Axencia Estatal Boletín Oficial do Estado






Contido non dispoñible en galego
LA COMISIÓN EUROPEA,
Visto el Tratado de Funcionamiento de la Unión Europea,
Visto el Reglamento (UE) n.o 1308/2013 del Parlamento Europeo y del Consejo, de 17 de diciembre de 2013, por el que se crea la organización común de mercados de los productos agrarios y por el que se derogan los Reglamentos (CEE) n.o 922/72, (CEE) n.o 234/79, (CE) n.o 1037/2001 y (CE) n.o 1234/2007 (1), y en particular su artículo 91, apartado 1, letra d);
Considerando lo siguiente:
(1) El Reglamento (CEE) n.o 2568/91 de la Comisión (2) define las características fisicoquímicas y organolépticas de los aceites de oliva y de los aceites de orujo de oliva, así como los métodos de evaluación de dichas características.
(2) Los métodos y los valores límite relativos a las características de los aceites se actualizan regularmente teniendo en cuenta la opinión de los expertos químicos y en consonancia con los trabajos efectuados en el marco del Consejo Oleícola Internacional (COI).
(3) A fin de garantizar la aplicación a nivel de la Unión de las normas internacionales más recientes establecidas por el COI, deben actualizarse determinados métodos de análisis establecidos en el Reglamento (CEE) n.o 2568/91.
(4) Se ha modificado la Norma Comercial del COI en lo que se refiere a la expresión del límite de acidez libre, el índice de peróxidos, la evaluación organoléptica (mediana de los defectos y mediana del atributo frutado) y la diferencia entre ECN42 (HPLC) y ECN42 (cálculo teórico) por coherencia con los valores de precisión del método analítico.
(5) De conformidad con el artículo 2 bis, apartado 5, del Reglamento (CEE) n.o 2568/91, los Estados miembros deben verificar si una muestra de aceite de oliva es coherente con la categoría declarada, mediante el control de las características establecidas en el anexo I de dicho Reglamento, en cualquier orden o siguiendo el orden establecido en un árbol de decisión que figura en el anexo I ter.
(6) A la vista de la evolución reciente, procede actualizar los cuadros que figuran en el anexo I ter del Reglamento (CEE) n.o 2568/91 y, en su caso, su apéndice. También parece que el término «diagrama de flujos» es más adecuado que el término «árbol de decisión» habida cuenta del contenido de dicho anexo I ter.
(7) El punto 9.4 del anexo XII del Reglamento (CEE) n.o 2568/91 define la mediana de los defectos como la mediana del defecto percibido con mayor intensidad. En el contexto de los análisis contradictorios y dado que los diferentes paneles tienen que evaluar la conformidad del aceite, debe aclararse que la decisión relativa a la conformidad de las características de un aceite con la categoría declarada está exclusivamente relacionada con el valor de la mediana del defecto principal, independientemente de su naturaleza.
(8) Procede, por tanto, modificar el Reglamento (CEE) n.o 2568/91 en consecuencia.
(9) Las medidas previstas en la presente Decisión se ajustan al dictamen del Comité de la Organización Común de Mercados Agrarios.
HA ADOPTADO EL PRESENTE REGLAMENTO:
El Reglamento (CEE) n.o 2568/91 queda modificado como sigue:
1) El artículo 2 se modifica como sigue:
a) en el apartado 1, la letra l) se sustituye por el texto siguiente:
«l) para determinar la composición y el contenido de esteroles y para determinar los compuestos alcohólicos, mediante cromatografía de gases con columna capilar, el método recogido en el anexo XIX»;
b) en el apartado 2, el párrafo tercero se sustituye por el texto siguiente:
«En caso de que el panel autorizado no confirme tal declaración en lo que respecta a las características organolépticas, las autoridades nacionales o sus representantes ordenarán que se proceda, a petición del interesado, a realizar sin dilación dos análisis contradictorios por otros paneles autorizados. Al menos uno de ellos deberá ser un panel de cata autorizado por el Estado miembro productor. Las características en cuestión se considerarán conformes a las declaradas si ambos análisis contradictorios confirman la clasificación declarada. En caso contrario, independientemente del tipo de defectos determinados durante los análisis contradictorios, la clasificación se declarará no coherente con las características y los gastos generados por los análisis contradictorios correrán por cuenta del interesado.».
2) En el artículo 2 bis, apartado 5, la letra b) se sustituye por el texto siguiente:
«b) según el orden establecido en el anexo I ter del diagrama de flujos hasta la adopción de una de las decisiones mencionadas en dicho diagrama.».
3) El cuadro «ANEXOS-Índice» se sustituye por el cuadro que figura en el anexo I del presente Reglamento.
4) El anexo I se sustituye por el texto que figura en el anexo II del presente Reglamento.
5) En el anexo I bis, el punto 2.1 se sustituye por el texto siguiente:
«2.1.
Cada muestra elemental debe subdividirse en muestras de laboratorio, con arreglo al punto 2.5 de la norma EN ISO 5555, y analizarse según el orden que se indica en el diagrama de flujos recogido en el anexo I ter o cualquier otro orden aleatorio.».
6) El anexo I ter se sustituye por el texto del anexo III del presente Reglamento.
7) Se suprime el anexo V.
8) En el anexo VII, el punto 4.2 se sustituye por el texto siguiente:
«4.2.
n-hexano (grado cromatográfico). El hexano podrá ser sustituido por isooctano (2,2,4-trimetilpentano en grado cromatográfico), siempre que se alcancen valores de precisión comparables.».
9) El anexo XII se modifica de conformidad con el anexo IV del presente Reglamento.
10) El anexo XVII se modifica de conformidad con el anexo V del presente Reglamento.
11) El anexo XVIII se modifica de conformidad con el anexo VI del presente Reglamento.
12) El anexo XIX se sustituye por el texto del anexo VII del presente Reglamento.
13) En el anexo XX, el punto 4.2 se sustituye por el texto siguiente:
«4.2.
n-hexano, para cromatografía o análisis de residuos. El hexano podrá ser sustituido por isooctano (2,2,4-trimetilpentano en grado cromatográfico), siempre que se alcancen valores de precisión comparables. Los disolventes con un punto de ebullición más elevado que el del n-hexano tardan más en evaporarse. No obstante, son preferibles debido a la toxicidad del hexano. El grado de pureza deberá comprobarse; por ejemplo, podrá controlarse el residuo tras la evaporación de 100 ml de disolvente.
ADVERTENCIA: Riesgo de inflamación de los vapores. Mantener lejos de fuentes de calor, chispas y llamas desnudas. Mantener en recipientes bien cerrados. Utilizar con la ventilación adecuada. Evitar la acumulación de vapores y eliminar toda posible causa de incendio, como calentadores o aparatos eléctricos no fabricados con material antiinflamable. Nocivo por inhalación: puede dañar las células del sistema nervioso. Evitar respirar los vapores. Utilizar, si es preciso, un aparato respirador adecuado. Evitar el contacto con los ojos y la piel.
El isooctano es un líquido inflamable que presenta un riesgo de incendio. Su explosión en el aire se sitúa entre los límites del 1,1 % y el 6,0 % (fracción en volumen). Es tóxico por ingestión y por inhalación. Utilizar una campana ventilada en buen estado para trabajar con este disolvente.».
El presente Reglamento entrará en vigor a los veinte días de su publicación en el Diario Oficial de la Unión Europea.
El presente Reglamento será obligatorio en todos sus elementos y directamente aplicable en todos los Estados miembros.
Hecho en Bruselas, el 27 de septiembre de 2019.
Por la Comisión,
El Presidente
Jean-Claude JUNCKER
(1) DO L 347 de 20.12.2013, p. 671.
(2) Reglamento (CEE) n.o 2568/91 de la Comisión, de 11 de julio de 1991, relativo a las características de los aceites de oliva y de los aceites de orujo de oliva y sobre sus métodos de análisis (DO L 248 de 5.9.1991, p. 1).
IMÁGENES OMITIDAS
«ANEXOS
ÍNDICE
|
Anexo I |
Características de los aceites de oliva |
|
Anexo I bis |
Muestreo de aceite de oliva o aceite de orujo de oliva entregado en envases inmediatos |
|
Anexo I ter |
Diagrama de flujos para la comprobación de la conformidad de una muestra de aceite de oliva con la categoría declarada |
|
Anexo II |
Determinación de los ácidos grasos libres, método en frío |
|
Anexo III |
Determinación del índice de peróxidos |
|
Anexo IV |
Determinación del contenido de ceras mediante cromatografía de gases con columna capilar |
|
Anexo VII |
Determinación del porcentaje de monopalmitato de 2-glicerilo |
|
Anexo IX |
Prueba espectrofotométrica en el ultravioleta |
|
Anexo X |
Determinación de ésteres metílicos de ácidos grasos mediante cromatografía de gases |
|
Anexo XI |
Determinación del contenido en disolventes halogenados volátiles en el aceite de oliva |
|
Anexo XII |
Método del Consejo Oleícola Internacional para la evaluación organoléptica de los aceites de oliva vírgenes |
|
Anexo XV |
Contenido de aceite de los orujos de aceituna |
|
Anexo XVI |
Determinación del índice de yodo |
|
Anexo XVII |
Método para la determinación de estigmastadienos en los aceites vegetales |
|
Anexo XVIII |
Determinación de la diferencia entre el contenido real y el contenido teórico de triglicéridos con ECN 42 |
|
Anexo XIX |
Determinación de la composición y contenido de los esteroles y compuestos alcohólicos mediante cromatografía de gases con columna capilar |
|
Anexo XX |
Método para la determinación del contenido en ceras y en ésteres metílicos y etílicos de los ácidos grasos mediante cromatografía de gases con columna capilar |
|
Anexo XXI |
Resultados de los controles de conformidad realizados en los aceites de oliva a que se refiere el artículo 8, apartado 2 |
»
«ANEXO I
CARACTERÍSTICAS DE LOS ACEITES DE OLIVA
Características de calidad
|
Ésteres etílicos de los ácidos grasos (mg/kg) |
≤ 35 |
— |
— |
— |
— |
— |
— |
— |
||||||||||||||||
|
Evaluación organoléptica |
Mediana del frutado (Mf) |
Mf > 0,0 |
Mf > 0,0 |
— |
— |
— |
— |
— |
— |
|||||||||||||||
|
Mediana del defecto (Md) (*) |
Md ≤ 0,0 |
Md ≤ 3,5 |
Md> 3,5 (1) |
|
|
|
|
|
||||||||||||||||
|
Delta-K |
≤ 0,01 |
≤ 0,01 |
— |
≤ 0,16 |
≤ 0,15 |
— |
≤ 0,20 |
≤ 0,18 |
||||||||||||||||
|
K268 o K270 |
≤ 0,22 |
≤ 0,25 |
— |
≤ 1,25 |
≤ 1,15 |
— |
≤ 2,00 |
≤ 1,70 |
||||||||||||||||
|
K232 |
≤ 2,50 |
≤ 2,60 |
— |
— |
— |
— |
— |
— |
||||||||||||||||
|
Índice de peróxidos (mEq O2/kg) |
≤ 20,0 |
≤ 20,0 |
— |
≤ 5,0 |
≤ 15,0 |
— |
≤ 5,0 |
≤ 15,0 |
||||||||||||||||
|
Acidez (%) (*) |
≤ 0,80 |
≤ 2,0 |
> 2,0 |
≤ 0,30 |
≤ 1,00 |
— |
≤ 0,30 |
≤ 1,00 |
||||||||||||||||
|
Categoría |
|
|
|
|
|
|
|
|
Características de pureza
|
Monopalmitato de 2-glicerilo (%) |
≤ 0,9 si el % total de ácido palmítico ≤ 14,00 % |
≤ 1,0 si el % total de ácido palmítico > 14 % |
≤ 0,9 si el % total de ácido palmítico ≤ 14,00 % |
≤ 1,0 si el % total de ácido palmítico > 14 % |
≤ 0,9 si el % total de ácido palmítico ≤ 14 % |
≤ 1,1 si el % total de ácido palmítico es > 14,00 % |
≤ 0,9 si el % total de ácido palmítico ≤ 14,00 % |
≤ 1,1 si el % total de ácido palmítico > 14 % |
≤ 0,9 si el % total de ácido palmítico ≤ 14 % |
≤ 1,0 si el % total de ácido palmítico es > 14,00 % |
≤ 1,4 |
≤ 1,4 |
≤ 1,2[ |
|
Diferencia: ECN 42 (HPLC) y ECN 42 (cálculo teórico) |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
≤ 0,30 |
≤ 0,30 |
≤ 0,60 |
≤ 0,50 |
≤ 0,50 |
|||||
|
Estigmastadienos (mg/kg) (3) |
≤ 0,05 |
≤ 0,05 |
≤ 0,50 |
— |
— |
— |
— |
— |
|||||
|
Sumas de los isómeros translinoleicos + translinolénicos (%) |
≤ 0,05 |
≤ 0,05 |
≤ 0,10 |
≤ 0,30 |
≤ 0,30 |
≤ 0,10 |
≤ 0,35 |
≤ 0,35 |
|||||
|
Sumas de los isómeros transoleicos (%) |
≤ 0,05 |
≤ 0,05 |
≤ 0,10 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,40 |
≤ 0,40 |
|||||
|
Composición de ácidos grasos (2) |
Lignocérico (%) |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
||||
|
Behénico (%) |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
≤ 0,30 |
≤ 0,30 |
|||||
|
Eicosenoico (%) |
≤ 0,50 |
≤ 0,50 |
≤ 0,50 |
≤ 0,50 |
≤ 0,50 |
≤ 0,50 |
≤ 0,50 |
≤ 0,50 |
|||||
|
Araquídico (%) |
≤ 0,60 |
≤ 0,60 |
≤ 0,60 |
≤ 0,60 |
≤ 0,60 |
≤ 0,60 |
≤ 0,60 |
≤ 0,60 |
|||||
|
Linolénico (%) |
≤ 1,00 |
≤ 1,00 |
≤ 1,00 |
≤ 1,00 |
≤ 1,00 |
≤ 1,00 |
≤ 1,00 |
≤ 1,00 |
|||||
|
Mirístico (%) |
≤ 0,03 |
≤ 0,03 |
≤ 0,03 |
≤ 0,03 |
≤ 0,03 |
≤ 0,03 |
≤ 0,03 |
≤ 0,03 |
|||||
|
Categoría |
1. Aceite de oliva virgen extra |
2. Aceite de oliva virgen |
3. Aceite de oliva lampante |
4. Aceite de oliva refinado |
5. Aceite de oliva compuesto de aceite de oliva refinado y aceites de oliva vírgenes |
6. Aceite de orujo de oliva crudo |
7. Aceite de orujo de oliva refinado |
8. Aceite de orujo de oliva |
|
Ceras (mg/kg) (**) |
C42 + C44 + C46 ≤ 150 |
C42 + C44 + C46 ≤ 150 |
C40 + C42 + C44 + C46 ≤ 300 (6) |
C40 + C42 + C44 + C46 ≤ 350 |
C40 + C42 + C44 + C46 ≤ 350 |
C40 + C42 + C44 + C46 > 350 (7) |
C40 + C42 + C44 + C46 > 350 |
C40 + C42 + C44 + C46 > 350 |
|
|
Eritrodiol y uvaol (%) (**) |
≤ 4,5 |
≤ 4,5 |
≤ 4,5 (6) |
≤ 4,5 |
≤ 4,5 |
> 4,5 (7) |
> 4,5 |
> 4,5 |
|
|
Esteroles totales (mg/kg) |
≥ 1 000 |
≥ 1 000 |
≥ 1 000 |
≥ 1 000 |
≥ 1 000 |
≥ 2 500 |
≥ 1 800 |
≥ 1 600 |
|
|
Composición de los esteroles |
Delta-7- estigmastenol (4) (%) |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
|
β-sitosterol aparente (5) (%) |
≥ 93,0 |
≥ 93,0 |
≥ 93,0 |
≥ 93,0 |
≥ 93,0 |
≥ 93,0 |
≥ 93,0 |
≥ 93,0 |
|
|
Estigmasterol (%) |
< Camp. |
< Camp. |
— |
< Camp. |
< Camp. |
— |
< Camp. |
< Camp. |
|
|
Campesterol (4) (%) |
≤ 4,0 |
≤ 4,0 |
≤ 4,0 |
≤ 4,0 |
≤ 4,0 |
≤ 4,0 |
≤ 4,0 |
≤ 4,0 |
|
|
Brasicasterol (%) |
≤ 0,1 |
≤ 0,1 |
≤ 0,1 |
≤ 0,1 |
≤ 0,1 |
≤ 0,2 |
≤ 0,2 |
≤ 0,2 |
|
|
Colesterol (%) |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
|
|
Categoría |
1. Aceite de oliva virgen extra |
2. Aceite de oliva virgen |
3. Aceite de oliva lampante |
4. Aceite de oliva refinado |
5. Aceite de oliva compuesto de aceite de oliva refinado y aceites de oliva vírgenes |
6. Aceite de orujo de oliva crudo |
7. Aceite de orujo de oliva refinado |
8. Aceite de orujo de oliva |
Notas:
a) Los resultados de los análisis deben expresarse indicando el mismo número de decimales que el previsto para cada característica. La última cifra expresada debe redondearse hacia arriba si la cifra siguiente es superior a 4.
b) Es suficiente con que una sola de las características no se ajuste a los valores indicados para que el aceite cambie de categoría o se declare no conforme a efectos del presente Reglamento.
c) En el caso del aceite de oliva lampante, las características cualitativas marcadas con un asterisco (*) pueden diferir al mismo tiempo de los límites establecidos para esa categoría.
d) Las características indicadas con dos asteriscos (**) implican que, en el caso del aceite de orujo de oliva crudo, pueden no respetarse simultáneamente los límites correspondientes. En el caso del aceite de orujo de oliva y el aceite de orujo de oliva refinado, uno de los límites pertinentes puede ser distinto de los valores declarados.
Apéndice
Árboles de decisión
Árbol de decisión del campesterol para los aceites de oliva virgen y virgen extra:

Los restantes parámetros deberán cumplir los límites que establece el presente Reglamento.
Árbol de decisión del delta-7-estigmastenol para:
— Aceites de oliva vírgenes extra y vírgenes
Los restantes parámetros se adaptarán a los límites que establece el presente Reglamento.
— Aceites de orujo de oliva (crudo y refinado)
Los restantes parámetros deberán cumplir los límites que establece el presente Reglamento.
».
(1) La mediana de los defectos puede ser inferior o igual a 3,5 cuando la mediana del frutado es igual a 0,0.
(2) Contenido de otros ácidos grasos (%): palmítico: 7,50-20,00. palmitoleico: 0,30-3,50. heptadecanoico: ≤ 0,40; heptadececenoico ≤ 0,60; esteárico: 0,50-5,00. oleico: 55,00- 83,00; linoleico: 2,50-21,00.
(3) Total de isómeros que han podido (o no han podido) separarse con columna capilar.
(4) Véase el apéndice del presente anexo.
(5) β-sitosterol aparente: delta-5,23-estigmastadienol + clerosterol + beta-sitosterol + sitostanol + delta-5-avenasterol + delta-5,24-estigmastadienol.
(6) Se considera que los aceites con un contenido de ceras entre 300 mg/kg y 350 mg/kg son aceites de oliva lampantes si el contenido total de alcoholes alifáticos es inferior o igual a 350 mg/kg o si el contenido de eritrodiol y uvaol es inferior o igual al 3,5 %.
(7) Los aceites con un contenido de ceras comprendido entre 300 mg/kg y 350 mg/kg se consideran aceite de orujo de oliva crudo si el contenido de alcoholes alifáticos totales es superior a 350 mg/kg y si el contenido de eritrodiol y uvaol es superior al 3,5 %.
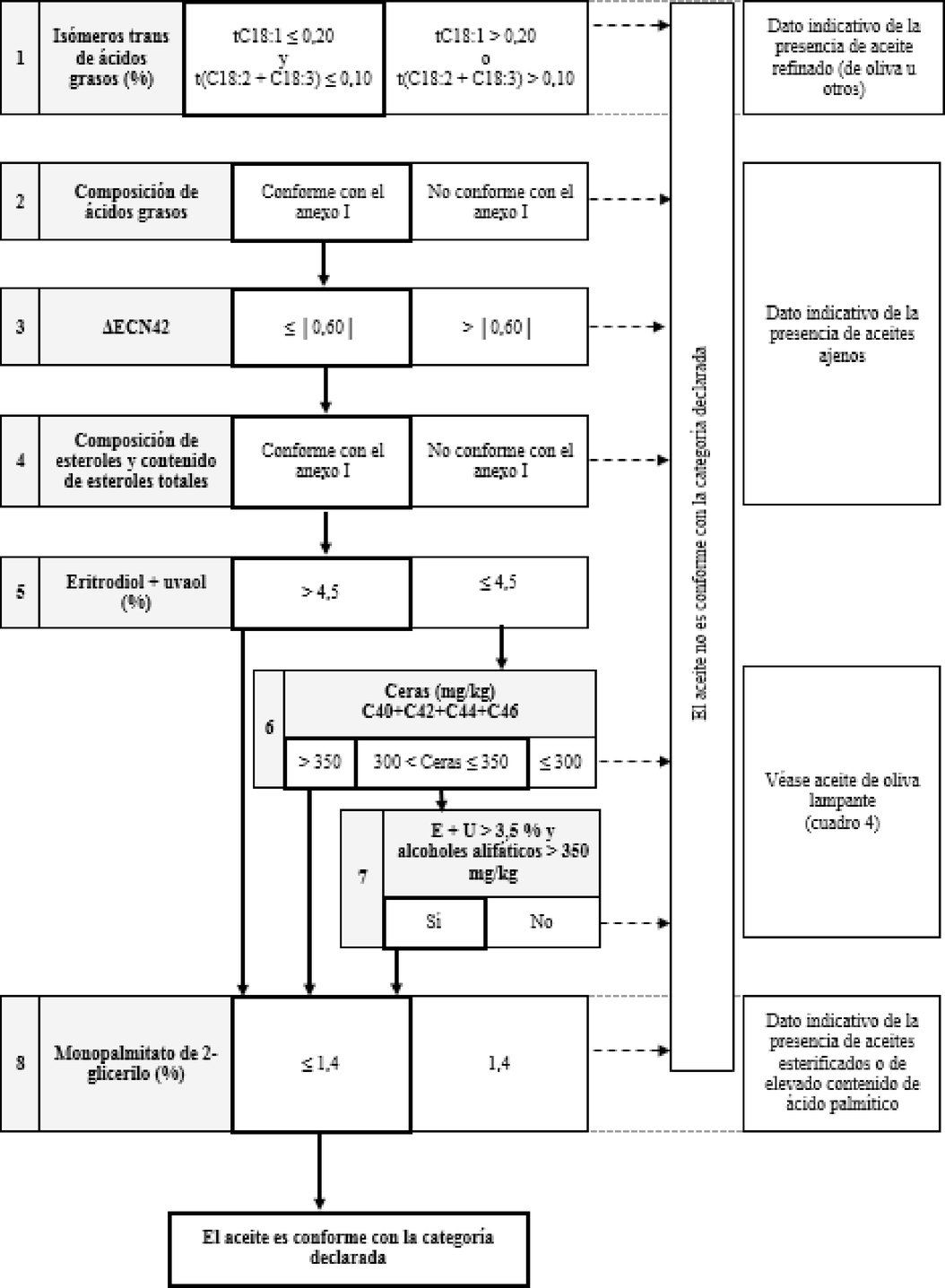
«ANEXO I ter
DIAGRAMA DE FLUJOS PARA LA COMPROBACIÓN DE LA CONFORMIDAD DE UNA MUESTRA DE ACEITE DE OLIVA CON LA CATEGORÍA DECLARADA
Cuadro general

