAYO
Las evaluaciones de los resultados de los ensayos se basan en diferencias relativamente pequeñas (es decir, un valor medio de ± 25 %) entre concentraciones de nitratos en las muestras tratadas y de control, de modo que grandes variaciones en las muestras de control pueden dar lugar a resultados falsos. Por ello, la variación entre muestras de control en paralelo debe ser inferior al ± 15 %.
1.6. DESCRIPCIÓN DEL MÉTODO DE ENSAYO
1.6.1. Equipo
Se utilizarán recipientes de ensayo hechos de material químicamente inerte. Estos recipientes deben ser de capacidad adecuada de acuerdo con el procedimiento utilizado para la incubación de suelos, es decir, incubación en una muestra única o en una serie de muestras separadas (véase el punto 1.7.1.2). Hay que procurar reducir al mínimo la pérdida de agua y permitir el intercambio de gas durante el ensayo (por ejemplo, los recipientes pueden cubrirse con láminas de polietileno perforado). Cuando se ensayen sustancias volátiles, deben usarse recipientes sellables y herméticos cuyo tamaño sea tal que aproximadamente un cuarto de su volumen esté ocupado por la muestra de suelo.
Se utilizará equipo de laboratorio estándar, incluyendo:
|
— |
mecanismo de agitación: agitador mecánico o equipo equivalente, |
|
— |
dispositivo de centrifugado (3 000 g) o filtrado (utilizando papel de filtro sin nitratos), |
|
— |
instrumental de sensibilidad y reproductibilidad adecuadas para el análisis de nitratos. |
1.6.2. Selección y número de suelos
Se utilizará un único suelo cuyas características recomendadas serán las siguientes:
|
— |
contenido de arena: entre el 50 % y el 75 %, |
|
— |
pH: 5,5-7,5, |
|
— |
contenido de carbono orgánico: 0,5-1,5 % |
|
— |
debe medirse la masa microbiana (8) (9), cuyo contenido de carbono tiene que ser al menos el 1 % del total de carbono orgánico en el suelo. |
En la mayoría de los casos, un suelo con estas características representa la situación más desfavorable, ya que la adsorción de la sustancia química de ensayo es mínima y su disponibilidad para la microflora es máxima. Por lo tanto, no son necesarios, por lo general, ensayos de otros suelos. Sin embargo, en algunas circunstancias, por ejemplo, cuando la sustancia de ensayo esté indicada principalmente para determinados suelos, como los suelos forestales ácidos, o en el caso de productos químicos cargados electrostáticamente, puede resultar necesario utilizar algún otro suelo.
1.6.3. Recogida y almacenamiento de muestras de suelo
1.6.3.1. Recogida
Deberá contarse con información detallada sobre la historia del campo de donde se recoja la tierra para el ensayo. Se necesitan datos, entre otras cosas, de la localización exacta, la cubierta vegetal, fechas de tratamiento con productos fitosanitarios, tratamientos con abonos orgánicos e inorgánicos, aplicación de sustancias biológicas o contaminación accidental. El lugar elegido para la recogida de suelo tiene que ser tal que permita un uso a largo plazo. Se consideran adecuados los pastos permanentes, los campos con cosechas anuales de cereales (excepto el maíz) o los abonos verdes sembrados densamente. El lugar donde se recojan las muestras no debe haberse tratado con productos fitosanitarios durante un mínimo de un año antes de la recogida de estas. Además, no debe haberse aplicado ningún abono orgánico durante al menos 6 meses. El uso de abonos minerales solo es aceptable cuando esté en concordancia con las necesidades del cultivo, en ese caso no deben tomarse muestras hasta transcurridos, al menos, 3 meses desde la aplicación del abono. Debe evitarse el uso de suelo tratado con abonos con efectos biocidas conocidos (por ejemplo, cianamida de calcio).
Hay que evitar la toma de muestras durante o inmediatamente después de períodos prolongados (de más de 30 días) de sequía o encharcamiento. En el caso de los suelos arados, deben tomarse muestras a una profundidad de 0 a 20 cm. Cuando se trate de pastizales u otros suelos que no se aren durante períodos más largos (al menos un ciclo de cultivo), la profundidad máxima del muestreo puede ser ligeramente superior a 20 cm (por ejemplo, 25 cm).
Las muestras de suelo deben transportarse en recipientes y en condiciones de temperatura que garanticen que las propiedades iniciales del suelo no se alteren significativamente.
1.6.3.2. Almacenamiento
Es preferible el uso de muestras de suelo recién recogidas. Si no se puede evitar el almacenamiento en el laboratorio, las muestras de suelo pueden guardarse en la oscuridad a 4 ± 2 oC durante un máximo de 3 meses. Durante el almacenamiento, debe asegurarse el mantenimiento de condiciones aerobias. Si se recogen muestras de suelo de zonas que estén congeladas durante, al menos, tres meses al año, puede considerarse la posibilidad de almacenarlas durante 6 meses a entre - 18 oC y - 22 oC. Se medirá la biomasa microbiana de las muestras almacenadas antes de cada experimento. La cantidad de carbono en la biomasa debe ser como mínimo el 1 % del total de carbono orgánico de la muestra de suelo (véase el punto 1.6.2).
1.6.4. Manejo y preparación del suelo para el ensayo
1.6.4.1. Preincubación
Cuando las muestras de suelo hayan estado almacenadas (véase el punto 1.6.3.2), se recomienda preincubar durante un período de entre 2 y 28 días. La temperatura y la humedad del suelo durante la preincubación deben ser semejantes a las que se den durante el ensayo (véanse los puntos 1.6.4.2 y 1.7.1.3).
1.6.4.2. Características fisicoquímicas
Se separarán manualmente los objetos grandes (por ejemplo, piedras, trozos de plantas, etc.) de la muestra de suelo y, a continuación, se tamizará la muestra por vía húmeda sin secar en exceso para obtener un tamaño de las partículas no superior a 2 mm. Deberá ajustarse el contenido de humedad de la muestra con agua destilada o desionizada para llegar a un valor entre el 40 % y el 60 % de la capacidad máxima de retención de agua.
1.6.4.3. Adición de sustrato orgánico
La muestra de suelo debe mezclarse con un sustrato orgánico adecuado, por ejemplo, harina de alfalfa-hierba-forraje verde (principal componente: Medicago sativa) con una proporción C/N entre 1271 y 16/1. La proporción alfalfa-muestra de suelo recomendada es 5 g de alfalfa por kilo de muestra (peso en seco).
1.6.5. Preparación de la sustancia de ensayo para la aplicación al suelo
La sustancia de ensayo se aplica normalmente utilizando un vehículo. El vehículo puede ser agua (para las sustancias solubles en el agua) o un sólido inerte como arena fina de cuarzo (tamaño de las partículas: 0,1-0,5 mm). Deben evitarse los vehículos líquidos distintos del agua (por ejemplo, disolventes orgánicos como la acetona o el cloroformo) porque pueden dañar la microflora. Si se usa arena como vehículo, puede recubrirse con la sustancia de ensayo disuelta o suspendida en un disolvente apropiado. En tal caso, debe eliminarse el disolvente por evaporación antes de mezclarse con la muestra de suelo. Para una distribución óptima de la sustancia de ensayo en el suelo, se recomienda una proporción de 10 g de arena por kilo de suelo (peso en seco). Las muestras de control se tratarán solo con una cantidad equivalente de agua y/o arena de cuarzo.
Cuando se ensayen sustancias químicas volátiles, deben evitarse pérdidas durante el tratamiento en la medida de lo posible y habrá que procurar conseguir una distribución homogénea en la muestra de suelo (por ejemplo, deberá inyectarse la sustancia de ensayo en la muestra en varios sitios).
1.6.6. Concentraciones de ensayo
Cuando se ensayen productos agroquímicos, deberán utilizarse al menos 2 concentraciones. La concentración más baja debe corresponder, al menos, a la cantidad máxima que se prevé que llegue al suelo en la práctica mientras la concentración más alta tiene que ser un múltiplo de la concentración más baja. Las concentraciones de la sustancia de ensayo añadida al suelo se calcula suponiendo una incorporación uniforme a una profundidad de 5 cm y una densidad aparente del suelo de 1,5. Para los productos agroquímicos que se aplican directamente al suelo o para las sustancias químicas para las cuales puede calcularse la cantidad que llega al suelo, las concentraciones de ensayo recomendadas son la máxima concentración ambiental prevista [Predicted Environmental Concentration (PEC)]. Las sustancias destinadas a ser aplicadas a los suelos varias veces a lo largo de un ciclo tiene que ensayarse a concentraciones resultantes de multiplicar la PEC por el número máximo de aplicaciones previsto. Sin embargo, la concentración superior ensayada no debe superar 10 veces el índice máximo para una aplicación. Si se ensayan productos químicos no agrícolas, se empleará una serie geométrica de, al menos, 5 concentraciones. Las concentraciones ensayadas han de cubrir el intervalo necesario para determinar los valores ECX.
1.7. REALIZACIÓN DEL ENSAYO
1.7.1. Condiciones de exposición
1.7.1.1. Tratamiento y control
Cuando se ensayen productos agroquímicos, la muestra de suelo se dividirá en 3 partes de igual peso. Dos partes se mezclarán con el vehículo que contenga el producto y la otra, con el vehículo sin el producto (control). Se recomienda un mínimo de 3 copias para ambos tipos de muestras, las tratadas y las no tratadas. Cuando se ensayen productos químicos no agrícolas, la muestra de suelo se dividirá en 6 partes de igual peso. 5 de las muestras se mezclarán con el vehículo que contenga la sustancia de ensayo y la sexta, con el vehículo sin la sustancia. Se recomienda preparar 3 copias tanto para las muestras tratadas como para las de control. Debe procurarse una distribución homogénea de la sustancia de ensayo en las muestras tratadas. Durante la mezcla, se evitará la compactación de la muestra o la formación de grumos.
1.7.1.2. Incubación de las muestras de suelo
La incubación de las muestras de suelo puede hacerse de dos maneras: como dos muestras únicas, una de suelo tratado y otra de suelo no tratado, o como una serie de submuestras separadas de igual tamaño de suelo tratado, por una parte, y de suelo no tratado, por otra. Sin embargo, cuando se ensayen sustancias volátiles, el ensayo solo debe hacerse con una serie de submuestras separadas. Cuando las muestras de suelo se incuben en una muestra única, se prepararán grandes cantidades de suelo tratado y no tratado, y se tomarán las submuestras que deban analizarse según se necesite a lo largo del ensayo. La cantidad preparada inicialmente para las muestras tratadas y para las de control dependerá del tamaño de las submuestras, el número de ejemplares utilizados para el análisis y el número máximo previsto de períodos de muestreo. Las muestras de suelo incubadas como muestra única deben mezclarse bien antes de preparar submuestras. Cuando la incubación se haga en una serie de muestras separadas, las dos muestras únicas, una de suelo tratado y otra de suelo no tratado, se dividirán en el número de submuestras requerido, y estas se utilizarán según se necesite. En los experimentos en los que puedan preverse más de dos tiempos de muestreo, habrán de prepararse las submuestras suficientes, teniendo en cuenta los ejemplares y los tiempos de muestreo. Deberán incubarse, al menos, 3 copias de las muestras de ensayo en condiciones aerobias (véase el punto 1.7.1.1). Durante todos los ensayos, se utilizarán recipientes adecuados con suficiente espacio libre para evitar la aparición de condiciones anaerobias. Sin embargo, cuando se ensayen sustancias volátiles, el ensayo solo debe hacerse con una serie de submuestras separadas.
1.7.1.3. Condiciones y duración del ensayo
El ensayo se hará en la oscuridad a una temperatura de 20 ± 2 oC. La humedad de las muestras se mantendrá durante el ensayo entre el 40 % y el 60 % de la capacidad máxima de retención de agua del suelo (véase el punto 1.6.4.2) con una variación máxima del ±5 %. Podrá añadirse agua destilada y desionizada según se necesite.
La duración mínima del ensayo es de 28 días. Cuando se ensayen productos agroquímicos, se compararán los índices de formación de nitrato en las muestras tratadas y de control. Si estos índices difieren en más del 25 % al 28o día, se continuará el ensayo hasta que se obtenga una diferencia igual o menor al 25 % o bien durante un máximo de 100 días. En el caso de los productos químicos no agrícolas, se pondrá fin al ensayo a los 28 días. Al 28o día, se determinarán las cantidades de nitratos en las muestras tratadas y de control y se calcularán los valores ECX.
1.7.2. Toma de muestras y análisis de suelos
1.7.2.1. Calendario de toma de muestras
Cuando se ensayen productos agroquímicos, se hará el análisis de nitratos en las muestras de suelo los días 0, 7, 14 y 28. Cuando se necesite un ensayo prolongado, se harán nuevas mediciones a intervalos de 14 de días a partir del 28o día.
Cuando se ensayen productos químicos no agrícolas, se utilizarán como mínimo 5 concentraciones de ensayo y se hará el análisis de nitratos en las muestras de suelo al principio (día 0) y al final del período de exposición (28 días). Si se considera necesario, puede añadirse una medición intermedia, por ejemplo, al séptimo día. Los datos obtenidos al 28o día se utilizarán para hallar el valor ECx de la sustancia química. Si se desea, pueden utilizarse los datos de las muestras de control obtenidos el día 0 para indicar la cantidad inicial de nitratos en el suelo.
1.7.2.2. Análisis de las muestras de suelo
La cantidad de nitratos formada en cada ejemplar de muestra tratado y de control se determinará en cada tiempo de muestreo. Los nitratos se extraerán de la muestra de suelo agitándola con un disolvente de extracción adecuado, por ejemplo, una solución 0,1 M de cloruro de potasio. Se recomienda una proporción de 5 ml de KCl por gramo equivalente de suelo en peso seco. Para optimizar la extracción, los recipientes que contengan la muestra de suelo y la solución de extracción no deberán llenarse más de la mitad. Las mezclas se agitarán a 150 rpm durante 60 minutos. A continuación, se centrifugarán o filtrarán y se analizará la fase líquida para hallar los nitratos. Los extractos líquidos sin partículas pueden almacenarse antes del análisis a menos 20 ± 5 oC hasta 6 meses.
2. RESULTADOS
2.1. TRATAMIENTO DE RESULTADOS
Si se hacen ensayos con productos agroquímicos, deberán registrarse las cantidades de nitratos formadas en cada copia de muestra de suelo y darse en forma de tabla los valores medios de todos los ejemplares. Se evaluarán los índices de transformación del nitrógeno mediante métodos estadísticos apropiados y generalmente aceptables (por ejemplo, test-F, nivel de significación del 5 %). Las cantidades de nitrato formadas se expresarán en mg de nitrato/kg de muestra (peso seco)/día. La tasa de formación de nitrato en cada tratamiento se comparará con el de la muestra de control y se calculará el porcentaje de desviación respecto a esta.
Cuando se ensayen productos químicos no agrícolas, se determinará la cantidad de nitratos formada en cada copia y se trazará una curva dosis-respuesta para el cálculo de los valores ECx. Las cantidades de nitratos (es decir, mg de nitratos/kg de muestra en peso seco) halladas en las muestras tratadas a los 28 días se compararán con las halladas en las muestras de control. A partir de estos datos, se calculará los valores porcentuales de inhibición para cada concentración de ensayo. Estos porcentajes se representarán en un gráfico en función de la concentración y, a continuación, se utilizarán procedimientos estadísticos para calcular los valores ECX. Los límites de confianza (p = 0,95) para el valor ECX calculado se hallarán también utilizando procedimientos estándar (10) (11) (12).
Las sustancias de ensayo que contengan grandes cantidades de nitrógeno pueden contribuir a las cantidades de nitratos formados durante el ensayo. Si estas sustancias se ensayan a concentraciones altas (por ejemplo, sustancias químicas que esté previsto utilizar en aplicaciones repetidas), deben incluirse en el ensayo muestras de control apropiadas (es decir, muestra de suelo más sustancia de ensayo, pero sin harina vegetal). Los datos de estas muestras de control deben tenerse en cuenta en los cálculos de la ECX.
2.2. INTERPRETACIÓN DE LOS RESULTADOS
Cuando se evalúen los resultados de los ensayos con productos agroquímicos y la diferencia en los índices de formación de nitratos entre el tratamiento más bajo (es decir, la concentración máxima prevista) y la muestra de control no supere el 25 % en cualquier tiempo de muestreo a partir del 28o día, el producto podrá considerarse que no tiene influencia a largo plazo en la transformación de nitrógeno en el suelo. Cuando se evalúen los resultados de ensayos con sustancias químicas no agrícolas, se utilizarán los valores EC50, EC25 y/o EC10.
3. INFORME
El informe del ensayo debe incluir los datos siguientes:
|
|
Identificación completa del suelo utilizado, incluyendo:
|
|
|
Sustancia de ensayo:
|
|
|
Sustrato:
|
|
|
Condiciones de ensayo:
|
|
|
Resultados:
|
4. REFERENCIAS
|
(1) |
EPPO (1994). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Chemicals. Chapter 7: Soil Microflora. EPPO Bulletin 24: 1-16, 1994. |
|
(2) |
BBA (1990). Effects on the Activity of the Soil Microflora. BBA Guidelines for the Official Testing of Plant Protection Products, VI, 1-1 (2nd eds., 1990). |
|
(3) |
EPA (1987). Soil Microbial Community Toxicity Test. EPA 40 CFR Part 797.3700. Toxic Substances Control Act Test Guidelines; Proposed rule. September 28, 1987. |
|
(4) |
SETAC-Europe (1995). Procedures for assessing the environmental fate and ecotoxicity of pesticides, Ed. M.R. Lynch, Pub. SETAC-Europe, Brussels. |
|
(5) |
ISO/DIS 14238 (1995). Soil Quality — Determination of Nitrogen Mineralisation and Nitrification in Soils and the Influence of Chemicals on these Processes. Technical Committee ISO/TC 190/SC 4: Soil Quality -Biological Methods. |
|
(6) |
OECD (1995). Final Report of the OECD Workshop on Selection of Soils/Sediments, Belgirate, Italy, 18-20 January 1995. |
|
(7) |
ISO 10381-6 (1993). Soil quality — Sampling. Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory. |
|
(8) |
ISO 14240-1 (1997). Soil quality — Determination of soil microbial biomass — Part 1: Substrate-induced respiration method. |
|
(9) |
ISO 14240-2 (1997). Soil quality — Determination of soil microbial biomass — Part 2: Fumigation-extraction method. |
|
(10) |
Litchfield, J.T. and Wilcoxon F. (1949). A simplified method of evaluating dose-effect experiments. Jour. Pharmacol, and Exper. Ther., 96, 99-113. |
|
(11) |
Finney, D.J. (1971). Probit Analysis. 3rd ed., Cambridge, London and New-York. |
|
(12) |
Finney, D.J. (1978). Statistical Methods in biological Assay. Griffin, Weycombe, UK. |
C.22. MICROORGANISMOS DEL SUELO: ENSAYO DE TRANSFORMACIÓN DEL CARBONO
1. MÉTODO
El presente método reproduce las directrices de ensayo de la OCDE TG 217 (2000).
1.1. INTRODUCCIÓN
En el presente método de ensayo se describe un método de laboratorio diseñado para investigar los efectos potenciales a largo plazo de una exposición única a productos fitosanitarios y posiblemente otras sustancias químicas sobre la actividad de transformación del carbono por los microorganismos del suelo. El ensayo se basa principalmente en las recomendaciones de la Organización Europea y Mediterránea para la Protección de las Plantas (1). Sin embargo, se han tenido en cuenta otras directrices, como las del Biologische Bundesanstalt (2) de Alemania, la Environmental Protection Agency de Estados Unidos (3) y la SETAC (4). En un seminario de la OCDE sobre selección de suelos y sedimentos celebrado en Belgirate (Italia) en 1995 (5) se alcanzó un acuerdo sobre el número y tipos de suelos que deben utilizarse en estos ensayos. Las recomendaciones sobre recogida, manipulación y conservación de muestras de suelo se basan en un documento orientativo de la ISO (6) y en las recomendaciones del seminario de Belgirate.
En la valoración y evaluación de las características de las sustancias de ensayo, puede resultar necesaria la determinación de los efectos de la actividad microbiana en el suelo, por ejemplo, cuando se requieran datos sobre los posibles efectos secundarios de los productos fitosanitarios en la microflora del suelo o cuando sea previsible la exposición de los microorganismos del suelo a productos químicos distintos de los fitosanitarios. El ensayo de transformación del carbono se lleva a cabo para determinar los efectos de estos productos químicos en la microflora del suelo. Si se ensayan productos agroquímicos (por ejemplo, productos fitosanitarios, abonos y productos químicos para la silvicultura), se llevarán a cabo tanto ensayos de transformación del carbono como del nitrógeno. Si se trata de productos no agroquímicos, es suficiente el ensayo de transformación del nitrógeno. Sin embargo, si los valores EC50 del ensayo de transformación del nitrógeno de estos productos químicos se sitúan dentro del intervalo encontrado para los inhibidores de la nitrificación disponibles en el comercio (por ejemplo, la nitrapirina), puede hacerse un ensayo de transformación del carbono para obtener más información.
Los suelos están formados por componentes vivos y no vivos que se dan en mezclas complejas y heterogéneas. Los microorganismos desempeñan un papel importante en la descomposición y transformación de la materia orgánica en los suelos fértiles. Existen muchas especies que contribuyen de diversas maneras a la fertilidad del suelo. Cualquier interferencia a largo plazo en estos procesos bioquímicos podría perturbar el ciclo de los nutrientes y alterar la fertilidad del suelo. La transformación del carbono y del nitrógeno se da en todos los suelos fértiles. Aunque las comunidades microbianas responsables de estos procesos difieren de unos suelos a otros, las rutas de transformación son esencialmente las mismas.
Este método de ensayo está pensado para detectar los efectos negativos a largo plazo de una sustancia en el proceso de transformación del carbono en los suelos aerobios de superficie. El ensayo es sensible a alteraciones de la envergadura y la actividad de las comunidades microbianas responsables de la transformación del carbono, al someterlas a estrés químico y privación de carbono. Se utiliza un suelo arenoso con bajo contenido de materia orgánica. Se trata este suelo con la sustancia de ensayo y se incuba en condiciones que permitan un rápido metabolismo microbiano. En estas condiciones, las fuentes de carbono de fácil obtención se agotan rápidamente en el suelo. Este hecho ocasiona una privación de carbono que causa la muerte de las células microbianas e induce el letargo y/o la esporulación. Si el ensayo se prolonga durante más de 28 días, puede medirse la suma de estas reacciones en controles (de suelo no tratado) como pérdida progresiva de biomasa microbiana metabólicamente activa (7). Si la biomasa del suelo sometido a estrés de carbono, en las condiciones del ensayo, se ve afectada por la presencia de un producto químico, puede que no regrese al mismo nivel que el control. Por ello, las perturbaciones causadas por la sustancia de ensayo en cualquier momento de la realización de este persistirán frecuentemente hasta su finalización.
Los ensayos a partir de los cuales se desarrolló este método estaban pensados principalmente para sustancias respecto a las cuales puede preverse la cantidad que llegará al suelo. Este es el caso, por ejemplo, de los productos fitosanitarios, cuyo índice de aplicación en el campo es conocido. Para los productos agroquímicos, es suficiente el ensayo de dos dosis con arreglo al índice de aplicación estimado o previsto. Los productos agroquímicos pueden someterse a ensayo en forma de principios activos (p.a.) o de productos formulados. Sin embargo, el ensayo no se limita a los productos químicos con concentraciones en el medio ambiente previsibles. Cambiando tanto las cantidades de la sustancia de ensayo aplicada al suelo como la manera en que se evalúan los datos, el ensayo puede usarse también para productos químicos de los cuales se desconozca la cantidad que llegará al suelo. De este modo, en el caso de los productos no agroquímicos, se determinan los efectos de una serie de concentraciones en la transformación del carbono. A continuación, se utilizan los datos de estos ensayos para trazar una curva dosis-respuesta y calcular los valores EC, donde x es el porcentaje de efecto definido.
1.2 DEFINICIONES
Transformación del carbono: es la degradación por microorganismos de la materia orgánica hasta formar dióxido de carbono como producto final.
ECX (concentración efectiva): es la concentración de la sustancia de ensayo en el suelo que da lugar a un porcentaje x de inhibición de la transformación del carbono en dióxido de carbono.
EC50 (concentración efectiva mediana): es la concentración de la sustancia de ensayo en el suelo que da lugar a un 50 % de inhibición de la transformación del carbono en dióxido de carbono.
1.3 SUSTANCIAS DE REFERENCIA
Ninguna.
1.4 PRINCIPIO DEL MÉTODO DE ENSAYO
Se trata parte del suelo tamizado con la sustancia de ensayo y se deja otra sin tratar (control). Cuando se ensayen productos agroquímicos, se recomienda un mínimo de dos concentraciones de ensayo, que deberán elegirse en relación con la concentración más alta prevista sobre el terreno. Después de 0, 7, 14 y 28 días de incubación, se mezclarán muestras de suelos tratados y de control con glucosa y se medirán las tasas de respiración inducidas por la glucosa durante 12 horas consecutivas. Las tasas de respiración se expresan como dióxido de carbono liberado (mg dióxido de carbono/kg suelo seco/h) u oxígeno consumido (mg oxígeno/kg suelo/h). Se comparará la tasa de respiración en las muestras de suelo tratado con la de control y se calculará el porcentaje de desviación respecto a esta. Todos los ensayos tendrán una duración mínima de 28 días. Si, al 28o día, la diferencia entre los suelos tratados y los no tratados es igual o superior al 25 %, se continuarán las mediciones a intervalos de 14 días hasta un máximo de 100 días. Cuando se ensayen productos no agroquímicos, se añadirá a las muestras de suelo una serie de concentraciones de la sustancia de ensayo y se medirán al cabo de 28 días las tasas de respiración inducidas por glucosa (es decir, la media de las cantidades de dióxido de carbono formado u oxígeno consumido). Se analizarán los resultados de los ensayos con una serie de concentraciones utilizando un modelo de regresión y se calcularán los valores ECX (es decir, EC50, EC25 y/o EC10). Véanse las definiciones.
1.5 VALIDEZ DEL ENSAYO
Las evaluaciones de los resultados de los ensayos con productos agroquímicos se basan en diferencias relativamente pequeñas (de un valor medio de ± 25 %) entre el dióxido de carbono liberado o el oxígeno consumido en (o por) las muestras de suelo tratado y de control, por lo que grandes variaciones en las muestras de control pueden dar lugar a resultados falsos. Por este motivo, la variación entre muestras de control en paralelo deberá ser inferior al ± 15 %.
1.6 DESCRIPCIÓN DEL MÉTODO DE ENSAYO
1.6.1 Equipo
Se utilizarán recipientes de ensayo hechos de material químicamente inerte. Estos recipientes deberán ser de capacidad adecuada de acuerdo con el procedimiento utilizado para la incubación de suelos, es decir, incubación en una muestra única o en una serie de muestras separadas (véase el punto 1.7.1.2). Hay que procurar reducir al mínimo la pérdida de agua y permitir el intercambio de gas durante el ensayo (por ejemplo, pueden cubrirse los recipientes con láminas de polietileno perforado). Cuando se ensayen sustancias volátiles, deberán usarse recipientes sellables y herméticos cuyo tamaño sea tal que aproximadamente un cuarto de su volumen esté ocupado por la muestra de suelo.
Para la determinación de la respiración inducida por glucosa, se precisarán sistemas e instrumentos de incubación que permitan medir la producción de dióxido de carbono o el consumo de oxígeno. Se encontrarán ejemplos de este tipo de sistemas e instrumentos en la bibliografía, referencias (8) (9) (10) (11).
1.6.2 Selección y número de suelos
Se utilizará un único suelo cuyas características recomendadas serán las siguientes:
|
— |
contenido de arena: no inferior al 50 % ni superior al 75 %, |
|
— |
pH: 5,5-7,5, |
|
— |
contenido de carbono orgánico: 0,5-1,5 % |
|
— |
deberá medirse la masa microbiana (12)(13), cuyo contenido de carbono deberá ser al menos el 1 % del total de carbono orgánico en el suelo. |
En la mayoría de los casos, un suelo de estas características representa la situación más desfavorable, ya que la adsorción de la sustancia química de ensayo es mínima y su disponibilidad para la microflora es máxima. Por lo tanto, no son necesarios, por lo general, ensayos de otros suelos. Sin embargo, en algunas circunstancias, por ejemplo cuando la sustancia de ensayo esté indicada principalmente para determinados suelos, como los suelos forestales ácidos, o en el caso de productos químicos cargados electrostáticamente, puede resultar necesario utilizar un suelo adicional.
1.6.3 Recogida y conservación de muestras de suelo
1.6.3.1 Recogida
Deberá contarse con información detallada sobre la historia del lugar donde se haya recogido el suelo de ensayo. En particular, localización precisa, cubierta vegetal, fechas de tratamiento con productos fitosanitarios, con abonos orgánicos e inorgánicos, adición de materiales biológicos o contaminación accidental. El lugar elegido para la recogida de suelo tiene que ser tal que permita un uso a largo plazo. Se consideran adecuados los pastos permanentes, los campos con cosechas anuales de cereales (excepto el maíz) o los abonos verdes sembrados densamente. El lugar donde se recojan las muestras no deberá haber sido tratado con productos fitosanitarios durante un mínimo de un año antes de la recogida de estas. Además, no deberá haberse aplicado ningún abono orgánico durante al menos seis meses. El uso de abonos minerales solo será aceptable cuando responda a las necesidades del cultivo, y en ese caso no deberán tomarse muestras hasta transcurridos, al menos, tres meses desde la aplicación del abono. Deberá evitarse el uso de suelo tratado con abonos de efectos biocidas conocidos (por ejemplo, cianamida de calcio).
Deberá evitarse la toma de muestras durante o inmediatamente después de períodos prolongados (de más de 30 días) de sequía o encharcamiento. En el caso de los suelos arados, deberán tomarse muestras a una profundidad de 0 a 20 cm. Cuando se trate de pastizales u otros suelos que no se aren durante períodos más largos (al menos un ciclo de cultivo), la profundidad máxima del muestreo podrá ser ligeramente superior a 20 cm (por ejemplo, 25 cm). Las muestras de suelo se transportarán en recipientes y en condiciones de temperatura que garanticen que las propiedades iniciales del suelo no se alteren significativamente.
1.6.3.2 Conservación
Es preferible el uso de muestras de suelo recién recogidas. Si no se puede evitar el almacenamiento en el laboratorio, las muestras de suelo podrán guardarse en la oscuridad a 4 ± 2 oC durante un máximo de tres meses. Durante el almacenamiento, deberá asegurarse el mantenimiento de unas condiciones aerobias. Si se recogen muestras de suelo de zonas que estén heladas durante, al menos, tres meses al año, podrá considerarse la posibilidad de almacenarlas durante seis meses a - 18 oC. Se medirá la biomasa microbiana de las muestras almacenadas antes de cada experimento. La cantidad de carbono en la biomasa deberá ser como mínimo el 1 % del total de carbono orgánico de la muestra de suelo (véase el punto 1.6.2).
1.6.4 Manipulación y preparación del suelo para el ensayo
1.6.4.1 Preincubación
Cuando las muestras de suelo hayan estado almacenadas (véanse los puntos 1.6.4.2 y 1.7.1.3), se recomienda preincubar durante un período de entre 2 y 28 días. La temperatura y el contenido de humedad del suelo durante la preincubación deberán ser semejantes a las que se den durante el ensayo (véanse los puntos 1.6.4.2 y 1.7.1.3).
1.6.4.2 Características fisicoquímicas
Se separarán manualmente los objetos grandes (por ejemplo, piedras, trozos de plantas, etc.) de la muestra de suelo y, a continuación, se tamizará la muestra por vía húmeda sin secar en exceso para obtener un tamaño de las partículas no superior a 2 mm. Deberá ajustarse el contenido de humedad de la muestra con agua destilada o desionizada para llegar a un valor entre el 40 % y el 60 % de la capacidad de retención de agua máxima.
1.6.5 Preparación de la sustancia de ensayo para la aplicación al suelo
La sustancia de ensayo se aplica normalmente utilizando un vehículo. El vehículo podrá ser agua (para las sustancias solubles en el agua) o un sólido inerte como arena fina de cuarzo (tamaño de las partículas: 0,1-0,5 mm). Se evitarán los vehículos líquidos distintos del agua (por ejemplo, disolventes orgánicos como la acetona o el cloroformo), porque pueden dañar la microflora. Si se usa arena como vehículo, podrá recubrirse con la sustancia de ensayo disuelta o suspendida en un disolvente apropiado. En tal caso, deberá eliminarse el disolvente por evaporación antes de mezclarse con la muestra de suelo. Para una distribución óptima de la sustancia de ensayo en el suelo, se recomienda una proporción de 10 g de arena por kilo de suelo (peso en seco). Las muestras de control se tratarán solo con la cantidad equivalente de agua y/o arena de cuarzo.
Cuando se ensayen sustancias químicas volátiles, deberán evitarse las pérdidas durante el tratamiento y se procurará conseguir una distribución homogénea en la muestra de suelo (por ejemplo, deberá inyectarse la sustancia de ensayo en la muestra en varios sitios).
1.6.6 Concentraciones de ensayo
Si se someten a ensayo productos fitosanitarios u otros productos químicos cuyas concentraciones en el medio ambiente sean predecibles, deberán utilizarse al menos dos concentraciones. La concentración más baja corresponderá, al menos, a la cantidad máxima que se prevé que llegue al suelo en la práctica, y la concentración más alta será un múltiplo de la concentración más baja. Las concentraciones de la sustancia de ensayo añadida al suelo se calcularán suponiendo una incorporación uniforme hasta una profundidad de 5 cm y una densidad aparente del suelo de 1,5. Para los productos agroquímicos que se aplican directamente al suelo o para las sustancias químicas para las cuales puede predecirse la cantidad que llega al suelo, las concentraciones de ensayo recomendadas son la concentración ambiental prevista (PEC) y cinco veces esa concentración. Las sustancias destinadas a ser aplicadas a los suelos varias veces en la misma temporada se ensayarán a las concentraciones resultantes de multiplicar la PEC por el número máximo de aplicaciones previsto. Sin embargo, la concentración máxima ensayada no deberá superar diez veces el índice máximo para una aplicación.
Si se ensayan productos no agroquímicos, se empleará una serie geométrica de, al menos, cinco concentraciones. Las concentraciones ensayadas deberán cubrir el intervalo necesario para determinar los valores ECX.
1.7 REALIZACIÓN DEL ENSAYO
1.7.1 Condiciones de exposición
1.7.1.1 Tratamiento y control
Cuando se ensayen productos agroquímicos, la muestra de suelo se dividirá en tres partes de igual peso. Dos partes se mezclarán con el vehículo que contenga el producto, y la otra, con el vehículo sin el producto (control). Se recomienda un mínimo de tres ejemplares para ambos tipos de muestras, las tratadas y las no tratadas. Cuando se ensayen productos no agroquímicos, la muestra de suelo se dividirá en seis partes de igual peso. Cinco de las muestras se mezclarán con el vehículo que contenga la sustancia de ensayo, y la sexta, con el vehículo sin la sustancia. Se recomienda preparar tres ejemplares tanto para las muestras tratadas como las de control. Deberá procurarse una distribución homogénea de la sustancia de ensayo en las muestras tratadas. Durante la mezcla, se evitará la compactación de la muestra o la formación de grumos.
1.7.1.2 Incubación de las muestras de suelo
La incubación de las muestras de suelo puede hacerse de dos maneras: como dos muestras únicas, una de suelo tratado y otra de suelo no tratado, o como una serie de submuestras de igual tamaño de suelo tratado, por una parte, y de suelo no tratado, por otra. Sin embargo, cuando se ensayen sustancias volátiles, solo deberá hacerse el ensayo con una serie de submuestras separadas. Cuando las muestras de suelo se incuben en una muestra única, se prepararán grandes cantidades de suelo tratado y no tratado, y se tomarán las submuestras que deban analizarse según se necesite a lo largo del ensayo. La cantidad preparada inicialmente para las muestras tratadas y para las de control dependerá del tamaño de las submuestras, el número de ejemplares utilizados para el análisis y el número máximo previsto de períodos de muestreo. Deberán mezclarse bien las muestras de suelo incubadas como muestra única antes de preparar las submuestras. Cuando la incubación se haga en una serie muestras separadas, se dividirán las dos muestras, de suelo tratado y de suelo no tratado, en el número de submuestras requerido, y estas se utilizarán según se necesite. En los experimentos en los que puedan preverse más de dos tiempos de muestreo, habrán de prepararse las submuestras suficientes, teniendo en cuenta los ejemplares y los tiempos de muestreo. Deberán incubarse, al menos, tres ejemplares de las muestras de ensayo en condiciones aerobias (véase el punto 1.7.1.1). Durante todos los ensayos, se utilizarán recipientes adecuados con suficiente espacio libre para evitar la aparición de condiciones anaerobias. Sin embargo, cuando se ensayen sustancias volátiles, solo deberá realizarse el ensayo con una serie de submuestras separadas.
1.7.1.3 Condiciones y duración del ensayo
El ensayo se hará en la oscuridad a una temperatura ambiente de 20± 2 oC. El contenido de humedad de las muestras se mantendrá durante el ensayo entre el 40 % y el 60 % de la capacidad de retención de agua máxima del suelo (véase el punto 1.6.4.2) con una variación máxima del ± 5 %. Podrá añadirse agua destilada y desionizada según se necesite.
La duración mínima del ensayo será de 28 días. Si se están ensayando productos agroquímicos, se compararán las cantidades de dióxido de carbono liberado u oxígeno consumido en las muestras tratada y de control. Si difieren en más del 25 % al 28o día, se continuará el ensayo hasta que se obtenga una diferencia igual o menor al 25 % o bien durante un máximo de 100 días. En el caso de los productos no agroquímicos, se pondrá fina al ensayo a los 28 días. Al 28o día, se determinarán las cantidades de dióxido de carbono liberado u oxígeno consumido en las muestras tratada y de control y se calcularán los valores ECX.
1.7.2 Toma de muestras y análisis de suelos
1.7.2.1 Calendario de toma de muestras
Cuando se ensayen productos agroquímicos, se hará el análisis de tasas de respiración inducida por glucosa en las muestras de suelo los días 0, 7, 14 y 28. Cuando se necesite un ensayo prolongado, se efectuarán nuevas mediciones a intervalos de 14 días a partir del 28o día.
Cuando se ensayen productos no agroquímicos, se utilizarán como mínimo cinco concentraciones de ensayo y se hará el análisis de la respiración inducida por glucosa en las muestras de suelo al principio (día 0) y al final del período de exposición (28 días). Si se considera necesario, podrá añadirse una medición intermedia, por ejemplo, al séptimo día. Se utilizarán los datos obtenidos al 28o día para hallar el valor ECX de la sustancia química. Si se desea, podrán utilizarse los datos de las muestras de control obtenidos el día 0 para estimar las cantidades iniciales de biomasa microbiana metabólicamente activa en el suelo (12).
1.7.2.2 Medida de las tasas de respiración inducida por glucosa
Se determinará la tasa de respiración inducida por glucosa en cada ejemplar de muestra tratada y de control en cada momento de muestreo. Se mezclarán las muestras de suelo con una cantidad de glucosa suficiente para suscitar una respuesta respiratoria máxima inmediata. Dicha cantidad de glucosa necesaria para suscitar una respuesta respiratoria máxima en un suelo dado puede determinarse en un ensayo preliminar utilizando una serie de concentraciones de glucosa (14). Sin embargo, en el caso de suelos arenosos con 0,5-1,5 % de carbono orgánico, suelen resultar suficientes de 2 000 mg a 4 000 mg de glucosa por kg de suelo (peso en seco). Es posible obtener un polvo mezclando la glucosa con arena de cuarzo limpia (10 g de arena/kg de suelo, peso en seco) y mezclarlo con el suelo de manera homogénea.
Se incubarán las muestras de suelo enmendadas con glucosa en un aparato adecuado para medir las tasas de respiración continuamente, cada hora o cada dos horas (véase el punto 1.6.1) a 20 ± 2 oC. Se medirá el dióxido de carbono liberado o el oxígeno consumido durante 12 horas consecutivas, iniciándose las medidas lo antes posible, es decir, de 1 a 2 horas después de añadido el suplemento de glucosa. Se medirán las cantidades totales de dióxido de carbono liberado u oxígeno consumido durante las 12 horas y se determinarán las tasas de respiración promedio.
2 RESULTADOS
2.1 TRATAMIENTO DE LOS RESULTADOS
Si se trata de productos agroquímicos, deberán registrarse las cantidades de dióxido de carbono liberado u oxígeno consumido en cada ejemplar de muestra de suelo, tabulándose además los valores promedio de todos los ejemplares. Se evaluarán los resultados mediante métodos estadísticos apropiados y generalmente aceptables (por ejemplo, prueba F, nivel de significación del 5 %). Las tasas de respiración inducida por glucosa se expresan en mg de dióxido de carbono/kg suelo, peso en seco/h o en mg oxígeno/suelo, peso en seco/h. Se comparará la tasa promedio de formación de dióxido de carbono o la tasa promedio de consumo de oxígeno en cada tratamiento con la de control, calculándose la desviación porcentual con respecto al control.