Cuadro 1 — Aceite de oliva virgen extra — Criterios de calidad

Cuadro 2 — Aceite de oliva virgen— Criterios de calidad

Cuadro 3 — Aceite de oliva virgen extra y aceite de oliva virgen — Criterios de pureza

Cuadro 4 — Aceite de oliva lampante — Criterios de pureza

Cuadro 5 — Aceite de oliva refinado — Criterios de calidad

Cuadro 6 — Aceite de oliva (compuesto de aceite de oliva refinado y aceites de oliva vírgenes) — Criterios de calidad

Cuadro 7 — Aceite de oliva refinado y aceite de oliva compuesto de aceite de oliva refinado y aceites de oliva vírgenes — Criterios de pureza
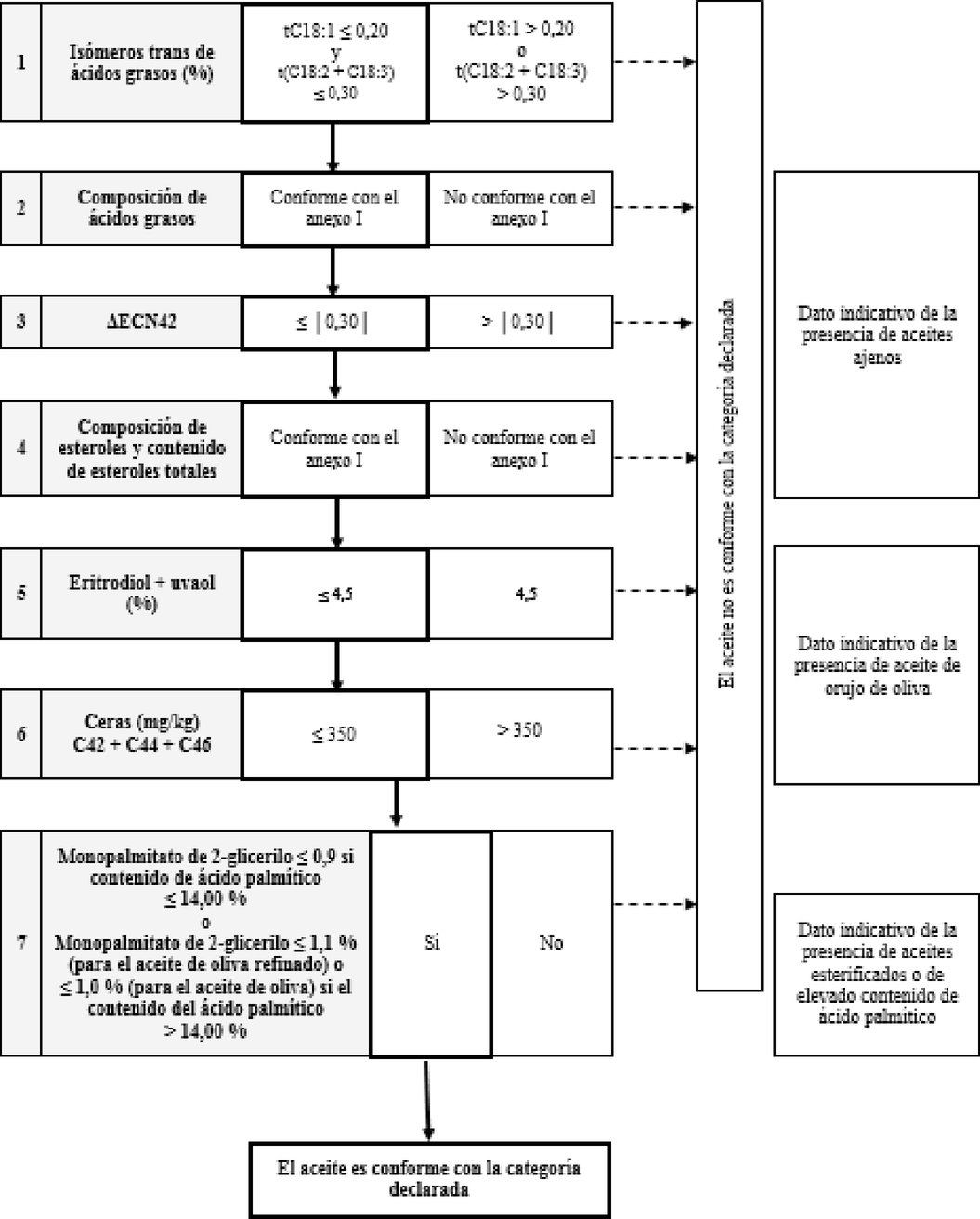
Cuadro 8 — Aceite de orujo de oliva crudo — Criterios de pureza
Cuadro 9 — Aceite de orujo de oliva refinado — Criterios de calidad