Cuando se ensayen productos no agroquímicos, se determinarán las cantidades de dióxido de carbono liberadas o de oxígeno consumidas por cada ejemplar y se trazará una curva dosis-respuesta para estimar los valores ECx. Se compararán las tasas de respiración inducida por glucosa (es decir, mg de dióxido de carbono/kg suelo, peso en seco/h o mg oxígeno/suelo, peso en seco/h) que presenten las muestras tratadas transcurridos 28 días con las que presenten las muestras de control. A partir de estos datos, se calcularán los valores porcentuales de inhibición para cada concentración de ensayo. Se representarán gráficamente estos porcentajes frente a la concentración, utilizándose luego procedimientos estadísticos para calcular los valores ECX. Utilizando asimismo procedimientos estándar, se determinarán los limites de confianza (p = 0,95) para los ECX calculados (10)(11)(12).
2.2 INTERPRETACIÓN DE LOS RESULTADOS
Cuando se evalúen los resultados de los ensayos con productos agroquímicos y la diferencia en las tasas de respiración entre el tratamiento más bajo (es decir, la concentración máxima prevista) y la muestra de control no supere el 25 % en ningún momento de muestreo a partir del 282 día, podrá considerarse que el producto no tiene influencia a largo plazo en la transformación de carbono en el suelo. Cuando se evalúen los resultados de ensayos con productos no agroquímicos, se utilizarán los valores EC50, EC25 y/o EC10
3 INFORME
INFORME DEL ENSAYO
El informe del ensayo deberá incluir la información siguiente:
|
|
Identificación completa del suelo utilizado, incluyendo:
|
|
|
Sustancia de ensayo:
|
|
|
Condiciones de ensayo:
|
|
|
Resultados:
|
4 REFERENCIAS
|
(1) |
EPPO (1994). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Chemicals. Chapter 7: Soil Microflora. EPPO Bulletin 24: 1-16, 1994. |
|
(2) |
BBA (1990). Effects on the Activity of the Soil Microflora. BBA Guidelines for the Official Testing of Plant Protection Products, VI, 1-1 (2nd eds., 1990). |
|
(3) |
EPA (1987). Soil Microbial Community Toxicity Test. EPA 40 CFR Part 797.3700. Toxic Substances Control Act Test Guidelines; Proposed rule. September 28, 1987. |
|
(4) |
SETAC-Europe (1995). Procedures for assessing the environmental fate and ecotoxicity of pesticides, Ed. M.R. Lynch, Pub. SETAC-Europe, Brussels. |
|
(5) |
OECD (1995). Final Report of the OECD Workshop on Selection of Soils/Sediments, Belgirate, Italy, 18-20 January 1995. |
|
(6) |
ISO 10381-6 (1993). Soil quality — Sampling. Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory. |
|
(7) |
Anderson, J.P.E. (1987). Handling and Storage of Soils for Pesticide Experiments, in «Pesticide Effects on Soil Microflora». Eds. L. Somerville and M.P. Greaves, Chap. 3:45-60. |
|
(8) |
Anderson, J.P.E. (1982). Soil Respiration, in «Methods of Soil Analysis — Part 2: Chemical and Microbiological Properties». Agronomy Monograph No 9. Eds. A.L. Page, R.H. Miller and D.R. Keeney. 41:831-871. |
|
(9) |
ISO 11266-1. (1993). Soil Quality — Guidance on Laboratory Tests for Biodegradation in Soil: Part 1. Aerobic Conditions. |
|
(10) |
ISO 14239 (1997E). Soil Quality — Laboratory incubation systems for measuring the mineralization of organic chemicals in soil under aerobic conditions. |
|
(11) |
Heinemeye, r O., Insam, H., Kaiser, E.A, and Walenzik, G. (1989). Soil microbial biomass and respiration measurements; an automated technique based on infrared gas analyses. Plant and Soil, 116: 77-81. |
|
(12) |
ISO 14240-1 (1997). Soil quality — Determination of soil microbial biomass — Part 1: Substrate-induced respiration method. |
|
(13) |
ISO 14240-2 (1997). Soil quality — Determination of soil microbial biomass — Part 2: Fumigation- extraction method. |
|
(14) |
Malkomes, H.-P. (1986). EinfluB von Glukosemenge auf die Reaktion der Kurzzeit-Atmung im Boden Gegenüber Pflanzenschutzmitteln, Dargestellt am Beispiel eines Herbizide. (Influence of the Amount of Glucose Added to the Soil on the Effect of Pesticides in Short-Term Respiration, using a Herbicide as an Example). Nachrichtenbl. Deut. Pflanzenschutzd., Braunschweig, 38: 113-120. |
|
(15) |
Litchfield, J.T. and Wilcoxon, F. (1949). A simplified method of evaluating dose-effect experiments. Jour. Pharmacol. and Exper. Ther., 96, 99-113. |
|
(16) |
Finney, D.J. (1971). Probit Analysis. 3rd ed., Cambridge, London and New-York. |
|
(17) |
Finney D.J. (1978). Statistical Methods in biological Assay. Griffin, Weycombe, UK. |
C.23. TRANSFORMACIÓN AEROBIA Y ANAEROBIA EN EL SUELO
1. MÉTODO
El presente método reproduce las directrices de ensayo de la OCDE TG 307 (2002).
1.1 INTRODUCCIÓN
El presente método se basa en las actuales directrices (1)(2)(3)(4)(5)(6)(7)(8)(9). El método está pensado para evaluar la transformación aerobia y anaerobia de las sustancias químicas en el suelo. Con los experimentos se pretende determinar: i) la tasa de transformación de la sustancia de ensayo, y ii) la naturaleza y las tasas de formación y disminución de los productos de transformación a los que pueden estar expuestos los vegetales y los organismos del suelo. Se precisa de dichos estudios para las sustancias químicas que se aplican directamente al suelo o es probable que alcancen el medio edáfico. Los resultados de los estudios pueden utilizarse igualmente para elaborar protocolos de muestreo y análisis para estudios de campo afines.
Por regla general, para evaluar las vías de transformación bastará efectuar estudios aerobios y anaerobios con un tipo de suelo (8)(10)(l 1). Las tasas de transformación, sin embargo, deberán determinarse por lo menos en otros tres suelos (8)(10).
En un seminario de la OCDE sobre selección de suelos y sedimentos celebrado en Belgirate (Italia) en 1995 (10) se alcanzó un acuerdo, en particular, sobre el número y tipos de suelos que deben utilizarse en estos ensayos. Los tipos de suelos sometidos a ensayo deben ser representativos de las condiciones ambientales en las que tendrá lugar el uso o la liberación. Por ejemplo, las sustancias químicas que vayan a liberarse en climas tropicales o subtropicales deben ensayarse en ferralsoles y nitosoles (sistema FAO). Dicho seminario formuló asimismo recomendaciones sobre recogida, manipulación y conservación de muestras de suelos (15). También se examina en este método el uso de suelos de (arroz) paddy.
1.2 DEFINICIONES
Sustancia de ensayo: cualquier sustancia, sea el compuesto original o los correspondientes productos de transformación.
Productos de transformación: todas las sustancias resultantes de las reacciones de transformación biótica o abiótica de la sustancia de ensayo, incluido el CO2 y los productos que se encuentran en los residuos ligados.
Residuos ligados: compuestos del suelo, la planta o el animal que persisten en la matriz en forma de sustancia original o de sus productos de transformación/metabolitos tras la extracción. El método de extracción no debe modificar sustancialmente ni los propios compuestos ni la estructura de la matriz. La naturaleza del vínculo puede aclararse parcialmente mediante métodos de extracción que alteren la matriz y técnicas analíticas sofisticadas. Así por ejemplo, hasta la fecha se han identificado de esta manera enlaces covalentes, iónicos y de sorción, así como atrapamientos. En general, la formación de residuos ligados reduce de manera significativa la bioaccesibilidad y la biodisponibilidad (12) [modificado de IUPAC 1984 (13)].
Transformación aerobia: reacciones que se producen en presencia de oxígeno molecular (14).
Transformación anaerobia: reacciones que se producen en ausencia de oxígeno molecular (14).
Suelo: mezcla de constituyentes químicos minerales y orgánicos; estos últimos contienen compuestos de elevado contenido en carbono y en nitrógeno y de peso molecular elevado, animados por pequeños organismos (principalmente microorganismos). El suelo puede manipularse en dos estados:
|
a) |
no perturbado, tal como se ha desarrollado en el tiempo, en capas características de diversos tipos de suelo; |
|
b) |
perturbado, como suele encontrarse en los campos cultivables o como se presenta cuando se toman muestras mediante excavación para utilizarlas en el presente método de ensayo (14). |
Mineralización: degradación completa de un compuesto orgánico a CO2 y H2O en condiciones aerobias y a CH4, CO2 y H2O en condiciones anaerobias. En el contexto del presente método, cuando se utiliza un compuesto marcado con 14C, mineralización significa la amplia degradación durante la que se oxida un átomo de carbono marcado con liberación de la cantidad apropiada de 14CO2 (14).
Semivida: to, s, es el tiempo que tarda la transformación del 50 % de una sustancia de ensayo cuando puede describirse dicha transformación mediante una cinética de primer orden; es independiente de la concentración.
DT 50 (tiempo de desaparición 50): es el tiempo que tarda la concentración de la sustancia de ensayo en reducirse en un 50 %; es distinto de la semivida t0,5 cuando la transformación no sigue una cinética de primer orden.
DT 75 (tiempo de desaparición 75): es el tiempo que tarda la concentración de la sustancia de ensayo en reducirse en un 75 %.
DT 90 (tiempo de desaparición 90): es el tiempo que tarda la concentración de la sustancia de ensayo en reducirse en un 90 %.
1.3 SUSTANCIAS DE REFERENCIA
Deberán utilizarse sustancias de referencia para la caracterización y/o identificación de los productos de transformación por métodos espectroscópicos o cromatográficos.
1.4 APLICABILIDAD DEL ENSAYO
El método es aplicable a todas las sustancias químicas (no marcadas o marcadas radiactivamente) para las que se dispone de un método analítico de suficiente exactitud y sensibilidad. Es aplicable a compuestos ligeramente volátiles, no volátiles, solubles en agua o insolubles en agua. El ensayo no deberá aplicarse a sustancias químicas que sean muy volátiles a partir del suelo (por ejemplo, fumigantes o disolventes orgánicos) y, por tanto, no se puedan mantener en el suelo en las condiciones experimentales del presente ensayo.
1.5 INFORMACIÓN SOBRE LA SUSTANCIA DE ENSAYO
Para medir la tasa de transformación podrá utilizarse una sustancia de ensayo marcada o no marcada. Para estudiar la vía de transformación y establecer un balance de materia se requiere material marcado. Se recomienda el marcado con 14C, pero también puede resultar de utilidad el uso de otros isótopos, tales como 13C, 15N, 3H o 32P. El marcador deberá ubicarse, en la medida de lo posible, en la parte o las partes más estables de la molécula (37). La pureza de la sustancia de ensayo deberá ser de al menos el 95 %.
Antes de realizar un ensayo sobre la transformación aerobia o anaerobia en el suelo, deberá contarse con la siguiente información relativa a la sustancia de ensayo.
|
a) |
solubilidad en agua (Método A.6); |
|
b) |
solubilidad en disolventes orgánicos; |
|
c) |
presión de vapor (Método A.4) y constante de la ley de Henry; |
|
d) |
coeficiente de reparto n-octanol/agua (Método A.8); |
|
e) |
estabilidad química en la oscuridad (hidrólisis) (Método C.7); |
|
f) |
pKa si una molécula es susceptible de protonación o desprotonación [Directriz 112 de la OCDE] (16). |
También pueden resultar de utilidad los datos sobre la toxicidad de la sustancia de ensayo para los microorganismos del suelo [Métodos de ensayo C.21 y C.22] (16).
Deberá contarse con métodos analíticos (incluidos métodos de extracción y depuración) para la cuantificación e identificación de la sustancia de ensayo y de sus productos de transformación.
1.6 PRINCIPIO DEL MÉTODO DE ENSAYO
Se tratan las muestras de suelo con la sustancia de ensayo y se incuban en la oscuridad en un matraz biométrico o en sistemas de flujo en condiciones de laboratorio controladas (a humedad del suelo y temperatura constantes). A intervalos de tiempo adecuados, se extraen muestras del suelo y se analizan en ellas la sustancia original y los productos de transformación. También se recogen los productos volátiles para su análisis con los dispositivos de absorción adecuados. Utilizando material marcado con C es posible medir las diversas tasas de mineralización de la sustancia de ensayo atrapando el 14CO2 resultante y establecer un balance de materia, incluyendo la formación de residuos ligados al suelo.
1.7 CRITERIOS DE CALIDAD
1.7.1 Recuperación
La extracción y el análisis de muestras de suelo cuando menos duplicadas inmediatamente después de la adición de la sustancia de ensayo dan una primera indicación de la repetibilidad del método analítico y de la uniformidad del procedimiento de aplicación de la sustancia de ensayo. Los valores de recuperación para fases posteriores del experimento los dan los balances másicos respectivos. Dichos valores deberán situarse entre el 90 % y el 110 % para sustancias químicas marcadas (8) y entre el 70 % y el 110 % para sustancias químicas no marcadas (3).
1.7.2 Repetibilidad y sensibilidad del método analítico
La repetibilidad del método analítico (excluida la eficiencia de extracción inicial) para cuantificar la sustancia de ensayo y los productos de transformación puede comprobarse realizando un análisis por duplicado del mismo extracto de suelo incubado durante tiempo suficiente para que se formen productos de transformación.
El límite de detección (LOD) del método analítico de la sustancia de ensayo y de los productos de transformación deberá alcanzar, como mínimo, el menor de los siguientes valores: 0,01 mg-kg-1 de suelo (como sustancia de ensayo) o un 1 % de la dosis aplicada. También deberá especificarse el límite de cuantificación (LOQ).
1.7.3 Exactitud de los datos de transformación
El análisis de regresión de las concentraciones de la sustancia de ensayo en función del tiempo facilita la información apropiada sobre, la fiabilidad de la curva de transformación y permite calcular los límites de confianza de las semividas (en el caso de la cinética de pseudo primer orden) o los valores DT50 y, si procede, DT75 y DT90.
1.8 DESCRIPCIÓN DEL MÉTODO DE ENSAYO
1.8.1 Equipos y reactivos químicos
Los sistemas de incubación consisten en sistemas cerrados estáticos o sistemas de flujo adecuados (7) (17). En las figuras 1 y 2 se muestran ejemplos de, respectivamente, aparatos de flujo de incubación de suelo y matraces biométricos adecuados. Los dos tipos de sistema de incubación tienen sus ventajas y sus limitaciones (7) (17).
Se precisa el equipo estándar de laboratorio y, en particular:
|
— |
instrumentos analíticos tales como equipos GLC, HPLC, TLC, incluidos los sistemas de detección adecuados para el análisis de sustancias marcadas o no marcadas o el método de dilución isotópica inversa, |
|
— |
instrumentos de identificación (por ejemplo, MS, GC-MS, HPLC-MS, NMR, etc.), |
|
— |
contador de centelleo de líquidos, |
|
— |
cámara de combustión oxidante de material radiactivo, |
|
— |
centrifugadora, |
|
— |
aparato de extracción (por ejemplo, tubos de centrifugación para extracción en frío y aparato Soxhlet para extracción continua a reflujo), |
|
— |
instrumental para concentrar soluciones y extractos (por ejemplo, evaporador rotatorio), |
|
— |
baño maria, |
|
— |
dispositivo mezclador mecánico (por ejemplo, amasadora, mezcladora rotatoria). |
Entre los reactivos químicos figuran, por ejemplo:
|
— |
NaOH, pureza de grado analítico, 2 mol dm-3, u otra base apropiada (por ejemplo, KOH, etanolamina), |
|
— |
H2SO4, pureza de grado analítico, 0,05 mol dm-3 |
|
— |
glicol etileno, pureza de grado analítico, |
|
— |
materiales sólidos de absorción, tales como cal sodada y piezas de poliuretano, |
|
— |
disolventes orgánicos, pureza de grado analítico, tales como acetona, metanol, etc., |
|
— |
líquido de centelleo. |
1.8.2 Aplicación de la sustancia de ensayo
Para incorporarla al suelo y distribuirla en él, podrá disolverse la sustancia de ensayo en agua (desionizada o destilada) o, si resulta necesario, en cantidades mínimas de acetona u otro disolvente orgánico (6) en el que la sustancia de ensayo sea suficientemente soluble y estable. No obstante, la cantidad de disolvente seleccionada no deberá tener una influencia significativa sobre la actividad microbiana del suelo (véanse los puntos 1.5 y 1.9.2-1.9.3. Deberá evitarse el uso de disolventes inhibidores de la actividad microbiana, tales como cloroformo, diclorometano y otros disolventes halogenados.
Podrá incorporarse asimismo la sustancia de ensayo en forma sólida, por ejemplo, mezclada con arena de cuarzo (6) o en una pequeña submuestra del suelo sometido a ensayo que se haya secado al aire y esterilizado. Si se incorpora la sustancia de ensayo utilizando un disolvente, deberá permitirse que este se evapore antes de incorporar la submuestra a la muestra de suelo original no estéril.
Para las sustancias químicas ordinarias, cuya principal vía de entrada en el suelo es a través de los lodos de depuración o la actividad agraria, deberá incorporarse primero la sustancia de ensayo al lodo, que luego se introducirá en la muestra de suelo, (véanse los puntos 1.9.2 y 1.9.3).
No se recomienda la utilización rutinaria de productos formulados. No obstante, su utilización podría constituir una alternativa apropiada, por ejemplo, en el caso de sustancias de ensayo poco solubles.
1.8.3 Suelos
1.8.3.1 Selección de suelos
Para determinar la vía de transformación, podrá utilizarse un suelo representativo; se recomienda un suelo franco arenoso o franco limoso o franco o arenoso franco [según la clasificación de FAO y USDA (18)] con un pH de 5,5-8,0, un contenido de carbono orgánico de 0,5-2,5 % y una biomasa microbiana de al menos 1 % del carbono orgánico total (10).
Para estudios de tasa de transformación deberán utilizarse al menos tres suelos adicionales representativos de la gama de suelos relevante. Estos suelos deben variar en cuanto a contenido de carbono orgánico, pH, contenido de arcilla y biomasa microbiana (10).
Deberán caracterizarse todos los suelos, cuando menos, en cuanto a textura ( % arena, % limo, % arcilla) [según la clasificación de FAO y USDA (18)], pH, capacidad de intercambio de cationes, carbono orgánico, densidad aparente, característica de retención de agua (38) y biomasa microbiana (solo para estudios aerobios). Cualquier otra información sobre las propiedades del suelo podría resultar de utilidad para la interpretación de los resultados. Para la determinación de las características del suelo, podrán utilizarse los métodos recomendados en las referencias (19)(20)(21)(22)(23). La biomasa microbiana se determinará utilizando el método de respiración inducida por el sustrato (SIR) (25)(26) o un método alternativo (20).
1.8.3.2 Recogida, manipulación y conservación de suelos
Deberá contarse con información detallada sobre la historia del lugar donde se haya recogido el suelo de ensayo. En particular, localización precisa, cubierta vegetal, tratamientos con sustancias químicas, tratamientos con abonos orgánicos e inorgánicos, adición de materiales biológicos u otro tipo de contaminación. No deberán utilizarse para estudios sobre transformación suelos que hayan sido tratados con la sustancia de ensayo o un análogo estructural de la misma dentro de los cuatro años precedentes (10)(15).
La muestra deberá haber sido recogida recientemente (del horizonte A o capa superior de 20 cm) y su contenido en agua deberá facilitar su tamizado. Salvo en el caso de los suelos de arrozales paddy, deberá evitarse la toma de muestras durante o inmediatamente después de un largo período (> 30 días) de sequía, helada o inundación (14). Las muestras deberán transportarse de manera que el contenido en agua del suelo varíe lo menos posible y mantenerse en la oscuridad y ventiladas en la mayor medida posible. Suele estar indicado el uso de una bolsa de polietileno sin cerrar completamente.
Una vez tomada la muestra, deberá procesarse el suelo lo antes posible. Deberá retirarse la vegetación, la fauna edáfica de mayor tamaño y las piedras antes de hacer pasar el suelo por un tamiz de 2 mm que permita retirar los restos de vegetales y animales y pequeñas piedras. Se procurará evitar el secado y aplastamiento excesivos del suelo antes del tamizado (15).
Cuando resulte difícil tomar muestras en invierno (por hallarse el suelo helado o cubierto por una capa de nieve), podrá extraerse de un lote de suelo almacenado en el invernadero bajo cubierta vegetal (por ejemplo, hierba o mezcla de hierba y trébol). Pese a preferirse claramente los estudios con suelos recién recogidos del campo, si es preciso almacenar el suelo recogido y procesado antes de comenzar el estudio, las condiciones de conservación deberán ser las adecuadas y el plazo de almacenamiento limitado (4 ± 2 oC durante un máximo de tres meses) para mantener la actividad microbiana (39). Se encontrarán instrucciones detalladas sobre la recogida, manipulación y conservación de suelos con vistas a su uso en experimentos de biotransformación en (8)(10)(15)(26)(27).