Cuadro 10 — Aceite de orujo de oliva— Criterios de calidad

Cuadro 11 — Aceite de orujo de oliva refinado y aceite de orujo de oliva — Criterios de pureza

».
El anexo XII queda modificado como sigue:
|
1) |
El texto del punto 3.3 se sustituye por el siguiente: «3.3. Terminología opcional para el etiquetado A petición expresa, el jefe de panel puede certificar que los aceites evaluados cumplen las definiciones e intervalos correspondientes únicamente a los términos siguientes en función de la intensidad y de la percepción de los atributos: Atributos positivos (frutado, amargo y picante): En función de la intensidad de la percepción: — Robusto, cuando la mediana del atributo sea superior a 6,0. — Medio, cuando la mediana del atributo sea superior a 3,0 e inferior o igual a 6,0; — Delicado, cuando la mediana del atributo sea inferior o igual a 3,0.
Lista de términos en función de la intensidad de la percepción:
|
|
2) |
El texto del punto 9.4 se sustituye por el siguiente: «9.4. Clasificación del aceite El aceite se clasifica en las categorías que se indican más adelante, en función de la mediana de los defectos y de la mediana del atributo frutado. Por mediana de los defectos se entiende la mediana del defecto percibido con mayor intensidad. La mediana de los defectos y la mediana del atributo frutado se expresarán con una sola cifra decimal. La clasificación del aceite se hace comparando el valor de la mediana de los defectos y de la mediana del atributo frutado con los intervalos de referencia expuestos a continuación. Los límites de estos intervalos han sido establecidos teniendo en cuenta el error del método, por lo que son considerados como absolutos. Los programas informáticos permiten visualizar la clasificación en un cuadro de datos estadísticos o gráficamente. a) Aceite de oliva virgen extra: la mediana de los defectos es igual a 0,0 y la del atributo frutado es superior a 0,0; b) Aceite de oliva virgen: la mediana de los defectos es superior a 0,0 pero inferior o igual a 3,5 y la del atributo frutado es superior a 0,0; c) Aceite de oliva virgen lampante: la mediana de los defectos es superior a 3,5, o bien la mediana de los defectos es inferior o igual a 3,5 y la del atributo frutado es igual a 0,0. Nota 1: Cuando las medianas del atributo amargo y/o del atributo picante sean superiores a 5,0, el jefe de panel lo señalará en el certificado de análisis del aceite. En el caso de los análisis efectuados en el marco de controles de conformidad, se realizará un ensayo. En el caso de análisis contradictorios, el análisis debe efectuarse por duplicado en diferentes sesiones de cata. En el caso de análisis contradictorios, el análisis debe efectuarse por duplicado en diferentes sesiones de cata. Los resultados de los análisis por duplicado deben ser estadísticamente homogéneos (véase el punto 9.5). En caso contrario, la muestra debe ser analizada de nuevo dos veces. El valor final de la mediana de los atributos de clasificación se calculará utilizando la media de ambas medianas.». |
El anexo XVII se modifica como sigue:
1) El texto del punto 5,1 se sustituye por el siguiente:
«5.1.
Hexano o una mezcla de alcanos con un intervalo de ebullición de 65-70 °C, destilados en columna rectificadora. El hexano podrá ser sustituido por isooctano (2,2,4-trimetil pentano en grado cromatográfico), siempre que se alcancen valores de precisión comparables. El residuo tras la evaporación de 100 ml de disolvente podrá ser controlado. Los disolventes con un punto de ebullición superior al del n-hexano tardan en evaporarse. No obstante, son preferibles debido a la toxicidad del hexano.».
2) en el punto 6.3.3 se añade el texto siguiente:
«Nota 10: En caso de que aparezcan los estigmastadienos en concentraciones superiores a 4 mg/kg, si se requiere la cuantificación deberá aplicarse el método del Consejo Oleícola Internacional para determinar los esterenos en los aceites vegetales refinados.».
El anexo XVIII se modifica de la siguiente manera:
1) El texto del punto 4.2.1 se sustituye por el siguiente:
«4.2.1.
Éter de petróleo 40-60 °C, de pureza de grado cromatográfico, o hexano. El hexano podrá ser sustituido por isooctano (2,2,4-trimetil pentano en grado cromatográfico), siempre que se alcancen valores de precisión comparables. Los disolventes con un punto de ebullición superior al del n-hexano tardan en evaporarse. No obstante, son preferibles debido a la toxicidad del hexano.».
2) Se añade el punto 4.2.12 siguiente:
«4.2.12.
Heptano, calidad cromatográfica. El heptano podrá ser sustituido por isooctano (2,2,4-trimetil pentano en grado cromatográfico).».
«ANEXO XIX
DETERMINACIÓN DE LA COMPOSICIÓN Y CONTENIDO DE LOS ESTEROLES Y COMPUESTOS ALCOHÓLICOS MEDIANTE CROMATOGRAFÍA DE GASES CON COLUMNA CAPILAR
1. ÁMBITO DE APLICACIÓN
El método describe un procedimiento para determinar el contenido alcohólico volumétrico total e individual de los aceites de oliva y de los aceites de orujo de oliva, así como de las mezclas de estos dos aceites.
Los compuestos alcohólicos de los aceites de oliva y los aceites de orujo de oliva comprenden alcoholes alifáticos, esteroles y dialcoholes triterpénicos.
2. PRINCIPIO
Los aceites, con α-colestanol y 1-eicosanol añadidos como patrones internos, se saponifican con hidróxido potásico en solución etanólica y la materia insaponificable se extrae a continuación con éter de etilo.
Las diferentes fracciones de compuestos alcohólicos se separan de la materia insaponificable, ya sea mediante cromatografía en capa fina sobre una placa de gel de sílice básica (método de referencia) o mediante HPLC con columna de gel de sílice. La fracción recuperada de la separación del gel de sílice se transforma en éteres de trimetilsililo y se analizan a continuación mediante cromatografía de gases con columna capilar.
PARTE 1
PREPARACIÓN DEL INSAPONIFICABLE
1. ÁMBITO DE APLICACIÓN
Esta parte describe la preparación y extracción del insaponificable. Incluye la preparación y extracción del insaponificable a partir de los aceites de oliva y de los aceites de orujo de oliva.
2. PRINCIPIO
Una porción de prueba se saponifica llevándola a ebullición por reflujo con una solución etanólica de hidróxido de potasio. El insaponificable se extrae mediante éter dietílico.
3. INSTRUMENTAL
El equipo de laboratorio habitual y, en particular, lo siguiente:
|
3.1. |
Matraz de fondo redondo de 250 ml provisto de refrigerante de reflujo con juntas esmeriladas. |
|
3.2. |
Embudo de decantación de 500 ml. |
|
3.3. |
Matraces de 250 ml. |
|
3.4. |
Microjeringas, de100 μl y 500 μl |
|
3.5. |
Embudo cilíndrico filtrante con filtro poroso G3 (porosidad 15-40 μm) con un diámetro aproximado de 2 cm y una profundidad de 5 cm, apropiado para filtraciones al vacío, con junta esmerilada macho. |
|
3.6. |
Matraz cónico de 50 ml, con junta esmerilada hembra acoplable al embudo filtrante (punto 3.5). |
|
3.7. |
Tubo de ensayo de 10 ml con fondo cónico y tapón de vidrio de cierre hermético. |
|
3.8. |
Desecador de dicloruro de calcio. |
4. REACTIVOS
|
4.1. |
Hidróxido potásico (pureza mínima del 85 %). |
|
4.2. |
Hidróxido potásico, solución etanólica 2 M, aproximadamente: Disolver, enfriando al mismo tiempo, 130 g de hidróxido potásico (4.1) en 200 ml de agua destilada y completar hasta un litro con etanol (4.7). Conservar la solución en botellas de vidrio oscuro bien cerradas y almacenarla un máximo de dos días. |
|
4.3. |
Éter etílico de calidad para análisis. |
|
4.4. |
Sulfato sódico anhidro, de calidad para análisis. |
|
4.5. |
Acetona, de calidad para cromatografía. |
|
4.6. |
Éter etílico, de calidad para cromatografía. |
|
4.7. |
Etanol, de calidad para análisis. |
|
4.8. |
Acetato de etilo, de calidad para análisis. |
|
4.9. |
patrón interno, α-Colestanol, de pureza superior al 99 % (hay que comprobar la pureza mediante cromatografía de gases). |
|
4.10. |
Solución de patrón interno de α-colestanol, solución 0,2 (m/V) en acetato de etilo (4.8). |
|
4.11. |
Solución de fenolftaleína, 10 g/l en etanol (4.7). |
|
4.12. |
1-eicosanol, solución al 0,1 % (m/v) en acetato de etilo (patrón interno). |
5. PROCEDIMIENTO
Utilizando una microjeringa de 500 μl (3.4), introducir en el matraz de 250 ml (3.1) un volumen de solución de patrón interno de α-colestanol (4.10) y un volumen de 1-eicosanol (4.12) que contengan una cantidad de colestanol y de eicosanol correspondiente a aproximadamente el 10 % del contenido de esteroles y alcohol de la muestra. Por ejemplo, para 5 g de aceite de oliva, añadir 500 μl de la solución de α-colestanol (4.10) y 250 μl de la solución de 1-eicosanol (4.12). Para los aceites de orujo de oliva, añadir 1 500 μl de la solución de α-colestanol (4.10) y 1 500 μl de la solución de 1-eicosanol (4.12). Se hace evaporar hasta secar mediante una ligera corriente de nitrógeno en un baño de agua templada. Después de enfriar el matraz, en el mismo matraz pesar 5,00 g ± 0,01 g de la muestra filtrada seca.
Nota 1: En el caso de aceites y grasas animales o vegetales con un alto contenido de colesterol puede producirse un pico cuyo tiempo de retención sea idéntico al del colestanol. En tal caso, el análisis de la fracción de esteroles debe realizarse dos veces: con patrón interno y sin él.
Añadir 50 ml de solución etanólica de hidróxido potásico 2 M (4.2) y algo de piedra pómez, ajustar el condensador de reflujo y calentar a ebullición suave hasta que tenga lugar la saponificación (la solución se vuelve incolora). Calentar durante 20 minutos más y, a continuación, añadir 50 ml de agua destilada por la parte superior del condensador; separar el condensador y enfriar el matraz a 30 °C aproximadamente.
Pasar el contenido del matraz cuantitativamente a una ampolla de decantación de 500 ml (3.2) usando varias fracciones de agua destilada (50 ml). Añadir aproximadamente 80 ml de éter etílico (4.6) y agitar con fuerza aproximadamente durante 60 segundos con liberación periódica de la presión invirtiendo la ampolla de decantación y abriendo la llave. Dejarla en reposo hasta que se produzca una completa separación en dos fases (nota 2). Retirar entonces la solución de jabón lo más exhaustivamente posible y pasar a una segunda ampolla de decantación. Efectuar otras dos extracciones de la fase hidroalcohólica por el mismo procedimiento, utilizando de 60 a 70 ml de éter etílico (4.6).
Nota 2: Las emulsiones pueden eliminarse añadiendo pequeñas cantidades de etanol (4.7).
Combinar las tres extracciones de éter en una sola ampolla de decantación que contenga 50 ml de agua. Continuar lavando con agua (50 ml) hasta que el agua de lavado no adquiera un color rosa al añadirle una gota de solución de fenolftaleína (4.11). Una vez eliminada el agua de lavado, filtrar con sulfato sódico anhidro (4.4) a un matraz de 250 ml previamente pesado, lavando el embudo y el filtro con pequeñas cantidades de éter etílico (4.6).
Evaporar el disolvente mediante su destilación en un rotavapor a 30 °C al vacío. Añadir 5 ml de acetona (4.5) y eliminar totalmente el disolvente volátil con una ligera corriente de aire. Secar el residuo en el horno a 103 ± 2 °C durante 15 min. Enfriar en desecador y pesar con precisión de 0,1 mg.
PARTE 2
SEPARACIÓN DE LAS FRACCIONES DE LOS COMPUESTOS ALCOHÓLICOS
1. ÁMBITO DE APLICACIÓN
El insaponificable preparado en la parte 1 está fraccionado en compuestos alcohólicos, alcoholes alifáticos, esteroles y dialcoholes triterpénicos (eritrodiol y uvaol).
2. PRINCIPIO
El insaponificable puede fraccionarse, mediante cromatografía de capa fina básica (método de referencia) y hacerse visible y las bandas correspondientes deben rasparse y extraerse. Como método alternativo, la separación puede realizarse mediante HPLC utilizando una columna de gel de sílice y un detector por luz ultravioleta y las diferentes fracciones recogidas. Los alcoholes alifáticos y triterpénicos, así como el esterol y los dialcoholes triterpénicos, se aíslan juntos.
3. INSTRUMENTAL
El equipo de laboratorio habitual y, en particular, lo siguiente:
|
3.1. |
Aparato completo para análisis mediante cromatografía en capa fina usando placas de vidrio de 20 × 20 cm. |
|
3.2. |
Lámpara ultravioleta con longitud de onda de 366 o 254 nm. |
|
3.3. |
Microjeringas, de100 μl y 500 μl |
|
3.4. |
Embudo cilíndrico filtrante con filtro poroso G3 (porosidad 15-40 μm) con un diámetro aproximado de 2 cm y una profundidad de 5 cm, apropiado para filtraciones al vacío, con junta esmerilada macho. |
|
3.5. |
Matraz cónico de 50 ml, con junta esmerilada hembra acoplable al embudo filtrante (punto 3.4). |
|
3.6. |
Tubo de ensayo de 10 ml con fondo cónico y tapón de vidrio de cierre hermético. |
|
3.7. |
Desecador de dicloruro de calcio. |
|
3.8. |
Sistema HPLC, compuesto por:
|
|
3.9. |
Columna de HPLC (25 cm × 4 mm de diámetro interior) con gel de sílice 60 (granulometría de 5 μm). |
|
3.10. |
Filtro de jeringa de 0,45 μm |
|
3.11. |
Matraz Erlenmeyer de 25 ml. |
4. REACTIVOS
|
4.1. |
Hidróxido potásico (pureza mínima del 85 %). |
|
4.2. |
Hidróxido potásico, solución etanólica 2 M, aproximadamente: Disolver, enfriando al mismo tiempo, 130 g de hidróxido potásico (punto 4.1) en 200 ml de agua destilada y completar hasta un litro con etanol (punto 4.9). Conservar la solución en botellas de vidrio oscuro bien cerradas y almacenarla un máximo de dos días. |
|
4.3. |
Éter etílico de calidad para análisis. |
|
4.4. |
Hidróxido potásico, solución etanólica 0,2 M, aproximadamente: Disolver 13 g de hidróxido potásico (4.1) en 20 ml de agua destilada y completar hasta un litro con etanol (4.9). |
|
4.5. |
Placas de vidrio de 20 × 20 cm recubiertas con gel de sílice, sin indicador de fluorescencia, de 0,25 mm de espesor (disponibles en el comercio y listas para su uso). |
|
4.6. |
Acetona, de calidad para cromatografía. |
|
4.7. |
N-hexano, de calidad para cromatografía. |
|
4.8. |
Éter etílico, de calidad para cromatografía. |
|
4.9. |
Etanol, de calidad para análisis. |
|
4.10. |
Acetato de etilo, de calidad para análisis. |
|
4.11. |
Solución de referencia para cromatografía en capa fina: colesterol, fitoesteroles, alcoholes y solución de eritrodiol al 5 % en acetato de etilo (4.10). |
|
4.12. |
Solución de 2,7-diclorofluoresceína al 0,2 % en etanol. Para hacerla ligeramente básica se añaden algunas gotas de solución alcohólica 2 M de hidróxido potásico (4.2). |
|
4.13. |
Mezcla de n-hexano (4.7)/éter etílico (4.8) 65:35 (V/V). |
|
4.14. |
Fase móvil HPLC n-hexano (4.7)/éter etílico (4.8) 1:1 (V/V). |
5. MÉTODO DE REFERENCIA: SEPARACIÓN DE LOS COMPUESTOS ALCOHÓLICOS MEDIANTE PLACA DE CROMATOGRAFÍA EN CAPA FINA BÁSICA
Preparación de placas para cromatografía en capa fina básicas. Sumergir las placas con gel de sílice (4.5) unos 4 cm en la solución etanólica 0,2 M de hidróxido potásico (4.4) durante 10 segundos; dejar secar las placas en una campana durante dos horas y, por último, mantenerlas en un horno a 100 °C durante una hora.
Sacarlas del horno y conservarlas en un desecador de cloruro de calcio (3.7) hasta el momento del uso (las placas sometidas a este tratamiento deben utilizarse en el plazo de quince días como máximo).
Colocar la mezcla de hexano y éter etílico (4.13) (nota 3) en la cubeta de desarrollo, con una profundidad aproximada de 1 cm. Cerrar la cubeta con la tapa adecuada y dejarla así durante al menos media hora, en un lugar frío, de manera que se establezca el equilibrio líquido-vapor. Se pueden colocar tiras de papel de filtro que se sumerjan en el eluyente en las caras interiores de la cubeta. De esta manera, el tiempo de desarrollo se reduce casi un tercio y se obtiene una elución más uniforme y regular de los componentes.
Nota 3: La mezcla de desarrollo debe sustituirse para cada ensayo, a fin de alcanzar unas condiciones de elución perfectamente reproducibles. Alternativamente, se puede utilizar una mezcla de n-hexano/éter etílico 50:50 (V/V).
Preparar una solución del insaponificable preparado en la parte 1 al 5 % aproximadamente en acetato de etilo (4.10) y, utilizando una microjeringa de 100 μl, depositar 0,3 ml de solución sobre una línea fina e uniforme en el extremo inferior (2 cm) de la placa cromatográfica (4.5). Paralela a esta línea, colocar de 2 a 3 μl de la solución de referencia (4.11), de manera que puedan identificarse las bandas de esterol, de dialcoholes triterpénicos y de alcoholes después del desarrollo.
Colocar la placa en la cámara de desarrollo (3.1). Deberá mantenerse a una temperatura ambiente entre 15 y 20 °C (nota 4). Tapar inmediatamente la cubeta y dejar que se produzca la elución hasta que el frente del disolvente se sitúe a 1 cm aproximadamente del borde superior de la placa. Sacar la placa de la cubeta y evaporar el disolvente en una corriente de aire caliente o dejando la placa bajo una campana unos minutos.
Nota 4: Una temperatura más alta podría perjudicar la separación.
Pulverizar sobre la placa de forma ligera y uniforme la solución de 2,7-diclorofluoresceína (4.12) y dejar secar. Al examinar la placa con ayuda de una lámpara ultravioleta (3.2), pueden identificarse las bandas de esteroles, dialcoholes triperpénicos y alcoholes mediante comparación con las manchas obtenidas a partir de la solución de referencia (4.11). Marcar con lápiz negro los límites de las bandas a lo largo de los márgenes de fluorescencia (véase la placa para cromatografía de placa fina del gráfico 1).
Rascar con una espátula metálica el gel de sílice contenido en el área delimitada. Introducir el material obtenido, finamente triturado, en el embudo filtrante (3.4); añadir 10 ml de acetato de etilo caliente (4.10), mezclar cuidadosamente con la espátula metálica y filtrar (en vacío si es necesario), recogiendo el filtrado en el matraz cónico (3.5) acoplado al embudo filtrante.
Lavar el residuo en el matraz tres veces con éter etílico (4.3) (empleando cada vez unos 10 ml), recogiendo el filtrado en el mismo matraz acoplado al embudo. Evaporar el filtrado hasta obtener un volumen de 4 a 5 ml, transvasar la solución residual al tubo de ensayo de 10 ml (3.6) previamente pesado, evaporar hasta secar mediante calentamiento suave en corriente ligera de nitrógeno, recubrir con algunas gotas de acetona (4.6), evaporar de nuevo hasta secar. El residuo contenido en el tubo de ensayo se compone de los esteroles y dialcoholes triperpénicos o los alcoholes y las fracciones de alcoholes triterpénicos y triterpénicos.
6. SEPARACIÓN DE LA FRACCIÓN ALCOHÓLICA POR HPLC
Disolver el insaponificable de la parte 1 en 3 ml de la fase móvil (4.14), filtrar la solución con un filtro de jeringa (3.10) y reservar.
Inyectar 200 μl de la solución filtrada insaponificable en el sistema HPLC (3.8).
Ejecutar la separación por HPLC a 0,8 ml/min, desechar los primeros 5 minutos y recoger en matraces Erlenmeyer de 25 ml (3.11) entre los minutos 5 y 10 para los alcoholes alifáticos y triterpénicos y entre los minutos 11 y 25 para los esteroles y el eritrodiol y el uvaol (nota 5).
La separación puede controlarse con un detector por luz ultravioleta a la longitud de onda de 210 nm o un detector de índice de refracción (véase el gráfico7).
Las fracciones se evaporan hasta sequedad y se preparan para cromatografía.
Nota 5: Controlar cuidadosamente la presión de la bomba de HPLC, el éter etílico puede aumentar la presión; ajustar el flujo para mantener la presión bajo control.
PARTE 3
ANÁLISIS POR CROMATOGRAFÍA DE GASES DE LAS FRACCIONES DE COMPUESTOS ALCOHÓLICOS
1. ÁMBITO DE APLICACIÓN
Esta parte ofrece orientaciones generales para la aplicación de la cromatografía de gases con columna capilar para determinar la composición cualitativa y cuantitativa de los compuestos alcohólicos aislados de acuerdo con el método especificado en la parte 2 de este método.
2. PRINCIPIO
Las fracciones que se recogen del insaponificable mediante cromatografía de capa fina o cromatografía líquida de alta resolución (HPLC) se transforman en éteres de trimetilsililo y se analizan con cromatografía de gases en columna capilar con inyector de fraccionamiento de flujo (split) y detector de ionización de llama.