Antes de utilizar en este ensayo el suelo procesado, deberá preincubarse este para hacer posible la germinación y eliminación de las semillas y restablecer el equilibrio del metabolismo microbiano tras el paso de las condiciones de muestreo o conservación a las de incubación. En general resulta adecuado un período de preincubación de entre 2 y 28 días en condiciones de temperatura y humedad próximas a las del ensayo real (15). La suma de los períodos de conservación y de preincubación no deberá exceder de tres meses.
1.9 REALIZACIÓN DEL ENSAYO
1.9.1 Condiciones de ensayo
1.9.1.1 Temperatura del ensayo
Durante la totalidad del período de ensayo, deberán incubarse los suelos en la oscuridad a una temperatura constante que sea representativa de las condiciones climáticas en que tendrá lugar el uso o la liberación. Se recomienda una temperatura de 20 ± 2 oC para todas las sustancias de ensayo que puedan llegar al suelo en climas templados. Deberá controlarse esta temperatura.
Si se trata de sustancias químicas aplicadas o liberadas en climas más fríos (por ejemplo, en países septentrionales, durante el otoño o el invierno), deberán incubarse muestras de suelo adicionales pero a una temperatura inferior (por ejemplo, 10± 2 oC).
1.9.1.2 Contenido de humedad
Para los ensayos de transformación en condiciones aerobias, deberá ajustarse el contenido de humedad (40) del suelo a un pF situado entre 2,0 y 2,5 y mantenerse en ese valor (3) El contenido de humedad del suelo se expresa como cociente entre masa de agua y masa de suelo seco y debe ser controlado periódicamente (por ejemplo, a intervalos de 2 semanas) pesando los recipientes de incubación y compensando las pérdidas de agua mediante la adición de, preferiblemente, agua de grifo esterilizada por filtración. Habrá que procurar evitar o reducir al mínimo las pérdidas de sustancia de ensayo y/o productos de transformación por volatilización y/o fotodegradación (en su caso) durante la adición de humedad.
Para los ensayos de transformación en condiciones anaerobias y de arrozal, se satura de agua el suelo mediante inundación.
1.9.1.3 Condiciones aerobias de incubación
En los sistemas de flujo, se mantendrán las condiciones aerobias mediante baldeo intermitente o ventilación continua con aire húmedo. En los matraces biométricos, se mantendrá el intercambio de aire mediante difusión.
1.9.1.4 Condiciones aerobias estériles
Con el fin de obtener información sobre la importancia de la transformación abiótica de una sustancia de ensayo, las muestras de suelo podrán ser esterilizadas [sobre métodos de esterilización, véanse las referencias (16) y (29)], tratadas con una sustancia de ensayo estéril (por ejemplo, adición de solución a través de un filtro estéril) y aireadas con aire humidificado estéril según se describe en el punto 1.9.1.3. En el caso de los suelos de arrozal, será preciso esterilizar el suelo y el agua, y efectuar la incubación según se describe en el punto 1.9.1.6.
1.9.1.5 Condiciones anaerobias de incubación
Para crear y mantener unas condiciones anaerobias, se procede a cubrir de agua (capa de 1-3 cm de agua) el suelo previamente tratado con la sustancia de ensayo e incubado en condiciones aerobias durante 30 días o una semivida o DT50 (el valor que sea más pequeño) y a inyectar un gas inerte (por ejemplo, nitrógeno o argón) en el sistema de incubación (41). El sistema de ensayo deberá permitir la medición del pH, la concentración de oxígeno y el potencial redox e incluir dispositivos de fijación de productos volátiles. El sistema biométrico deberá estar cerrado, para evitar la entrada de aire por difusión.
1.9.1.6 Condiciones de incubación de arrozal
Para estudiar la transformación en suelos de arrozal, se inundará el suelo con una capa de agua de 1-5 cm y se aplica la sustancia de ensayo a la fase acuosa (9). Se recomienda que la profundidad del suelo sea de al menos 5 cm. El sistema se ventila con aire como en condiciones aerobias. Deberán controlarse y registrarse el pH, la concentración de oxígeno y el potencial redox de la capa acuosa. Será necesario un período de preincubación de al menos dos semanas antes de dar comienzo a los estudios de transformación (véase el punto 1.8.3.2).
1.9.1.7 Duración del ensayo
Los estudios de tasa y vía no deberán exceder normalmente de 120 días (42) (3)(6)(8), porque transcurrido ese plazo es de esperar un decrecimiento de la actividad microbiana del suelo con el tiempo en un sistema artificial de laboratorio sin posibilidad de reconstitución natural. Cuando resulte necesario para caracterizar el declive de la sustancia de ensayo y la formación y declive de los principales productos de transformación, podrán prolongarse los estudios durante períodos más largos (por ejemplo, 6 o 12 meses) (8). El uso de un período de incubación más largo deberá quedar justificado en el informe del ensayo y acompañarse de medidas de biomasa durante y al final del período.
1.9.2 Realización del ensayo
Se colocan de 50 a 200 g de suelo (peso seco) en cada recipiente de incubación (véanse las figuras 1 y 2 del anexo 3) y se trata el suelo con la sustancia de ensayo mediante uno de los métodos descritos en el punto 1.8.2. Cuando se utilicen disolventes orgánicos para la aplicación de la sustancia de ensayo, deberán retirarse del suelo por evaporación. A continuación, se mezcla bien el suelo con una espátula y/o agitando el recipiente. Si el estudio se realiza en condiciones de arrozal, se mezclarán bien el suelo y el agua tras la aplicación de la sustancia de ensayo. Se analizarán pequeñas partes alícuotas (por ejemplo, 1 g) de los suelos tratados para comprobar la distribución uniforme de la sustancia de ensayo. Más adelante se presentan métodos alternativos.
La tasa de tratamiento debe corresponder a la tasa de aplicación más elevada de un producto fitosanitario recomendada en las instrucciones de empleo y a incorporación uniforme a una profundidad apropiada en el campo [por ejemplo, capa superior (43) de 10 cm de suelo]. Por ejemplo, para productos químicos aplicados sobre el follaje o el suelo sin incorporación, la profundidad apropiada para calcular la cantidad de producto químico que debe añadirse a cada recipiente es de 2,5 cm. Para productos químicos incorporados al suelo, la profundidad apropiada es la profundidad de incorporación especificada en las instrucciones de empleo. Para productos químicos ordinarios, deberá calcularse la tasa de aplicación sobre la base de la vía de penetración más importante; por ejemplo, cuando la vía principal de penetración en el suelo sean los lodos de depuración, deberá dosificarse el producto químico en el lodo a una concentración que corresponda a la concentración prevista en el lodo y deberá añadirse al suelo una cantidad de lodo que corresponda a su carga normal en los suelos agrícolas. Si esta concentración no es suficientemente elevada para permitir la identificación de los principales productos de transformación, podrá proceder la incubación de muestras de suelo independientes que contengan tasas superiores, pero deben evitarse las tasas excesivas que influyan en las funciones microbianas del suelo (véanse los puntos 1.5 y 1.8.2).
Alternativamente, se podrá tratar un lote de suelo mayor (de 1 a 2 kg) con la sustancia de ensayo, mezclándolo bien en una máquina mezcladora adecuada y transfiriéndolo luego en porciones pequeñas de 50 a 200 g a los recipientes de incubación (por ejemplo utilizando separadores de muestras). Se analizarán pequeñas partes alícuotas (por ejemplo, 1 g) del lote de suelo tratado para comprobar la distribución uniforme de la sustancia de ensayo. Resulta preferible este procedimiento porque hace posible una distribución más uniforme de la sustancia de ensayo en el suelo.
Por otra parte, se incuban muestras de suelo no tratadas en las mismas condiciones (aerobias) de las muestras tratadas con la sustancia de ensayo. Estas muestras se utilizan para medición de biomasa durante los estudios y al finalizar estos.
Cuando se aplica al suelo la sustancia de ensayo disuelta en uno o más disolventes orgánicos, se incuban muestras de suelo tratadas con la misma cantidad de disolvente(s) en las mismas condiciones (aerobias) que las muestras tratadas con la sustancia de ensayo. Estas muestras se utilizan para medir la biomasa al comienzo, durante y al final de los estudios, con el fin de comprobar los efectos del disolvente o los disolventes en la biomasa microbiana.
Los recipientes con el suelo tratado serán conectados al sistema de flujo descrito en la figura 1 o cerrados con la columna de absorción mostrada en la figura 2 (véase el anexo 3).
1.9.3 Muestreo y medidas
Se retirarán los recipientes de incubación duplicados a intervalos de tiempo apropiados, extrayéndose muestras de suelo con disolventes adecuados de distinta polaridad y analizándose para medir la sustancia de ensayo y/o los productos de transformación. Un estudio bien diseñado deberá incluir recipientes suficientes, de manera que puedan sacrificarse dos en cada toma de muestras. De la misma manera, se retirarán soluciones de absorción o materiales sólidos de absorción a distintos intervalos temporales (de 7 días durante el primer mes y de 17 días posteriormente) durante y al final de la incubación de cada muestra de suelo y se analizan para medir productos volátiles. Además de la muestra de suelo tomada directamente tras la aplicación (muestra del día cero), deberán incluirse al menos 5 puntos de muestreo adicionales. Se escogerán los intervalos de tiempo de manera que se pueda determinar el patrón de declive de la sustancia de ensayo y los patrones de formación y declive de los productos de transformación (por ejemplo, 0, 1, 3, 7 días; 2,3 semanas; 1, 2, 3 meses, etc.).
Cuando se use una sustancia de ensayo marcada con 14C, se cuantificará la radiactividad no extraíble mediante combustión y se calculará un balance de materia para cada intervalo de muestreo.
En caso de incubación anaerobia o de arrozal, se podrán analizar conjuntamente las fases de suelo y agua para medir la sustancia de ensayo y los productos de transformación, o separarlas por filtración o centrifugado antes de la extracción y el análisis.
1.9.4 Ensayos facultativos
Podría resultar de utilidad la realización de estudios aerobios no estériles para otros valores de temperatura y humedad del suelo para estimar la influencia de la temperatura y la humedad del suelo sobre la tasa de transformación de una sustancia de ensayo y/o de sus productos de transformación en el suelo.
Puede intentarse otra caracterización de la radiactividad no extraíble utilizando, por ejemplo, extracción mediante fluido supercrítico.
2 RESULTADOS
2.1 TRATAMIENTO DE LOS RESULTADOS
Las cantidades de sustancia de ensayo, productos de transformación, sustancias volátiles (solo en porcentaje) y no extraíbles se expresarán en porcentaje de la concentración inicial aplicada y, cuando proceda, en mg.kg-1 de suelo (sobre la base del peso del suelo en seco) para cada intervalo de muestreo. El balance de materia se expresará en porcentaje de la concentración inicial aplicada para cada intervalo de muestreo. La representación gráfica de las concentraciones de la sustancia de ensayo en función del tiempo permitirá estimar la semivida de su transformación o DT50. Deberán identificarse los principales productos de transformación y se representarán gráficamente sus concentraciones en función del tiempo para mostrar sus tasas de formación y declive. Se considerará producto de transformación principal cualquier producto que represente ≥ 10 % de la dosis aplicada en cualquier momento del estudio.
Los productos volátiles fijados dan una indicación de la volatilidad potencial de la sustancia de ensayo y de sus productos de transformación a partir del suelo.
Se obtendrán determinaciones más exactas de las semividas o valores DT50 y, si procede, valores DT75 y DT90, mediante cálculos con los modelos cinéticos apropiados. Se consignarán los valores de semivida y DT50 junto con la descripción del modelo utilizado, el orden de la cinética y el coeficiente de determinación (r2). Se prefiere la cinética de primer orden salvo que r2 < 0,7. Si procede, se aplicarán los cálculos asimismo a los principales productos de transformación. En las referencias (31) a (35) se describen ejemplos de modelos adecuados.
Si se trata de estudios de tasas efectuados a varias temperaturas, las tasas de transformación deberán describirse como función de la temperatura dentro del intervalo de temperaturas experimental utilizando la relación de Arrhenius de la forma:
 o
o 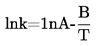 ,
,
donde ln A y B son constantes de regresión que corresponden a la ordenada en el origen y la pendiente, respectivamente, de una recta de mejor ajuste generada por regresión lineal de ln k frente a 1/T, k es la constante de tasa a temperatura T y T es la temperatura en Kelvin. Deberá prestarse atención al intervalo limitado de temperaturas en el que será válida la relación de Arrhenius en el caso de que la transformación esté gobernada por la acción microbiana.
2.2 EVALUACIÓN E INTERPRETACIÓN DE LOS RESULTADOS
Aunque los estudios se realizan en un sistema artificial de laboratorio, los resultados permitirán estimar la tasa de transformación de la sustancia de ensayo e igualmente la tasa de formación y declive de los productos de transformación en condiciones de campo (36)(37).
El estudio de la vía de transformación de una sustancia de ensayo aporta información sobre la transformación estructural de la sustancia aplicada en el suelo por reacciones químicas y microbianas.
3 INFORME
INFORME DEL ENSAYO
El informe del ensayo debe incluir la información siguiente:
|
|
Sustancia de ensayo:
|
|
|
Sustancias de referencia:
|
|
|
Suelos de ensayo:
|
|
|
Condiciones de ensayo:
|
|
|
Resultados:
|
4 REFERENCIAS
|
(1) |
US- Environmental Protection Agency (1982). Pesticide Assessment Guidelines, Subdivision N. Chemistry: Environmental Fate. |
|
(2) |
Agriculture Canada (1987). Environmental Chemistry and Fate. Guidelines for registration of pesticides in Canada. |
|
(3) |
Unión Europea (UE) (1995). Directiva 95/36/CE de la Comisión, de 14 de julio de 1995, por la que se modificà la Directiva 91/414/CEE del Consejo relativa a la comercialización de productos Fitosanitarios. Anexo II, parte A y anexo III, parte A: Destino y comportamiento en el medio ambiente. |
|
(4) |
Dutch Commission for Registration of Pesticides (1995). Application for registration of a pesticide. Section G: Behaviour of the product and its metabolites in soil, water and air. |
|
(5) |
BBA (1986). Richtlinie für die amtliche Prüfung von Pflanzenschutzmitteln, Teil IV, 4-1. Verbleib von Pflanzenschutzmitteln im Boden — Abbau, Umwandlung und Metabolismus. |
|
(6) |
ISO/DIS 11266-1 (1994). Soil Quality -Guidance on laboratory tests for biodegradation of organic chemicals in soil — Part 1: Aerobic conditions. |
|
(7) |
ISO 14239 (1997). Soil Quality — Laboratory incubation systems for measuring the mineralization of organic chemicals in soil under aerobic conditions. |
|
(8) |
SETAC (1995). Procedures for Assessing the Environmental Fate and Ecotoxicity of Pesticides. Mark R. Lynch, Ed. |
|
(9) |
MAFF — Japan 2000 — Draft Guidelines for transformation studies of pesticides in soil — Aerobic metabolism study in soil under paddy field conditions (flooded). |
|
(10) |
OECD (1995). Final Report of the OECD Workshop on Selection of Soils/Sediments. Belgirate, Italy, 18-20 January 1995. |
|
(11) |
Guth, J.A. (1980). The study of transformations. In Interactions between Herbicides and the Soil (R.J. Hance, Ed.), Academic Press, 123-157. |
|
(12) |
DFG: Pesticide Bound Residues in Soil. Wiley — VCH (1998). |
|
(13) |
T.R. Roberts: Non-extractable pesticide residue in soils and plants. Pure Appl. Chem. 56,945-956 (IUPAC 1984) |
|
(14) |
OECD Test Guideline 304 A: Inherent Biodegradability in Soil (adopted 12 May 1981) |
|
(15) |
ISO 10381-6 (1993). Soil Quality — Sampling — Part 6: Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory. |
|
(16) |
Anexo V de la Dir. 67/548/CEE. |
|
(17) |
Guth, J.A. (1981). Experimental approaches to studying the fate of pesticides in soil. In Progress in Pesticide Biochemistry. D.H. Hutson, T.R. Roberts, Eds. J. Wiley & Sons. Vol 1, 85-114. |
|
(18) |
Soil Texture Classification (US and FAO systems): Weed Science, 33, Suppl. 1 (1985) and Soil Sci. Soc. Amer. Proc. 26:305 (1962). |
|
(19) |
Methods of Soil Analysis (1986). Part 1, Physical and Mineralogical Methods. A. Klute, Ed.) Agronomy Series No 9, 2nd Edition. |
|
(20) |
Methods of Soil Analysis (1982). Part 2, Chemical and Microbiological Properties. A.L. Page, R.H. Miller and D.R. Kelney, Eds. Agronomy Series No 9, 2nd Edition. |
|
(21) |
ISO Standard Compendium Environment (1994). Soil Quality — General aspects; chemical and physical methods of analysis; biological methods of analysis. First Edition. |
|
(22) |
Mückenhausen, E. (1975). Die Bodenkunde und ihre geologischen, geomorphologischen, mineralogischen und petrologischen Grundlagen. DLG-Verlag, Frankfurt, Main. |
|
(23) |
Scheffer, F., Schachtschabel, P. (1975). Lehrbuch der Bodenkunde. F. Enke Verlag, Stuttgart. |
|
(24) |
Anderson, J.P.E., Domsch, K.H. (1978) A physiological method for the quantitative measurement of microbial biomass in soils. Soil Biol. Biochem. 10, 215-221. |
|
(25) |
ISO 14240-1 and 2 (1997). Soil Quality — Determination of soil microbial biomass — Part 1: Substrate-induced respiration method. Part 2: fumigation-extraction method. |
|
(26) |
Anderson, J.P.E. (1987). Handling and storage of soils for pesticide experiments. In Pesticide Effects on Soil Microflora. L. Somerville, M.P. Greaves, Eds. Taylor & Francis, 45-60. |
|
(27) |
Kato, Yasuhiro. (1998). Mechanism of pesticide transformation in the environment: Aerobic and biotransformation of pesticides in aqueous environment. Proceedings of the 16th Symposium on Environmental Science of Pesticide, 105-120. |
|
(28) |
Keuken O., Anderson J.P.E. (1996). Influence of storage on biochemical processes in soil. In Pesticides, Soil Microbiology and Soil Quality, 59-63 (SETAC-Europe). |
|
(29) |
Stenberg B., Johansson M., Pell M., Sjödahl-Svensson K., Stenström J., Torstensson L. (1996). Effect of freeze and cold storage of soil on microbial activities and biomass. In Pesticides, Soil Microbiology and Soil Quality, 68-69 (SETAC-Europe). |
|
(30) |
Gennari, M., Negre, M., Ambrosoli, R. (1987). Effects of ethylene oxide on soil microbial content and some chemical characteristics. Plant and Soil 102, 197-200. |
|
(31) |
Anderson, J.P.E. (1975). Einfluss von Temperatur und Feuchte auf Verdampfung, Abbau und Festlegung von Diallat im Boden. Z. PflKrankh Pflschutz, Sonderheft VU, 141-146. |
|
(32) |
Hamaker, J.W. (1976). The application of mathematical modelling to the soil persistence and accumulation of pesticides. Proc. BCPC Symposium: Persistence of Insecticides and Herbicides, 181-199. |
|
(33) |
Goring, C.A.I., Laskowski, D.A., Hamaker, J.W., Meikle, R.W. (1975). Principles of pesticide degradation in soil. In «Environmental Dynamics of Pesticides». R. Haque and V.H. Freed, Eds., 135-172. |
|
(34) |
Timme, G., Frehse, H., Laska, V. (1986). Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues. II. Pflanzenschutz — Nachrichten Bayer 39, 188-204. |
|
(35) |
Timme, G., Frehse, H. (1980). Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues. I. Pflanzenschutz — Nachrichten Bayer 33, 47-60. |
|
(36) |
Gustafson D.I., Holden L.R. (1990). Non-linear pesticide dissipation in soil; a new model based on spatial variability. Environm. Sci. Technol. 24, 1032-1041. |
|
(37) |
Hurle K., Walker A. (1980). Persistence and its prediction. In Interactions between Herbicides and the Soil (R.J. Hance, Ed.), Academic Press, 83-122. |
Anexo 1
TENSIÓN DE AGUA, CAPACIDAD DE CAMPO (FC) Y CAPACIDAD DE RETENCIÓN DE AGUA (WHC) (44)
|
Altura de la columna de agua [cm] |
PF (45) |
bar (46) |
Observaciones |
|
10' |
7 |
104 |
Suelo seco |
|
1,6 .104 |
4,2 |
16 |
Punto de marchitamiento |
|
104 |
4 |
10 |
|
|
103 |
3 |
1 |
|
|
6.102 |
2,8 |
0,6 |
|
|
3,3 • 102 |
2,5 |
0,33 (47) |
Gama de Capacidad de campo (48) |
|
102 |
2 |
0,1 |
|
|
60 |
1,8 |
0,06 |
|
|
33 |
1,5 |
0,033 |
|
|
10 |
1 |
0,01 |
WHC (aproximación) |
|
1 |
0 |
0,001 |
Suelo saturado de agua |
La tensión de agua se mide en cm de columna de agua o en bar. Dada la amplia gama de valores posibles, la tensión de succión se expresa sencillamente como el valor pF equivalente al logaritmo de la altura en cm de la columna de agua.