3. INSTRUMENTAL
El equipo de laboratorio habitual y, en particular, lo siguiente:
|
3.1. |
Tubo de ensayo de 10 ml con fondo cónico y tapón de vidrio de cierre hermético. |
|
3.2. |
Cromatógrafo de gases adecuado para su uso con columna capilar con inyector de fraccionamiento de flujo (split) formado por:
|
|
3.3. |
Columna capilar de sílice fundida, de 20 a 30 m de longitud y de 0,25 a 0,32 mm de diámetro interno, recubierta con difenil (5 %) y dimetilpolisiloxano (95 %) (fase estacionaria SE 52 o SE 54 o equivalente), con un espesor uniforme que oscile entre 0,10 y 0,30 μm. |
|
3.4. |
Microjeringa, de 10 μl de capacidad, para cromatografía de gases, con aguja fija apropiada para división de flujo. |
4. REACTIVOS
|
4.1. |
Piridina anhidra para calidad de cromatografía. |
|
4.2. |
Hexametildisilazano, de calidad para análisis. |
|
4.3. |
Trimetilclorosilano, de calidad para análisis. |
|
4.4. |
Solución patrón de los trimetilsililéteres de esteroles: Preparar en el momento del uso a partir de esteroles y eritrodiol obtenidos de aceites que los contengan. |
|
4.5. |
Solución patrón de trimetilsililéteres de los alcoholes alifáticos de C20 a C28; debe prepararse en el momento de utilización a partir de mezclas de alcoholes puros. |
|
4.6. |
Gas portador: hidrógeno o helio, de calidad para cromatografía de gases. |
|
4.7. |
Gases auxiliares: hidrógeno, helio, nitrógeno y aire, de calidad para cromatografía de gases. |
|
4.8. |
Reactivo de sililación, consiste en una mezcla 9:3:1 (V/V/V) de piridina/hexametildisilazano/trimetilclorosilano. |
|
4.9. |
n-Hexano, de calidad para cromatografía. |
5. PREPARACIÓN DE LOS ÉTERES DE TRIMETILSILILO
Añadir al tubo (3.1.) que contiene la fracción de compuesto alcohólico el reactivo de sililación (4.8) (nota 6) a razón de 50 μl por miligramo de compuesto alcohólico, evitando toda absorción de humedad (nota 7).
|
Nota 6: |
Existen soluciones comerciales listas para usar. Además, también existen otros reactivos de sililación, como la bis-trimetilsililtrifluoroacetamida + trimetilclorosilano al 1 %, que se diluye en el mismo volumen de piridina anhidra. La piridina se puede sustituir por la misma cantidad de acetonitrilo. |
|
Nota 7: |
La eventual formación de una ligera opalescencia es normal y no ocasiona ninguna anomalía. La formación de una floculación blanca o la aparición de una coloración rosa es indicativa de presencia de humedad o de deterioro del reactivo. En este caso deberá repetirse la prueba (solo cuando se utilice hexametildisilazano/trimetilclorosilano). |
Tapar el tubo (3.1.) y agitar cuidadosamente (sin invertir) hasta la completa disolución de los compuestos. Dejarlo en reposo durante al menos 15 minutos a temperatura ambiente y centrifugar durante algunos minutos. La solución límpida está lista para el análisis mediante cromatografía de gases.
6. ANÁLISIS DE CROMATOGRAFÍA DE GASES
6.1. Operaciones preliminares: acondicionamiento de la columna capilar
Colocar la columna (3.3) en el cromatógrafo de gases conectando el extremo de entrada al inyector de fraccionamiento y el extremo de salida al detector.
Efectuar los controles generales del equipo para cromatografía de gases (estanquidad de los circuitos de gases, eficacia del detector, eficacia del sistema de división de flujo y del sistema de registro, etc.).
Si la columna se utiliza por vez primera, es conveniente acondicionarla previamente: hacer pasar un ligero flujo de gas a través de la columna, encender el equipo de cromatografía de gases e iniciar un calentamiento gradual hasta alcanzar una temperatura al menos 20 °C superior a la temperatura de trabajo (nota 8). Mantener dicha temperatura durante 2 horas como mínimo; a continuación, poner el equipo completo en condiciones de funcionamiento [regulación del flujo de gases y del fraccionamiento (split), ignición de la llama, conexión con el sistema informático, regulación de la temperatura de la columna, del detector y del inyector, etc.] y registrar la señal con una sensibilidad al menos dos veces superior a la prevista para el análisis. El trazado de la línea de base debe ser lineal, exento de picos de cualquier tipo y no debe presentar deriva. Una deriva rectilínea negativa indica que las conexiones de la columna no son totalmente estancas; una deriva positiva indica que el acondicionamiento de la columna es insuficiente.
Nota 8: La temperatura de acondicionamiento deberá ser siempre como mínimo 20 °C inferior a la temperatura máxima especificada para la fase estacionaria utilizada.
6.2. Condiciones de funcionamiento
Optimizar el programa de temperatura y el flujo del gas portador de tal manera que se obtengan los cromatogramas similares a los de los gráficos 3 a 6.
Los siguientes parámetros se sometieron a prueba y se consideraron útiles:
6.2.1. Alcoholes alifáticos
|
Programa del horno |
180 °C (8 min) → 260 °C (a 5 °C/min) → 260 °C (15 min) |
|
Temperatura del inyector |
280 °C |
|
Temperatura del detector |
290 °C |
|
Velocidad lineal del gas portador |
Helio (20 a 30 cm/s); hidrógeno (30 a 50 cm/s); |
|
Relación de fraccionamiento |
de 1/50 a 1/100 |
|
Volumen inyectado |
0,5 a 1 μl de solución de TMSE |
6.2.2. Esteroles y dialcoholes triterpénicos
|
Programa del horno |
260 ± 5 °C isotérmico |
|
Temperatura del inyector |
280 – 300 °C |
|
Temperatura del detector |
280 – 300 °C |
|
Velocidad lineal del gas portador |
Helio (20 a 30 cm/s); hidrógeno (30 a 50 cm/s); |
|
Relación de fraccionamiento |
de 1/50 a 1/100 |
|
Volumen inyectado |
0,5 a 1 μl de solución de TMSE |
Estas condiciones pueden modificarse en función de las características de la columna y del cromatógrafo de gases, de modo que se obtengan cromatogramas que cumplan los siguientes requisitos:
— el tiempo de retención del alcohol C26 debe ser de 18 ± 5 minutos.
— el pico del alcohol C22 debe ser 80 ± 20 % de la escala de fondo en el caso del aceite de oliva y 40 ± 20 % de la escala de fondo en el caso del aceite de orujo de oliva.
— el tiempo de retención del pico del ß-sitosterol debe ser de 20 ± 5 min.
— el pico del campesterol debe ser: para el aceite de oliva (contenido medio del 3 %), 20 ± 5 % del fondo de escala.
— se deben separar todos los esteroles presentes; es necesario que los picos no solo se separen sino que se resuelvan completamente, es decir, que el trazo del pico llegue a la línea de base antes de que se inicie el pico siguiente. No obstante, podrá admitirse una resolución incompleta si el pico a TRR 1,02 (sitostanol) puede cuantificarse utilizando la perpendicular.
6.3. Realización del análisis
Con la microjeringa de 10 μl (3.4.) tomar 1 μl de hexano, aspirar 0,5 μl de aire y, a continuación, entre 0,5 y 1 μl de la solución de muestra; elevar el émbolo de la jeringa de modo que la aguja quede vacía. Introducir la aguja a través de la membrana del inyector y, después de 1 o 2 segundos, inyectar rápidamente; transcurridos unos 5 segundos, extraer la aguja lentamente. También puede emplearse un inyector automático.
Realizar la grabación hasta la completa elución de los TMSE de los correspondientes compuestos alcohólicos presentes. La línea de base debe seguir cumpliendo los requisitos de las condiciones de funcionamiento correspondientes (6.2.1 o 6.2.2).
6.4. Identificación de los picos
Identificar los picos individuales sobre la base de los tiempos de retención y en comparación con la mezcla de alcoholes alifáticos y triterpénicos o de los esteroles y dialcoholes triperpénicos TMSE, analizados en las mismas condiciones. En el gráfico 3 se muestra un cromatograma de la fracción de alcoholes alifáticos y triterpénicos, y, en el gráfico 2, los cromatogramas correspondientes de los esteroles y dialcoholes triterpénicos.
La elución de los alcoholes alifáticos se efectúa en el orden siguiente: C20-ol (I.S.), C22-ol, C23-ol, C24-ol, C25-ol, C26-ol, C27-ol y C28-ol.
La elución de los esteroles y dialcoholes triperpénicos se efectúa en el orden siguiente: colesterol, brasicasterol, ergosterol, 24-metilencolesterol, campesterol, campestanol, estigmasterol, Δ-7-campesterol, Δ-5,23-estigmastadienol, clerosterol, ß-sitosterol, sitostanol, Δ-5-avenasterol, Δ-5,24-estigmastadienol, Δ-7-estigmastenol, Δ-7-avenasterol, eritrodiol y uvaol.
6.5. Determinación cuantitativa
Calcular las áreas de los picos del 1-eicosanol y de los alcoholes alifáticos C22, C24, C26 y C28 mediante un sistema apropiado. El factor de respuesta para el 1-eicosanol debe considerarse igual a 1.
Calcular las áreas de los picos del α-colestanol y de los esteroles y dialcoholes triperpénicos utilizando el sistema informático. No se tomarán en cuenta los picos de aquellos componentes que no figuren en el cuadro 1 (el ergosterol no debe calcularse). El factor de respuesta para el α-colestanol debe considerarse como 1.
Calcular del modo siguiente el contenido de cada uno de compuestos alcohólicos, expresado en mg/kg de materia grasa:
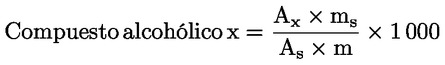
donde:
Ax = área bajo el pico para el compuesto alcohólico x, en unidades de cuenta del sistema informático.
As = área bajo el pico del 1-eicosanol/α-cholestanol, en unidades de cuenta del sistema informático;
ms = peso del 1-eicosanol/α-colestanol añadido, en miligramos.
m = peso de la muestra tomada para la determinación, en gramos.
7. EXPRESIÓN DE LOS RESULTADOS
Indicar las concentraciones de alcoholes alifáticos y triterpénicos em mg/kg de materia grasa y su suma como «contenido de alcoholes alifáticos totales». El contenido total es la suma de C22, C24, C26 y C28.
La composición de cada uno de los compuestos alcohólicos se expresará con una precisión decimal.
La concentración de esteroles totales debe expresarse sin ningún decimal.
El contenido porcentual de cada uno de los esteroles simples se calcula como la razón entre el área bajo el pico correspondiente y la suma de las áreas bajo el pico de los esteroles:

donde:
Ax = área bajo el pico para el esterol x.
ΣΑ = suma de las áreas bajo el pico de los esteroles.
β-sitosterol aparente: Δ5,-23-estigmastadienol+ clerosterol + β-sitosterol + sitostanol + Δ5-avenasterol + Δ5,-24-estigmastadienol.
Cálculo del porcentaje de eritrodiol y uvaol:

donde:
AEr = área del eritrodiol en unidades de cuenta del sistema informático.
AUv = área del uvaol en unidades de cuenta del sistema informático.
ΣAT = suma de las áreas de esterol + eritrodiol + uvaol en unidades de cuenta del sistema informático.
Además del cálculo del porcentaje relativo de esteroles y dialcoholes triterpénicos y de la concentración total de esteroles, debe calcularse la concentración de eritrodiol y uvaol y su suma, en mg/kg de material graso, con arreglo a las siguientes expresiones:


donde:
AEr = área bajo el pico del eritrodiol en unidades de cuenta del sistema informático.
AUv = área del uvaol en unidades de cuenta del sistema informático.
As = área bajo el pico del α-colestanol, en unidades de cuenta del sistema informático.
ms = peso del α-colestanol añadido, en miligramos;
m = peso de la muestra tomada para la determinación, en gramos.
Apéndice
1 Hidrocarburos
2 α-Tocoferol
3 Prenoles
4 alcoholes triterpénicos
5 Alcoholes alifáticos
6 Metil-esteroles
7 Esteroles
8 Dialcoholes triterpénicos
Gráfico 1 — TLC de la fracción insaponificable del aceite de orujo de oliva eluido en dos ocasiones con hexano/éter dietílico (65:35), desarrollada con SO4H2 (50 %) y calentada. Las bandas que deben rasparse son las que figuran dentro del rectángulo, «1» son las bandas para alcoholes alifáticos y «2» las bandas para los esteroles y dialcoholes triterpénicos.
Cuadro I — Plazos de retención relativa de esteroles
|
Pico |
Identificación |
Tiempo de retención relativo |
||
|
Columna SE 54 |
Columna SE 52 |
|||
|
1 |
Colesterol |
Δ-5-Colesten-3ß-ol |
0,67 |
0,63 |
|
2 |
Colestanol |
5α-Colestan-3ß-ol |
0,68 |
0,64 |
|
3 |
Brasicasterol |
[24S]-24-Metil-Δ-5,22-colestadien-3β-ol |
0,73 |
0,71 |
|
* |
Ergosterol |
[24S]-24-Metil-Δ-5,7-22 colestatrien-3β-ol |
0,78 |
0,76 |
|
4 |
24-metilencolesterol |
24-Metilen-Δ-5,24-colestadien-3ß-ol |
0,82 |
0,80 |
|
5 |
Campesterol |
(24R)-24-Metil-Δ-5-colesten-3ß-ol |
0,83 |
0,81 |
|
6 |
Campestanol |
(24R)-24-Metil-colestan-3ß-ol |
0,85 |
0,82 |
|
7 |
Estigmasterol |
[24S]-24-Etil-Δ-5,22-colestadien-3ß-ol |
0,88 |
0,87 |
|
8 |
Δ-7-campesterol |
(24R)-24-Metil-Δ-7-colesten-3ß-ol |
0,93 |
0,92 |
|
9 |
Δ-5,23-Estigmastadienol |
[24R,S]-24-Etil-Δ-5,23-colestadien-3ß-ol |
0,95 |
0,95 |
|
10 |
Clerosterol |
[24S]-24-Etil-Δ-5,25-colestadien-3ß-ol |
0,96 |
0,96 |
|
11 |
ß-sitosterol |
(24R)-24-Etil-Δ-5-colesten-3ß-ol |
1,00 |
1,00 |
|
12 |
Sitostanol |
24-Etil-colestan-3ß-ol |
1,02 |
1,02 |
|
13 |
Δ-5-avenasterol |
(24Z)-24-Etiliden-Δ-colesten-3ß-ol |
1,03 |
1,03 |
|
14 |
Δ-5,24-Estigmastadienol |
(24R,S)-24-Etil-Δ-5,24-colestadien-3ß-ol |
1,08 |
1,08 |
|
15 |
Δ-7-Estigmastenol |
(24R,S)-24-Etil-Δ-7-colesten-3ß-ol |
1,12 |
1,12 |
|
16 |
Δ-7-avenasterol |
(24Z)-24-Etiliden-Δ-7-colesten-3ß-ol |
1,16 |
1,16 |
|
17 |
Eritrodiol |
5α-Olean-12-en-3β.28-diol |
1,41 |
1,41 |
|
18 |
Uvaol |
Δ12-Ursen-3β.28-diol |
1,52 |
1,52 |
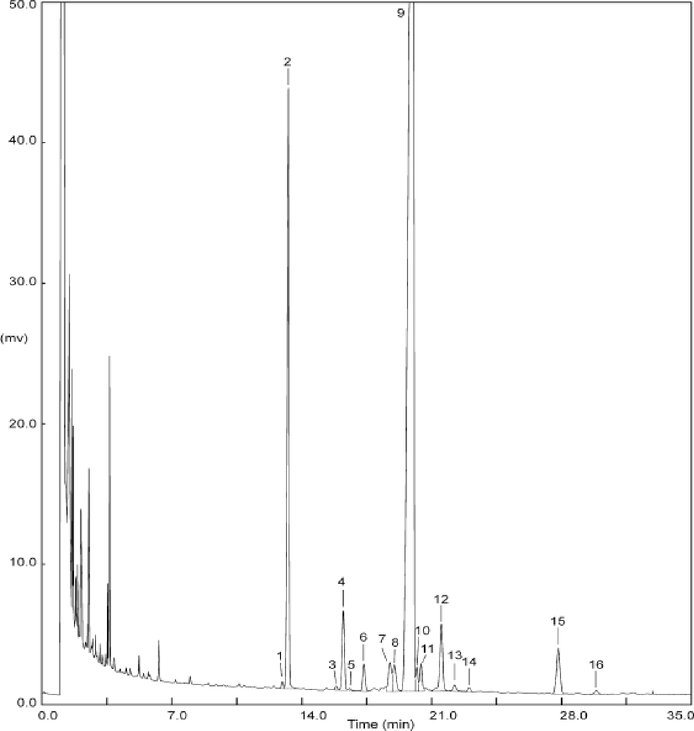
Gráfico 2 — Perfil cromatográfico GC-FID de los esteroles y de los dialcoholes triterpénicos derivados del aceite de oliva refinado. 1) colesterol, 2) α-colestanol (I.S.), 3) 24-metilencolesterol, 4) campesterol, 5) campestanol, 6) estigmasterol, 7) Δ5,23-estigmastadienol, 8) clerosterol, 9) β-sitosterol, 10) sitostanol, 11) Δ5-avenasterol, 12) Δ5,24-estigmastadienol, 13) Δ7-estigmastenol, 14) Δ7-avenastol, 15) eritrodiol, 16) uvaol.

Gráfico 3 — Perfil cromatográfico GC-FID de los esteroles y de los dialcoholes triterpénicos derivados de un aceite de oliva lampante. 1) colesterol, 2) α-colestanol, 3) brasicasterol, 4) 24-metilencolesterol, 5) campesterol, 6) campeestanol, 7) estigmasterol, 8) Δ7-campesterol, 9) Δ5,23-estigmastadienol, 10) clerosterol, 11) β-sitosterol, 12) sitostanol, 13) Δ5-avenasterol, 14) Δ5,24-estigmastadienol, 15) Δ7-estigmastenol, 16) Δ7-avenastol, 17) eritrodiol, 18) uvaol.

Gráfico 4 – Perfil cromatográfico GC-FID de los alcoholes alifáticos y alcoholes triterpénicos procedentes del aceite de oliva. (I.S.) C20-ol, 1) C22-ol, 2) C24-ol, 3) C26-ol, 4) C28-ol, 5) alcoholes triterpénicos.
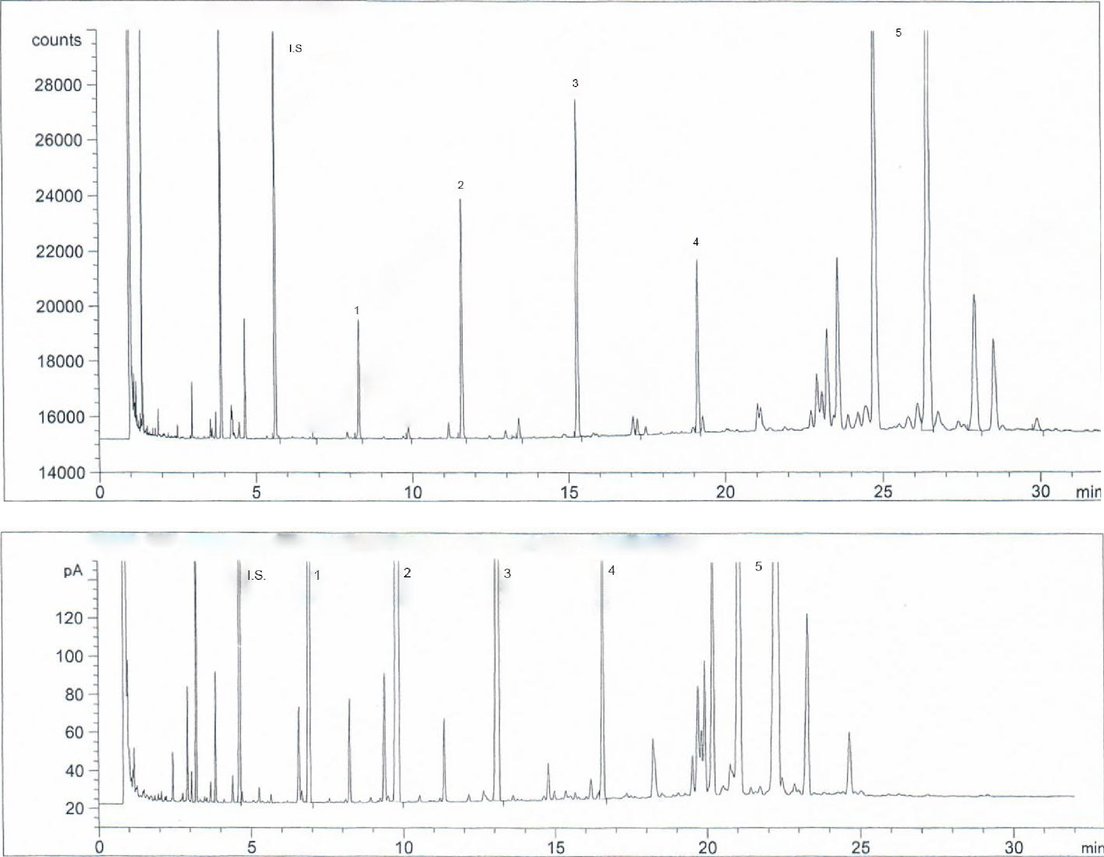
Gráfico 5 – Perfil cromatográfico GC-FID de los alcoholes alifáticos y alcoholes triterpénicos procedentes de un aceite de oliva refinado y un aceite de oliva de segunda centrifugación. (I.S.) C20-ol, 1) C22-ol, 2) C24-ol, 3) C26-ol, 4) C28-ol, 5) alcoholes triterpénicos.
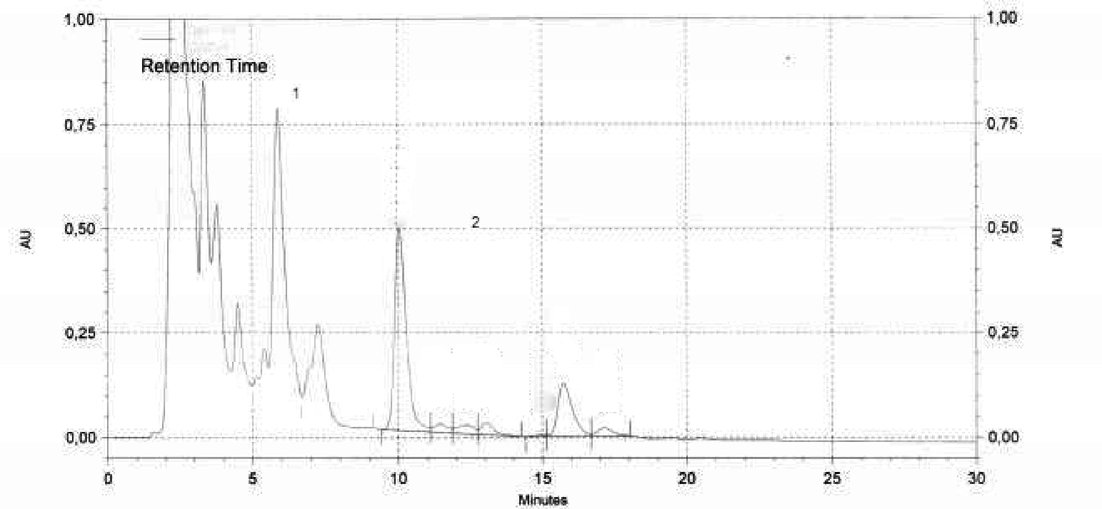
Gráfico 6 — Chromatograma de HPLC de un aceite de oliva insaponificable separado por HPLC con un detector por luz ultravioleta. 1) alcoholes alifáticos y alcoholes triterpénicos. 2) esteroles y dialcoholes triperpénicos.
»
Axencia Estatal Boletín Oficial do Estado
Avda. de Manoteras, 54 - 28050 Madrid