La capacidad de campo se define como la cantidad de agua que puede almacenar un suelo natural contra la gravedad 2 días después de un largo período de lluvia o tras una irrigación suficiente. Se determina sobre el terreno en un suelo no perturbado. Por ello, la medida no es aplicable a muestras de suelo de laboratorio perturbadas. Los valores de FC determinados en suelos perturbados pueden mostrar grandes varianzas sistemáticas.
La capacidad de retención de agua (WHC) se determina en el laboratorio con suelo perturbado y no perturbado mediante saturación con agua de una columna de suelo por transporte capilar. Resulta especialmente útil para suelos perturbados y puede ser hasta un 30 % superior a la capacidad de campo (1). Además, experimentalmente es más fácil determinar la WHC que valores de FC fiables.
Notas
Anexo 2
CONTENIDO DE HUMEDAD DEL SUELO (g de agua por 100 g de suelo seco) DE VARIOS TIPOS DE SUELO DE DIVERSOS PAÍSES
|
|
|
Contenido de humedad del suelo a |
||
|
Tipo de suelo |
País |
|||
|
|
|
WHC (49) |
pF = 1,8 |
pF = 2,5 |
|
Arena |
Alemania |
28,7 |
8,8 |
3,9 |
|
Suelo arenoso franco |
Alemania |
50,4 |
17,9 |
12,1 |
|
Suelo arenoso franco |
Suiza |
44,0 |
35,3 |
9,2 |
|
Suelo franco limoso |
Suiza |
72,8 |
56,6 |
28,4 |
|
Suelo franco arcilloso |
Brasil |
69,7 |
38,4 |
27,3 |
|
Suelo franco arcilloso |
Japón |
74,4 |
57,8 |
31,4 |
|
Suelo franco arenoso |
Japón |
82,4 |
59,2 |
36,0 |
|
Suelo franco limoso |
EE.UU. |
47,2 |
33,2 |
18,8 |
|
Suelo franco arenoso |
EE.UU. |
40,4 |
25,2 |
13,3 |
Anexo 3
Figura 1
Ejemplo de aparato de flujo para estudiar la transformación de las sustancias químicas en el suelo (50) (51)
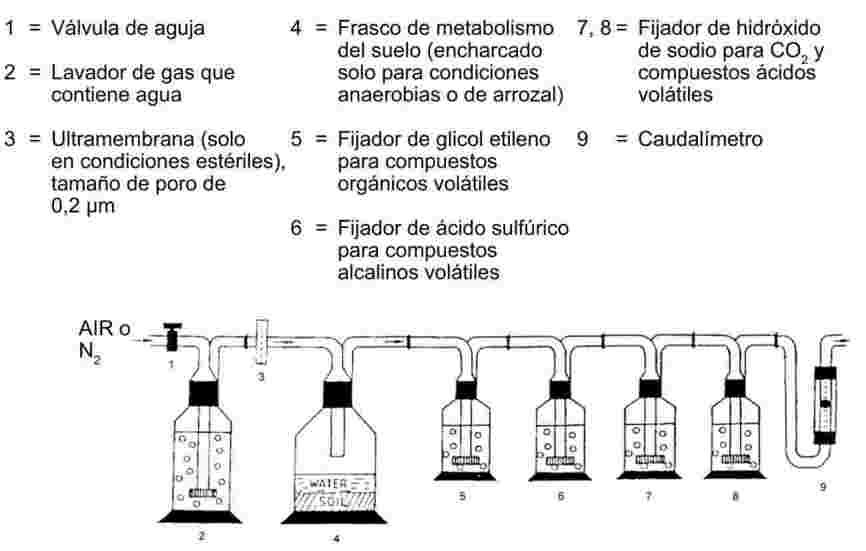
Figura 2
Ejemplo de matraz biométrico para el estudio de la transformación de las sustancias químicas en el suelo (52)
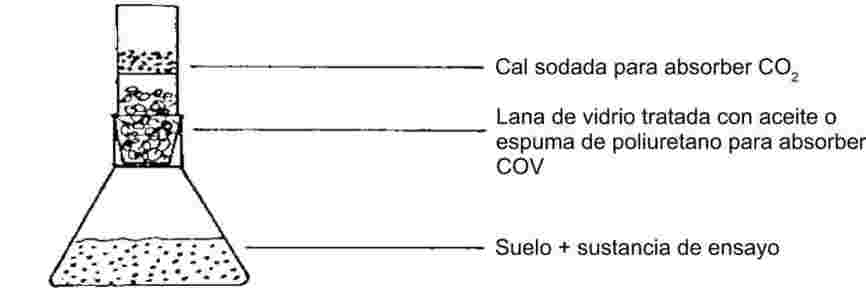
C.24. TRANSFORMACIÓN AEROBIA Y ANAEROBIA EN SISTEMAS DE SEDIMENTOS ACUÁTICOS
1. MÉTODO
El presente método reproduce las directrices de ensayo de la OCDE TG 308 (2002).
1.1 INTRODUCCIÓN
Los productos químicos pueden llegar a las aguas superficiales a nivel profundo o poco profundo por vías como la aplicación directa, los aerosoles erráticos, la escorrentía, la eliminación de residuos, los efluentes domésticos o agrícolas y la deposición atmosférica. Este método de ensayo consiste en un método de laboratorio para evaluar la transformación aerobia y anaerobia de las sustancias químicas orgánicas en los sistemas de sedimentos acuáticos. El método se basa en directrices ya existentes (1)(2)(3)(4)(5)(6). En un seminario de la OCDE sobre selección de suelos y sedimentos celebrado en Belgirate (Italia) en 1995 (7) se alcanzó un acuerdo, en particular, sobre el número y tipos de sedimentos que deben utilizarse en este ensayo. Dicho seminario formuló asimismo recomendaciones sobre recogida, manipulación y conservación de muestras de sedimentos, basadas en la Directriz ISO correspondiente (8). Estos estudios son necesarios para las sustancias químicas que se apliquen directamente al agua o que tengan probabilidades de llegar al medio ambiente acuático por las vías mencionadas anteriormente.
Las condiciones en los sistemas de sedimentos acuáticos naturales son a menudo aerobias en la fase acuosa superior. La capa superficial del sedimento puede ser o bien aerobia o bien anaerobia, mientras que el sedimento más profundo normalmente es anaerobio. Para abarcar todas estas posibilidades, en el presente documento se describen tanto ensayos aerobios como anaerobios. El ensayo aerobio simula una columna de agua aerobia sobre una capa de sedimento aerobio a la que subyace una gradiente anaerobio. El ensayo anaerobio simula un sistema de sedimento acuático completamente anaerobio. Si las circunstancias indican que es necesario desviarse significativamente de estas recomendaciones, por ejemplo utilizando núcleos de sedimentos intactos o sedimentos que haya estado expuestos a las sustancia de ensayo, se dispone de otros métodos con este fin (9).
1.2 DEFINICIONES
En cualquier caso deben utilizarse unidades internacionales (Standard International (SI) units).
Sustancia de ensayo: cualquier sustancia, sea el compuesto original o los correspondientes productos de transformación.
Productos de transformación: todas las sustancias resultantes de las reacciones de transformación biótica o abiótica de la sustancia de ensayo, incluido el CO2 y los residuos ligados.
Residuos ligados: compuestos del suelo, la planta o el animal que persisten en la matriz en forma de sustancia original o de sus productos de transformación/metabolitos tras la extracción. El método de extracción no debe modificar sustancialmente ni los propios compuestos ni la estructura de la matriz. La naturaleza del vínculo puede aclararse parcialmente mediante métodos de extracción que alteren la matriz y técnicas analíticas sofisticadas. Así por ejemplo, hasta la fecha se han identificado de esta manera enlaces covalentes, iónicos y de sorción, así como atrapamientos. En general, la formación de residuos ligados reduce de manera significativa la bioaccesibilidad y la biodisponibilidad (10) [modificado de TUPAC 1984 (11)].
Transformación aerobia: (oxidación) reacciones que se producen en presencia de oxígeno molecular (12).
Transformación anaerobia: (reducción) reacciones que se producen en ausencia de oxígeno molecular (12).
Aguas naturales: aguas superficiales obtenidas de estanques, ríos, arroyos, etc.
Sedimento: mezcla de constituyentes químicos minerales y orgánicos; estos últimos contienen compuestos de elevado contenido en carbono y en nitrógeno y de peso molecular elevado. El sedimento es depositado por las aguas naturales y forma una interfaz con el agua.
Mineralización: degradación completa de un compuesto orgánico a CO2 y H2O en condiciones aerobias y a CH4, CO2 y H2O en condiciones anaerobias. En el contexto del presente método, cuando se utiliza un compuesto radiomarcado, mineralización significa la amplia degradación de una molécula durante la cual se oxida un átomo de carbono marcado o se reduce cuantitativamente con liberación de la cantidad apropiada de 14CO2 o 14CH4, respectivamente.
Semivida: t0,5, es el tiempo que tarda la transformación del 50 % de una sustancia de ensayo cuando puede describirse dicha transformación mediante una cinética de primer orden; es independiente de la concentración inicial.
DT 50 (tiempo de desaparición SO): es el tiempo que tarda la concentración inicial de la sustancia de ensayo en reducirse en un 50 %.
DT 75 (tiempo de desaparición 75): es el tiempo que tarda la concentración inicial de la sustancia de ensayo en reducirse en un 75 %.
DT 90 (tiempo de desaparición 90): es el tiempo que tarda la concentración inicial de la sustancia de ensayo en reducirse en un 90 %.
1.3 COMPUESTOS DE REFERENCIA
Deben utilizarse sustancias de referencia para la cuantificación e identificación de los productos de transformación por métodos espectroscópicos o cromatográficos.
1.4 INFORMACIÓN SOBRE LA SUSTANCIA DE ENSAYO
Para medir la tasa de transformación podrá utilizarse una sustancia de ensayo no marcada o marcada con un isótopo, aunque se prefiere el material marcado. Para estudiar la vía de transformación y establecer un balance de materia se requiere material marcado. Se recomienda el marcado con 14C, pero también puede resultar de utilidad el uso de otros isótopos, tales como 13C, 15N, 3H o 32P. El marcador deberá ubicarse, en la medida de lo posible, en la parte o las partes más estables de la molécula (53). La pureza química y/o radioquímica de la sustancia de ensayo debe ser de al menos el 95 %.
Antes de llevar a cabo el ensayo, debe disponerse de la información siguiente sobre la sustancia de ensayo:
|
a) |
solubilidad en agua (Método A.6); |
|
b) |
solubilidad en disolventes orgánicos; |
|
c) |
presión de vapor (Método A.4) y constante de la ley de Henry; |
|
d) |
coeficiente de reparto n-octanol/agua (Método A.8); |
|
e) |
coeficiente de adsorción (Kd, Kf o Koc, en su caso) (Método C. 18); |
|
f) |
hidrólisis (Método C.7); |
|
g) |
constante de disociación (pKa) [Directriz OECD 112] (13); |
|
h) |
estructura química de la sustancia de ensayo y posición de la marca o marcas isotópicas, en su caso. |
Nota: Debe indicarse la temperatura a la que se hicieron estas mediciones.
Además, puede incluirse otra información de utilidad como datos sobre la toxicidad de la sustancia de ensayo para los microorganismos, sobre la biodegrabilidad inherente o inmediata, y sobre la transformación aerobia y anaerobia en el suelo.
Debe contarse con métodos analíticos (incluidos métodos de extracción y depuración) para la cuantificación e identificación de la sustancia de ensayo y de sus productos de transformación en el agua y los sedimentos (véase el punto 1.7.2).
1.5 PRINCIPIO DEL MÉTODO DE ENSAYO
El método descrito en este ensayo emplea un sistema de sedimento acuático aerobio y anaerobio (véase el anexo 1) que permite:
|
i) |
la medición de la tasa de transformación de la sustancia de ensayo en un sistema de sedimento-agua, |
|
ii) |
la medición de la tasa de transformación de la sustancia de ensayo en el sedimento, |
|
iii) |
la medición de la tasa de mineralización de la sustancia de ensayo y/o sus productos de transformación (cuando se utiliza una sustancia de ensayo marcada con 14C), |
|
iv) |
la identificación y cuantificación de los productos de transformación en las fases acuática y de sedimentaria incluido el balance de materia (cuando se utilice una sustancia de ensayo marcada), |
|
v) |
la medición de la distribución de la sustancia de ensayo y sus productos de transformación entre las dos fases durante un período de incubación en la oscuridad (para evitar, por ejemplo, floraciones de algas) a temperatura constante; las semividas y los valores DT50, DT75 y DT90 se determinarán cuando los datos lo permitan, pero no deben extrapolarse mucho más allá del período experimental (véase el punto 1.2). |
Se requieren al menos dos sedimentos y sus aguas asociadas para cada uno de los estudios aerobio y anaerobio (7). Sin embargo, puede haber casos en los que se usen más de dos sedimentos acuáticos, por ejemplo, para una sustancia química que pueda estar presente en entornos de aguas dulces y/o marinas.
1.6 APLICABILIDAD DEL ENSAYO
El método es aplicable en general a todas las sustancias químicas (no marcadas o marcadas) para las que se disponga de un método analítico de suficiente exactitud y sensibilidad. Es aplicable a compuestos ligeramente volátiles, no volátiles, solubles en agua o poco solubles en agua. El ensayo no deberá aplicarse a sustancias químicas que sean muy volátiles a partir del agua (por ejemplo, fumigantes o disolventes orgánicos) y, por tanto, no se puedan mantener en el agua y/o el sedimento en las condiciones experimentales del presente ensayo.
Este método se ha aplicado hasta ahora para estudiar la transformación de sustancias químicas en aguas dulces y sedimentos, pero, en principio, se puede aplicar también a sistemas marinos/de estuarios. No es adecuado para simular las condiciones de aguas corrientes (por ejemplo, ríos) o el mar abierto.
1.7 CRITERIOS DE CALIDAD
1.7.1 Recuperación
La extracción y el análisis de muestras de agua y sedimento, cuando menos duplicadas, inmediatamente después de la adición de la sustancia de ensayo dan una primera indicación de la repetibilidad del método analítico y de la uniformidad del procedimiento de aplicación de la sustancia de ensayo. Los valores de recuperación para fases posteriores del experimento los dan los balances de materia respectivos (cuando se utiliza material marcado). Dichos valores deben situarse entre el 90 % y el 110 % para sustancias químicas marcadas (6) y entre el 70 % y el 110 % para sustancias químicas no marcadas.
1.7.2 Repetibilidad y sensibilidad del método analítico
La repetibilidad del método analítico (excluida la eficiencia de extracción inicial) para cuantificar la sustancia de ensayo y los productos de transformación puede comprobarse realizando un análisis por duplicado del mismo extracto de las muestras de agua o sedimento que fueron incubadas durante tiempo suficiente para que se formaran productos de transformación.
El límite de detección (LOD) del método analítico de la sustancia de ensayo y de los productos de transformación debe alcanzar, como mínimo, el menor de los siguientes valores: 0,01 mg.kg-1 de agua o sedimento (como sustancia de ensayo) o un 1 % de la cantidad inicial aplicada a un sistema de ensayo. También debe especificarse el límite de cuantificación (LOQ).
1.7.3 Exactitud de los datos de transformación
El análisis de regresión de las concentraciones de la sustancia de ensayo en función del tiempo facilita la información apropiada sobre la fiabilidad de la curva de transformación y permite calcular los límites de confianza de las semividas (en el caso de la cinética de pseudo primer orden) o los valores DT50 y, si procede, DT75 y DT90.
1.8 DESCRIPCIÓN DEL MÉTODO
1.8.1 Sistema de ensayo y equipo
El estudio debe hacerse en recipientes de vidrio (por ejemplo, frascos, tubos de centrifugación), a menos que se disponga de información preliminar (como el coeficiente de reparto n-octanol/agua, datos sobre adsorción, etc.) que indiquen que la sustancia de ensayo puede adherirse al vidrio, en este caso tendrá que estudiarse la posibilidad de emplear un material alternativo (como Teflon). Cuando se sepa que la sustancia de ensayo se adhiere al vidrio, podrá paliarse el problema recurriendo a uno o varios de los métodos siguientes:
|
— |
determinar la masa de la sustancia de ensayo y los productos de transformación que adsorbe el vidrio, |
|
— |
hacer un lavado con disolvente de todos los recipientes de vidrio al final del ensayo, |
|
— |
utilizar productos formulados (véase también el punto 1.9.2), |
|
— |
utilizar una cantidad mayor de co-disolvente para añadir la sustancia de ensayo al sistema; si se emplea un co-disolvente debe tratarse de uno que no solvate la sustancia de ensayo. |
En los anexos 2 y 3, respectivamente, se muestran ejemplos de aparatos de ensayo habituales, por ejemplo, sistemas de flujo de gas y biométricos (14). En la referencia 15 se describen otros sistemas útiles de incubación. El diseño del aparato experimental debe permitir el intercambio de aire o nitrógeno y la fijación de los productos volátiles. Las dimensiones del aparato deben ser tales que se cumplan los requisitos del ensayo (véase el punto 1.9.1). Podrá ventilarse o bien mediante un burbujeo suave o bien haciendo pasar aire o nitrógeno sobre la superficie del agua. En este último caso puede ser aconsejable agitar suavemente el agua desde arriba para una mejor distribución del oxígeno o el nitrógeno en esta. No debe usarse aire sin CO2 ya que puede aumentar el pH del agua. En cualquiera de los dos casos, no es conveniente remover el sedimento, cosa que debe evitarse todo lo posible. Las sustancias químicas ligeramente volátiles deben ensayarse en sistema biométrico con una agitación suave de la superficie del agua. También pueden utilizarse recipiente cerrados con un espacio libre o bien de aire atmosférico o bien de nitrógeno y frascos internos para la fijación de productos volátiles (16). En el ensayo aerobio se requiere un intercambio regular del gas del espacio libre a fin de compensar el consumo de oxígeno de la biomasa.
Entre los fijadores adecuados para recoger productos de transformación volátiles cabe citar, de manera no exhaustiva, las soluciones lmol.dm-3 de hidróxido de potasio o hidróxido de sodio para el dióxido de carbono (54) y el etileno glicol, la etanolamina o la parafina al 2 % en xileno para los compuestos orgánicos. Los compuestos volátiles formados en condiciones anaerobias, como el metano, pueden recogerse, por ejemplo, mediante tamices moleculares. Estos compuestos volátiles pueden convertirse, por ejemplo, en CO2 por combustión haciendo pasar el gas por un tubo de cuarzo lleno de CuO a una temperatura de 900 oC y captando el CO2 formado en un absorbedor con una base (17).
Se requiere un equipo de laboratorio para el análisis químico de la sustancia de ensayo y los productos de transformación (por ejemplo, cromatografía líquida de gases (GLC), cromatografía líquida de alta resolución (HPLC), cromatografía de capa fina (TLC), espectroscopia de masas (MS), cromatografía de gases-espectroscopia de masas (GC-MS), espectrometría de masas-cromatografía líquida (LC-MS), resonancia magnética nuclear (RMN), etc.), incluidos sistemas de detección para sustancias químicas radiomarcadas o no radiomarcadas, según corresponda. Cuando se utilice material radiomarcado se necesitará también un contador de centelleo de líquidos y una cámara de combustión oxidante (para la combustión de muestras de sedimento antes del análisis de radiactividad).
Además se necesitará, según corresponda, otro equipo de laboratorio estándar para análisis fisicoquímicos y biológicos (véase el cuadro 1, punto 1.8.2.2), así como recipientes de vidrio, sustancias químicas y reactivos.
1.8.2 Selección y número de sedimentos acuáticos
Los lugares donde se tomen las muestras deberán seleccionarse con arreglo a la finalidad del ensayo en cualquier situación dada. Al seleccionar los lugares donde se tomen las muestras, se tendrá en cuenta su historia en cuanto a posibles aportaciones de origen agrícola, industrial o doméstico a la cuenca o los ríos o arroyos aguas arriba. No deben usarse sedimentos si han sido contaminados con la sustancia de ensayo o sus análogos estructurales durante los 4 años anteriores.
1.8.2.1 Selección de sedimentos
Para los estudios aerobios se utilizarán normalmente dos sedimentos (7). Los dos sedimentos seleccionados tienen que ser distintos en cuanto a contenido de carbono orgánico y textura. Un sedimento ha de tener un elevado contenido de carbono orgánico (2,5-7,5 %) y una textura fina, y el otro, un bajo contenido de carbono orgánico (0,5-2,5 %) y una textura gruesa. La diferencia en cuanto al contenido de carbono orgánico debe ser normalmente, al menos, del 2 %. Se entiende por «textura fina» un contenido de [arcilla+limo] (55) >50 % y por «textura gruesa», un contenido de [arcilla+limo] de <50 %. La diferencia en cuanto al contenido de [arcilla+limo] debe ser normalmente, al menos, del 20 %. En los casos en que una sustancia química pueda llegar también a las aguas marinas, al menos uno de los sistemas de sedimentos acuáticos deberá ser de origen marino.
Para el estudio estrictamente anaerobio, deben tomarse muestras de dos sedimentos (incluidas sus aguas asociadas) de las zonas anaerobias de las masas de agua superficiales (7). Tanto el sedimento como las fases acuáticas tienen que manejarse y transportarse cuidadosamente en condiciones de exclusión de oxígeno.
Puede haber otros parámetros que sean importantes para la selección de los sedimentos y que deban considerarse caso por caso. Por ejemplo, el pH de los sedimentos sería importante para ensayar sustancias químicas cuando su transformación y/o adsorción pueda depender de este. La dependencia de la adsorción con respecto al pH podría venir dada por la pKa de la sustancia de ensayo.
1.8.2.2 Caracterización de las muestras sedimento-agua
En el cuadro a continuación se resumen los parámetros clave que deben medirse e indicarse (con referencia al método utilizado) tanto para el agua como para el sedimento, y la fase del ensayo en la que deben determinarse. En la bilbiografía, referencias (18)(19)(20) y (21), se dan, a título informativo, los métodos de determinación de estos parámetros.
Además, puede ser necesario medir e indicar caso por caso otros parámetros (por ejemplo, para las aguas dulces: partículas, alcalinidad, dureza, conductividad y NO3/PO4 (proporción y valores de cada compuesto); para los sedimentos: capacidad de intercambio de cationes, capacidad de retención de agua, carbonates, fósforo y nitrógeno totales; y para los sistemas marinos: salinidad. El análisis de los sedimentos y el agua para detectar nitratos, sulfatos, hierro biodisponible y posiblemente otros aceptadores de electrones puede ser útil también al valorar las condiciones redox, especialmente en relación con la transformación anaerobia.
Medición de parámetros para la caracterización de las muestras agua-sedimentos (7)(22)(23)
|
Parametro |
Fase del procedimiento de ensayo |
|||||
|
muestreo in situ |
manejo posterior |
inicio de la aclimatación |
al inicio del ensayo |
durante el ensayo |
al final del ensayo |
|
|
Agua |
||||||
|
Origen/fuente |
x |
|
|
|
|
|
|
Temperatura |
x |
|
|
|
|
|
|
pH* |
x |
|
x |
x |
x |
x |
|
COT |
|
|
x |
x |
|
x |
|
Concentración de O2* |
x |
|
x |
x |
x |
x |
|
Potencial redox* |
|
|
x |
x |
x |
x |
|
Sedimento |
||||||
|
Origen/fuente |
x |
|
|
|
|
|
|
Profundidad de la capa |
x |
|
|
|
|
|
|
pH (56) |
|
x |
x |
x |
x |
x |
|
Distribución del tamaňo de las partículas |
|
x |
|
|
|
|
|
COT |
|
x |
x |
x |
|
x |
|
Biomasa microbiana (57) |
|
x |
|
x |
|
x |
|
Potencial redox (56) |
Observación (color/olor) |
|
x |
x |
x |
x |
1.8.3 Recogida, manejo y almacenamiento
1.8.3.1 Recogida
Para la toma de muestras de sedimentos, deberá utilizarse el proyecto de Directriz ISO sobre la toma de muestras de sedimentos de fondo (8). Las muestras de sedimento se tomarán de toda la capa superior de 5 a 10 cm del sedimento. El agua asociada debe recogerse del mismo lugar y al mismo tiempo que el sedimento. Para el estudio anaerobio, las muestras de sedimento y agua asociada deben tomarse en condiciones de exclusión de oxígeno (28) (véase el punto 1.8.2.1). En la bibliografía se describen algunos aparatos de toma de muestras (8) (23).
1.8.3.2 Manejo
El sedimento se separa del agua por filtración y se tamiza por va húmeda a través de una criba de 2 mm utilizando el agua del lugar de recogida sobrante que luego se tira. A continuación, las cantidades conocidas de sedimentos y agua se mezclan en la proporción deseada (véase el punto 1.9.1) en los recipientes de incubación y se preparan para el período de aclimatación (véase el punto 1.8.4). Para el estudio anaerobio, todas las etapas del manejo tiene que llevarse a cabo en condiciones de exclusión de oxígeno (29) (30) (31) (32) y (33).
1.8.3.3 Almacenamiento
Se recomienda encarecidamente utilizar sedimentos y agua recién recogidos, pero, si es necesario el almacenamiento, el sedimento y el agua tiene que tamizarse como se ha descrito anteriormente y almacenarse juntos, empapados en agua (en una capa de agua de 6-10 cm), en la oscuridad y a 4 ± 2 oC4 durante un máximo de 4 semanas (7) (8) (23). Las muestras utilizadas par estudios aerobios tienen que almacenarse de manera que el aire pueda acceder libremente (por ejemplo, en recipientes abiertos), mientras que las de estudios anaerobios tiene que estar en condiciones de exclusión de oxígeno. No debe haber congelación del sedimento y el agua ni secado del sedimento durante el transporte y el almacenamiento.
1.8.4 Preparación de las muestras de sedimento/agua para el ensayo
Tiene que haber un período de aclimatación antes de añadir la sustancia de ensayo, teniendo en cuenta que cada muestra de sedimento/agua ha de colocarse en el recipiente de incubación que se vaya a usar en el ensayo principal, y que la aclimatación ha de llevarse a cabo exactamente en las mismas condiciones que la incubación para el ensayo (véase el punto 1.9.1). El período de aclimatación es el tiempo necesario para alcanzar una estabilidad razonable del sistema, expresada mediante el pH, la concentración de oxígeno en el agua, el potencial redox del sedimento y el agua, y la separación macroscópica de las fases. El período de aclimatación debe durar normalmente entre una y dos semanas y no debe superar las cuatro semanas. Se indicarán los resultados de las determinaciones efectuadas durante este período.
1.9 REALIZACIÓN DEL ENSAYO
1.9.1 Condiciones del ensayo
El ensayo deberá hacerse en el aparato de incubación (véase el punto 1.8.1) con una proporción agua/sedimento en volumen entre 3:1 y 4:1, y urta capa de sedimento de 2,5 cm (±0,5 cm). Se recomienda una cantidad mínima de sedimento de 50 g (en peso seco) por recipiente de incubación.
El ensayo se hará en la oscuridad a una temperatura constante dentro de un intervalo de 10 a 30 oC. Se considera apropiada una temperatura de 20 ± 2 oC. En su caso, podría considerarse otra temperatura más baja (por ejemplo, 10 oC) en determinados casos concretos, según la información que desee obtenerse del ensayo. Deberá controlarse e indicarse la temperatura de incubación.
1.9.2 Tratamiento y aplicación de la sustancia de ensayo
Se utilizará una única concentración para el ensayo. Para productos fitosanitarios aplicados directamente a las masas de agua, la dosificación máxima indicada en la etiqueta debe tomarse como la tasa de aplicación máxima calculada basándose en la superficie del agua en el recipiente de ensayo. En todos los demás casos, la concentración que ha de emplearse tiene que basarse en las previsiones de emisiones medioambientales. Se procurará aplicar una concentración adecuada de la sustancia de ensayo a fin de caracterizar la vía de transformación y la formación y la desaparición de productos de transformación. Puede resultar necesario aplicar dosis superiores (por ejemplo, 10 veces más) cuando las concentraciones de la sustancia de ensayo estén próximas a los límites de detección al inicio del estudio y/o cuando no puedan detectarse fácilmente productos de transformación importantes que estén presentes al 10 % de la tasa de aplicación de la sustancia de ensayo. Sin embargo, si se usan concentraciones de ensayo más altas, no deberán tener un efecto perjudicial significativo en la actividad microbiana del sistema sedimento-agua. Para conseguir una concentración constante de la sustancia de ensayo en recipientes de diferentes dimensiones puede considerarse adecuado un ajuste según la cantidad del material aplicado, basándose en la altura de la columna de agua en el recipiente en relación con la profundidad del agua en el campo (que se supone que es 100 cm, aunque pueden utilizarse otras alturas). En el anexo 4 se da un ejemplo de cálculo.
El procedimiento ideal es aplicar la sustancia de ensayo como una solución acuosa en la fase acuosa del sistema de ensayo. Si es inevitable, se permite el uso de cantidades pequeñas de disolventes miscibles con agua (como la acetona y el etanol) para la aplicación y distribución de la sustancia de ensayo, pero estas cantidades no deben superar el 1 % v/v ni tener efectos perjudiciales para la actividad microbiana del sistema de ensayo. Debe procederse con cuidado al generar la solución acuosa de la sustancia de ensayo: puede resultar necesario utilizar aparatos de columnas y pre-mezclado para asegurar una homogeneidad completa. Tras añadir la solución acuosa al sistema de ensayo, se recomienda mezclar suavemente la fase acuosa, removiendo el sedimento lo menos posible.
No se recomienda por lo general el uso de productos formulados ya que los ingredientes de la fórmula pueden afectar a la distribución de la sustancia de ensayo y/o los productos de transformación entre las fases acuosa y sedimentaria. No obstante, en el caso de sustancias de ensayo poco solubles en agua, su utilización puede constituir una alternativa apropiada.
El número de recipientes de incubación depende del número de tiempos de muestreo (véase el punto 1.9.3). Debe incluirse un número suficiente de sistemas de ensayo de manera que puedan sacrificarse dos sistemas en cada tiempo de muestreo. Cuando se empleen unidades de control de cada sistema de sedimento acuático, estas no deben tratarse con la sustancia de ensayo. Las unidades de control pueden utilizarse para determinar la biomasa microbiana del sedimento y el carbono orgánico total del agua y del sedimento al final del estudio. Dos de las unidades de control (es decir, una unidad de control de cada sedimento acuático) pueden utilizarse para controlar los parámetros requeridos en el sedimento y el agua durante el período de aclimatación (véase el cuadro del punto 1.8.2.2). Hay que incluir dos unidades de control adicionales en caso de que la sustancia de ensayo se aplique mediante un disolvente para medir los efectos negativos del sistema de ensayo en la actividad microbiana.
1.9.3 Duración del ensayo y muestreo
La duración del experimento no superará normalmente los 100 días (6). El experimento ha de continuar hasta que queden establecidas la vía de degradación y el modelo de distribución sedimento/agua o cuando el 90 % de la sustancia de ensayo se haya disipado por transformación y/o volatilización. El número de tiempos de muestreo debe ser seis, como mínimo, (incluido el tiempo 0), teniendo en cuenta que deberá hacerse un estudio opcional preliminar (véase el punto 1.9.4) para establecer un régimen de muestreo adecuado y la duración del ensayo, a menos que se disponga de datos suficientes sobre la sustancia de ensayo a partir de estudios previos. Para las sustancias de ensayo hidrofóbicas, puede ser necesaria la inclusión de otros puntos de muestreo durante el período inicial del estudio a fin de determinar la tasa de distribución entre las fases acuosa y sedimentaria.
Puede ser útil un ensayo con una segunda concentración para sustancias químicas que lleguen a las aguas superficiales por diferentes vías de entrada en concentraciones significativamente diferentes, siempre que la concentración más baja pueda analizarse con suficiente exactitud. A los tiempos de muestreo adecuados, se separan los recipientes de incubación completos (copias) para su análisis. Se analizan por separado el sedimento y el agua que sobrenada (58). El agua superficial tiene que separarse con cuidado procurando remover el sedimento lo menos posible. La extracción y caracterización de la sustancia de ensayo y los productos de transformación deben hacerse conforme a procedimientos analíticos adecuados. Se pondrá cuidado en separar el material que pueda haber sido adsorbido en el recipiente de incubación o los tubos de interconexión utilizados para atrapar sustancias volátiles.
1.9.4 Ensayo preliminar opcional
Si no puede calcularse la duración ni el régimen de muestreo a partir de otros estudios sobre la sustancia de ensayo, puede resultar adecuado un ensayo preliminar opcional, que debe hacerse en las mismas condiciones de ensayo propuestas para el estudio definitivo. Si se lleva a cabo el ensayo preliminar, se informará brevemente sobre sus condiciones experimentales y sus resultados.
1.9.5 Mediciones y análisis
Se medirá y se indicará la concentración de la sustancia de ensayo y los productos de transformación en cada tiempo de muestreo en el agua y el sedimento (como, concentración y como porcentaje de la sustancia aplicada). En general, deberán identificarse los productos de transformación detectados a >10 % de la radiactividad aplicada en todo el sistema agua-sedimento en cualquier tiempo de muestreo, a no ser que esté razonablemente justificado no hacerlo. Habrá de tomarse en consideración la identificación de los productos de transformación cuyas concentraciones aumenten continuamente durante el estudio, aunque las concentraciones no superen los límites indicados anteriormente, ya que este aumento puede indicar persistencia. Este aspecto se considerará caso por caso, justificándolo debidamente en el informe.
Los resultados de los sistemas de fijación de gases/sustancias volátiles (CO2 y otros, por ejemplo, compuestos orgánicos volátiles) deberán indicarse en cada tiempo de muestreo. Se indicarán las tasas de mineralización. Asimismo, se indicarán los residuos no extraíbles (ligados) en el sedimento en cada punto de muestreo.
2 RESULTADOS
2.1 TRATAMIENTO DE LOS RESULTADOS
En cada tiempo de muestreo se calculará el balance total de materia o la recuperación (véase el punto 1.7.1) de la radiactividad añadida. Los resultados deberán indicarse en porcentaje de la radiactividad añadida. La distribución de la radiactividad entre el agua y el sedimento se expresará en concentraciones y porcentajes en cada tiempo de muestreo.
La semivida, el DT50 y, en su caso, el DT75 y el DT90 deberán calcularse junto con sus límites de confianza (véase el punto 1.7.3) Podrá obtenerse información sobre la tasa de disipación de la sustancia de ensayo en el agua y el sedimento mediante el uso de instrumentos de evaluación adecuados. Estos instrumentos pueden ir desde la aplicación de cinética de pseudo primer orden a técnicas empíricas de ajuste de curvas con soluciones gráficas o numéricas y evaluaciones más complejas utilizando; por ejemplo, modelos unicompartimentales o multicompartimentales. Para más información puede consultarse la bibliografía correspondiente (35) (36) (37).
Todos los planteamientos al respecto tienen sus ventajas e inconvenientes y varían considerablemente en cuanto a complejidad. El supuesto de una cinética de primer orden puede ser una simplificación excesiva de los procesos de degradación y distribución, pero, cuando es posible, da un término (constante de velocidad o semivida) que es fácilmente comprensible y valioso en la modelización por simulación y en los cálculos de las concentraciones previstas en el medio ambiente. Los planteamientos empíricos o las transformaciones lineales pueden dar lugar a un mejor ajuste de las curvas a los datos y, por tanto, permitir una mejor estimación de las semividas, los valores DT50 y, en su caso, los DT75 y DT90. Sin embargo, el uso de las constantes derivadas es limitado. Los modelos compartimentales pueden generar una serie de constantes de valor útiles en la evaluación de riesgos que describen la velocidad de degradación en diferentes compartimentos y la distribución de la sustancia química. Asimismo, estos modelos deben utilizarse para el cálculo de constantes de velocidad en la formación y degradación de los principales productos de transformación. En todos los casos, el método elegido debe justificarse y el experimentador ha de demostrar gráfica o estadísticamente la corrección del ajuste.
3 INFORME
3.1 INFORME DEL ENSAYO
El informe debe comprender la siguiente información:
|
|
Sustancia de ensayo:
|
|
|
Sustancias de referencia:
|
|
|
Sedimentos y aguas del ensayo:
|
|
|
Condiciones de ensayo:
|
|
|
Resultados:
|
REFERENCIAS
|
(1) |
BBA-Guidelines for the examination of plant protectors in the registration process. (1990). Part IV, Section 5-1: Degradability and fate of plant protectors in the water/sediment system. Germany. |
|
(2) |
Commission for registration of pesticides: Application for registration of a pesticide. (1991). Part G. Behaviour of the product and its metabolites in soil, water and air, Section G.2.1 (a). The Netherlands. |
|
(3) |
MAFF Pesticides Safety Directorate. (1992). Preliminary guideline for the conduct of biodegradability tests on pesticides in natural sediment/water systems. Ref No SC 9046. United-Kingdom. |
|
(4) |
Agriculture Canada: Environmental chemistry and fate. (1987). Guidelines for registration of pesticides in Canada. Aquatic (Laboratory) — Anaerobic and aerobic. Canada. pp 35-37. |
|
(5) |
US-EPA: Pesticide assessment guidelines, Subdivision N. Chemistry: Environmental fate (1982). Section 162-3, Anaerobic aquatic metabolism. |
|
(6) |
SETAC-Europe publication. (1995). Procedures for assessing the environmental fate and ecotoxicity of pesticides. Ed. Dr Mark R. Lynch. SETAC-Europe, Brussels. |
|
(7) |
OECD Test Guidelines Programme. (1995). Final Report of the OECD Workshop on Selection of Soils/sediments, Belgirate, Italy, 18-20 January 1995. |
|
(8) |
ISO/DIS 5667-12. (1994). Water quality — Sampling — Part 12: Guidance on sampling of bottom sediments. |
|
(9) |
US-EPA (1998a). Sediment/water microcosm biodegradation test. Harmonised Test Guidelines (OPPTS 835.3180). EPA 712-C-98-080. |
|
(10) |
DFG: Pesticide Bound Residues in Soil. Wiley-VCH (1998). |
|
(11) |
T.R. Roberts: Non-extractable pesticide residues in soils and plants. Pure Appl. Chem. 56, 945-956 (IUPAC 1984). |
|
(12) |
OECD Test Guideline 304A: Inherent Biodegradability in Soil (adopted 12 May 1981). |
|
(13) |
OECD (1993): Guidelines for Testing of Chemicals. Paris. OECD (1994-2000): Addenda 6-11 to Guidelines for the Testing of Chemicals. |
|
(14) |
Scholz, K., Fritz R., Anderson C. and Spiteller M. (1988) Degradation of pesticides in an aquatic model ecosystem. BCPC — Pests and Diseases, 3B-4, 149-158. |
|
(15) |
Guth, J.A. (1981). Experimental approaches to studying the fate of pesticides in soil. In Progress in Pesticide Biochemistry (D.H. Hutson, T.R. Roberts, Eds.), Vol. 1, 85-114. J. Wiley & Sons. |
|
(16) |
Madsen, T., Kristensen, P. (1997). Effects of bacterial inoculation and non-ionic surfactants on degradation of polycyclic aromatic hydrocarbons in soil. Environ. Toxicol. Chem. 16, 631-637. |
|
(17) |
Steber, J., Wierich, P. (1987). The anaerobic degradation of detergent range fatty alcohol ethoxylates. Studies with 14C-labelled model surfactants. Water Research 21, 661-667. |
|
(18) |
Black, C.A. (1965). Methods of Soil Analysis. Agronomy Monograph No. 9. American Society of Agronomy, Madison. |
|
(19) |
APHA (1989). Standard Methods for Examination of Water and Wastewater (17th edition). American Public Health Association, American Water Works Association and Water Pollution Control Federation, Washington D.C. |
|
(20) |
Rowell, D.L. (1994). Soil Science Methods and Applications. Longman. |
|
(21) |
Light, T.S. (1972). Standard solution for redox potential measurements. Anal. Chemistry 44, 1038-1039. |
|
(22) |
SETAC-Europe publication (1991). Guidance document on testing procedures for pesticides in freshwater mesocosms. From the Workshop «A Meeting of Experts on Guidelines for Static Field Mesocosms Tests», 3-4 July 1991. |
|
(23) |
SETAC-Europe publication. (1993). Guidance document on sediment toxicity tests and bioassays for freshwater and marine environments. From the Workshop On Sediment Toxicity Assessment (WOSTA), 8-10 November 1993.Eds.: I.R. Hill, P. Matthiessen and F. Heimbach. |
|
(24) |
Vink, J.P.M., van der Zee, S.E.A.T.M. (1997). Pesticide biotransformation in surface waters: multivariate analyses of environmental factors at field sites. Water Research 31, 2858-2868. |
|
(25) |
Vink, J.P.M., Schraa, G., van der Zee, S.E.A.T.M. (1999). Nutrient effects on micTobial transformation of pesticides in nitrifying waters. Environ. Toxicol, 329-338. |
|
(26) |
Anderson, T.H., Domsch, K.H. (1985). Maintenance carbon requirements of actively-metabolising microbial populations under in-situ conditions. Soil Biol. Biochem. 17, 197-203. |
|
(27) |
ISO-14240-2. (1997). Soil quality — Determination of soil microbial biomass — Part 2: Fumigation-extraction method. |
|
(28) |
Beelen, P. Van and F. Van Keulen. (1990), The Kinetics of the Degradation of Chloroform and Benzene in Anaerobic Sediment from the River Rhine. Hydrobiol. Bull. 24 (1), 13-21. |
|
(29) |
Shelton, D.R. and Tiedje, J.M. (1984). General method for determining anaerobic biodegradation potential. App. Environ. Microbiol. 47, 850-857. |
|
(30) |
Birch, R.R., Biver, G, Campagna, R., Gledhill, W.E., Pagga, U., Steber, J., Reust, H. and Bontinck, W.J. (1989). Screening of chemicals for anaerobic biodegradation. Chemosphere 19, 1527-1550. |
|
(31) |
Pagga, U. and Beimborn, D.B. (1993). Anaerobic biodegradation tests for organic compounds. Chemoshpere 27, 1499-1509. |
|
(32) |
Nuck, B.A. and Federle, T.W. (1986). A batch test for assessing the mineralisation of 14C-radiolabelled compounds under realistic anaerobic conditions. Environ. Sci. Technol. 30, 3597-3603. |
|
(33) |
US-EPA (1998b). Anaerobic biodegradability of organic chemicals. Harmonised Test Guidelines (OPPTS 835.3400). EPA 712-C-98-090. |
|
(34) |
Sijm, Haller and Schrap (1997). Influence of storage on sediment characteristics and drying sediment on sorption coefficients of organic contaminants. Bulletin Environ. Contam. Toxicol. 58, 961-968. |
|
(35) |
Timme, G., Frehse H. and Laska V. (1986) Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues II. Pflanzenschutz — Nachrichten Bayer, 39, 187-203. |
|
(36) |
Timme, G., Frehse, H. (1980) Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues I. Pflanzenschutz — Nachrichten Bayer, 33, 47-60. |
|
(37) |
Carlton, R.R. and Allen, R. (1994). The use of a compartment model for evaluating the fate of pesticides in sediment/water systems. Brighton Crop Protection Conference — Pest and Diseases, pp 1349-1354. |
Anexo 1
DIRECTRIZ SOBRE LOS SISTEMAS DE ENSAYO AEROBIOS Y ANAEROBIOS
Sistema de ensayo aerobio
El sistema de ensayo aerobio descrito en este método de ensayo consiste en una capa de agua aerobia (cuyas concentraciones de oxígeno oscilan normalmente entre 7 y 10 mg l-1) y una capa de sedimento, aerobio en la superficie y anaerobio por debajo de la superficie (cuyos potenciales redox medios (Eh) oscilan normalmente en la zona anaerobia del sedimento entre - 80 y - 190 mV). Se hará circular aire húmedo sobre la superficie del agua en cada unidad de incubación para mantener suficiente oxígeno en el espacio libre.
Sistema de ensayo anaerobio
Para el sistema de ensayo anaerobio, el procedimiento del ensayo es esencialmente el mismo que el descrito para el sistema aerobio con la excepción de que el nitrógeno humedecido se pasa por la superficie del agua en cada unidad de incubación para mantener un espacio libre de nitrógeno. El sedimento y el agua se consideran anaerobios cuando el potencial redox (Eh) sea inferior a - 100 mV.
En el ensayo anaerobio, la valoración de la mineralización incluye la medición del dióxido de carbono y el metano liberados.
Anexo 2
EJEMPLO DE APARATO DE RUJO
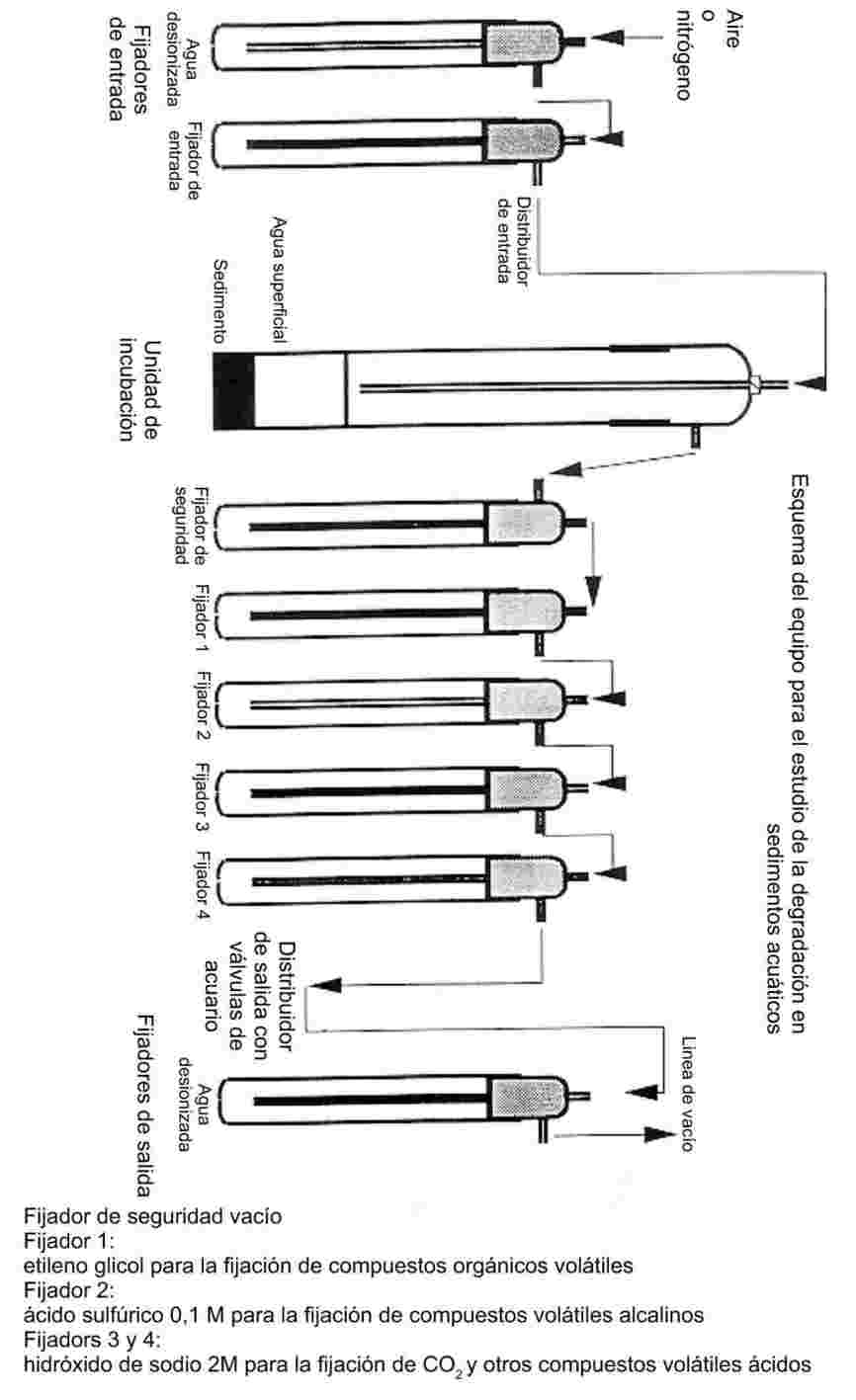
Anexo 3
EJEMPLO DE APARATO BIOMÉTRICO

Anexo 4
EJEMPLO DE CÁLCULO DE LA DOSIS QUE DEBE APLICARSE A LOS RECIPIENTES DE ENSAYO
|
Diámetro interno del cilindro: |
= 8 cm |
|
Altura de la columna de agua sin incluir el sedimento: |
= 12 cm |
|
Área de la superficie: 3,142 × 42 |
= 50,3 cm2 |
|
Tasa de aplicación: 500 g de la sustancia de ensayo/ha corresponde a 5 μg/cm2 |
|
|
Total μg: 5 × 50,3 |
= 251,5 μg |
|
Ajuste de la cantidad en relación con una altura de 100 cm. 12 × 251,5 ÷ 100 |
= 30,18 μg |
|
Volumen de la columna de agua: 50,3 × 12 |
= 603 ml |
|
Concentración en el agua: 30,18 ÷ 603 |
= 0,050 |μg/ml o 50 μg/1 |
(1) Los resultados de estos ensayos interlaboratorios y unas correcciones técnicas de la norma ISO 6341 indican una CE50-24 h de dicromato potásico (K2Cr2O7) en la banda de 0,6 mg/l a 1,7 mg/l.
(2) Agua de pureza adecuada como, por ejemplo, agua desionizada, destilada o tratada mediante ósmosis inversa con una conductividad que de preferencia no exceda de 10 μS.cm-1.
(3) Si existe una diferencia considerable entre D1 y D2, no debe calcularse el término medio.
(4) Si existe una diferencia considerable entre D1 y D2, no debe calcularse el término medio.
(5) O al final de la incubación.
(6) Si existe una diferencia considerable entre D1 y D2, no debe calcularse el término medio.
(7) O al final de la incubación.
(8) No tener en cuenta si hay una diferencia considerable en las repeticiones.
(9) No tener en cuenta si hay una diferencia considerable en las repeticiones.
(10) Mabey y Mill recomiendan los amortiguadores de borato o acetato en lugar de los de fosfato (11).
(11) Esta información puede proceder de fuentes distintas, como datos bibliográficos de la hidrólisis de compuestos similares estructuralmente o mediante otros ensayos preliminares semicuantitativos de hidrólisis de la sustancia problema en una fase anterior del desarrollo.
(12) Si la representación gráfica del logaritmo de los datos frente al tiempo no indica una función lineal (identificada con una velocidad de reacción de primer orden), no es apropiado utilizar la ecuación [3] para determinar la constante de la velocidad de hidrólisis de la sustancia problema.
(13) Los valores de pH indicados en estos cuadros se han calculado a partir de mediciones del potencial utilizando las ecuaciones tipo de Sörensen (1909). Los valores correspondientes de pH son 0,04 unidades superiores a los valores de los cuadros.
(14) Se añaden pequeños cristales de timol u otra sustancia similar para evitar el crecimiento de mohos.
(15) Valores medios de determinaciones por triplicado.
(16) Meyer A., Orti G. (1993) Proc. Royal Society of London, Series B, Vol. 252, p. 231.
(17) Tomar el agua después de transferir al menos 3 volúmenes de recinto.
Los valores entre paréntesis son los números de muestras (agua, peces) que se toman si se procede a un muestreo suplementario.
|
Nota |
: |
La estimación antes del ensayo de kn para un log Pow de 4,0 es de 0,652 días-1 La duración total de la experiencia se fija en: 3 x abs. = 3 x 4,6 días, o sea 14 días. Remitirse al anexo 3 para el cálculo de «abs.». |
(18) Puede seleccionarse una serie de 5 (o más) concentraciones sucesivas en una columna. Los puntos medios entre las concentraciones de la columna (x) se encuentran en la columna (2x + 1)l Los valores recogidos pueden representar concentraciones expresadas como porcentaje en volumen o en peso (mg/l o μg/l). Los valores pueden multiplicarse o dividirse por la potencia de 10 adecuada. Puede emplearse la primera columna si el grado de toxicidad es muy incierto.
(19) OECD, París. 1992. Directrices de ensayo 210. «Ensayo de toxicidad en las primeras fases de la vida en peces».
(20) Para los embriones.
(21) Para las larvas.
(22) Embriones y larvas en la oscuridad hasta una semana después de la eclosión, salvo durante las observaciones. Después, luz tenue hasta el final del ensayo.
(23) Según el sistema de la FAO y los Estados Unidos de América (85).
(24) Las variables respectivas deben mostrar preferentemente valores dentro de la gama considerada. Si, no obstante, hay dificultades para encontrar material de suelo adecuado, se aceptarán valores por debajo del mínimo indicado.
(25) Los suelos con menos del 0,3 % de carbono orgánico pueden perturbar la correlación entre el contenido orgánico y la adsorción. Así pues, se recomienda el uso de suelos con un contenido mínimo de carbono orgánico del 0,3 %.
(26) 
(27) También pueden utilizarse gráficas de la concentración de la sustancia problema en la fase acuosa ![]() frente al tiempo para estimar la llegada a la meseta de equilibrio (véase la figura 2 del apéndice 5).
frente al tiempo para estimar la llegada a la meseta de equilibrio (véase la figura 2 del apéndice 5).
(28) DT-50: tiempo necesario para la degradación del 50 % de la sustancia problema.
(29) W Kördel, D. Hennecke, M. Herrmann (1997). Application of the HPLC-screening method for the determination of the adsorption coefficient on sewage sludges. Chemosphere, 35(1/2), pp. 121-128.
(30) W. Kördel, D. Hennecke, C. Franke (1997). Determination of the adsorption-coefficients of organic substances on sewage sludges Chemosphere, 35 (1/2), pp. 107-119.
(31) W. Kördel, G. Kotthoff, J. Müller (1995). HPLC-screening method for the determination of the adsorption coefficient on soil-results of a ring test. Chemosphere, 30(7), pp. 1373-1384.
(32) W. Kördel. J. Müller (1994). Bestimmung des Adsorptionskoeffizienten organischer Chemikalien mit der HPLC. UBA R & D Report No 106 01044 (1994).
(33) B.V. Oepen. W. Kördel, W. Klein (1991). Chemosphere, 22, pp. 285-304.
(34) Datos facilitados por la industria.
(35) Las camadas abortadas se registran escribiendo «AB» en la casilla correspondiente.
(36) La mortalidad de los animales adultos se registra escribiendo «M» en la casilla correspondiente.
(37) Por ejemplo, si la sustancia de ensayo contiene un anillo, es preciso marcar ese anillo: si contiene dos o más anillos, puede resultar necesario efectuar estudios separados para evaluar el destino de cada anillo marcado y obtener información adecuada sobre la formación de los productos de transformación.
(38) La característica de retención de agua de un suelo puede medirse como capacidad de campo, como capacidad de retención de agua o como tensión de succión de agua (pF). Más explicaciones en el anexo 1. Deberá consignarse en el informe del ensayo si las características de retención de agua y la densidad aparente de los suelos se determinaron en muestras no perturbadas o en muestras perturbadas (procesadas).
(39) Según los resultados de recientes investigaciones, los suelos de las zonas templadas pueden almacenarse también a - 20 oC durante más de tres meses (28) (29) sin pérdida significativa de actividad microbiana.
(40) El suelo no deberá estar ni demasiado húmedo ni demasiado seco para mantener una aireación y nutrición adecuadas de la microflora edáfica. El contenido de humedad recomendado para un crecimiento microbiano óptimo se sitúa entre 40-60 % de capacidad de retención de agua (WHC) y entre 0,1-0,33 bar (6). Este último intervalo equivale a un pF comprendido entre 2,0 y 2,5. En el anexo 2 se dan los contenidos de humedad típicos de varios tipos de suelo.
(41) Las condiciones aerobias son dominantes en los suelos superficiales e incluso en los situados debajo de la superficie, según demuestra un proyecto de investigación patrocinado por la UE [K. Takagi et al. (1992). Microbial diversity and activity in subsoils: Methods, field site, seasonal variation in subsoil temperatures and oxygen contents. Proc. Internat. Symp. Environm. Aspects Pesticides Microbiol., 270-277, 17-21 August 1992, Sigtuna, Sweden]. Las condiciones anaerobias se presentan tan solo ocasionalmente con motivo de la inundación del suelo tras lluvias intensas o cuando se establecen las condiciones de arrozal.
(42) Los estudios aerobios podrían terminar mucho antes de estos 120 días siempre que se haya establecido manifiestamente la vía de transformación última y la mineralización última. Es posible asimismo la terminación del ensayo a los 120 días, o cuando se haya transformado el 90 % de la sustancia de ensayo, pero solo si se ha formado al menos un 5 % de CO2.
(43) La concentración inicial en una superficie se calcula utilizando la ecuación siguiente:

|
Csoil |
= |
concentración inicial en el suelo [mg.kg-1] |
|
A |
= |
tasa de aplicación [kg.ha-1]; 1 = espesor de la capa de suelo en el campo [m]; d = densidad aparente en seco del suelo [kg-m-3]. |
A título indicativo, una tasa de aplicación de 1 kg.ha-1 da por resultado una concentración en el suelo de aproximadamente 1 mg.kg-1 en una capa de 10 cm (supuesta una densidad aparente de 1 g . cm-3).
(44) Mückenhausen, E. (1975). Die Bodenkunde und ihre geologischen, geomorphologischen, mineralogischen und petrologischen Grundlagen. DLG-Verlag, Frankfurt, Main.
(45) pF = log de Ja altura en cm de la columna de agua.
(46) 1 bar= 105 Pa.
(47) Corresponde a un contenido de agua aproximado de 10 % en arena, 35 % en suelo franco y 45 % en arcilla.
(48) La capacidad de campo no es constante, sino que varía con el tipo de suelo entre pF 1,5 y 2,5.
(49) Capacidad de retención de agua.
(50) Guth, J.A. (1980). The study of transformations. In Interactions between Herbicides and the Soi] (R.J. Hance, Ed.), Academic Press, 123-157.
(51) Guth, J.A. (1981). Experimental approaches to studying the fate of pesticides in soil. In Progress in Pesticide Biochemistry. D.H. Hutson, T.R. Roberts, Eds. J. Wiley & Sons. Vol 1, 85-114.
(52) Anderson, J.P.E. (1975). Einfluss von Temperatur und Feuchte auf Verdampfung, Abbau und Festlegung von Diallat im Boden. Z. PflKrankh Pflschutz, Sonderheft VU, 141-146.
(53) Por ejemplo, si la sustancia de ensayo contiene un anillo, es preciso marcar ese anillo: si contiene dos o más anillos, puede resultar necesario efectuar estudios separados para evaluar el destino de cada anillo marcado y obtener información adecuada sobre la formación de los productos de transformación.
(54) Como estas soluciones de absorción alcalinas absorben también el dióxido de carbono del aire de la ventilación y el formado por la respiración en los experimentos aerobios, tienen que cambiarse a intervalos regulares para evitar su saturación y, así, la pérdida de su capacidad de absorción.
(55) [Arcilla+limo] es la fracción mineral del sedimento con un tamaño de partícula < 50 μm.
(56) Los resultados de investigaciones recientes han mostrado que las mediciones de las concentraciones de oxígeno en el agua y los potenciales redox no tienen un valor mecáno ni predictivo on lo que se refiere al crecimiento y desarrollo de poblaciones microbianas en aguas superficiales (24)(25). La determinacíon de la demanda bioquímica de oxígeno (DBO en la toma de muestras, el inicio y el final del ensayo) y de las concentraciones de los micro/macronutrientes Ca, Mg y Mn (al inicio y al final del ensayo) en el agua, y la medición de N y el P totales en los sedimentos (en la toma de muestras y al final del ensayo) pueden ser instrumentos mejores para interpretar y evaluar las tasas y las vías de biotransformación aerobia.
(57) Método de la tasa de respiración microbiana (26), método de la fumigación (27) o mediciones de recuento en placas (por ejemplo, bacterias, actinomicetos, hongos y colonias totales) para estudios anaerobios; tasa de metanogénesis para estudios anaerobios
(58) Cuando pueda darse fácilmente una reoxidación rápida de los productos de la transformación anaerobia, se mantendrán las condiciones anaerobias durante el muestreo y el análisis.
Axencia Estatal Boletín Oficial do Estado
Avda. de Manoteras, 54 - 28050 Madrid


 (mN/m)
(mN/m)


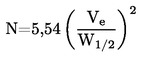

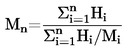
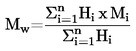





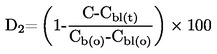

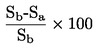







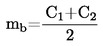





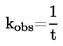













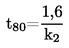

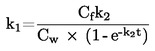















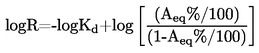

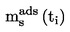


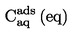



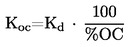
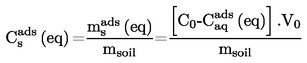


















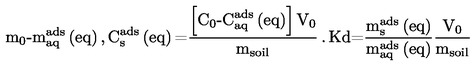

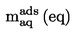
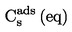

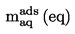 ,
,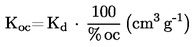





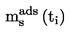








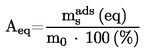




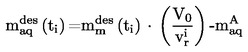

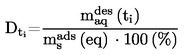

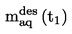



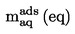


 =
= 



 and
and