Estatuko Aldizkari Ofiziala Estatu Agentzia






Edukia ez dago euskaraz
Excelentísimos señores:
La experiencia adquirida desde la publicación de las Ordenes ministeriales de 30 de noviembre de 1976 («Boletín Oficial del Estado» de 4 de enero de 1977), de 31. de enero de 1977 («Boletín Oficial del Estado» de 14 de julio), 31 de julio de 1979 («Boletín Oficial del Estado» de 29 de agosto) y 17 de septiembre de 1981 («Boletín Oficial del Estado» de 14 de octubre), por las que se declaraban como oficiales diversos métodos de análisis, aconsejan la inmediata aprobación de los nuevos métodos que van siendo estudiados y puestos a punto por los diferentes grupos de trabajo, con miras a la agilización en la actuación de la Administración,
En su virtud, a propuesta de los Ministerios de Defensa, de Hacienda, de Administración Territorial, de Trabajo, Sanidad y Seguridad Social, de Industria y Energía, de Economía y Comercio, de Educación y Ciencia, de Obras Públicas y Urbanismo y de Agricultura y Pesca, esta Presidencia del Gobierno dispone:
Se, aprueban como oficiales los métodos de análisis de aceites y grasas, aguas, carne y productos cárnicos, fertilizantes, productos fitosanitarios, leche y productos lácteos, productos orgánicos, fertilizantes, suelos y productos derivados de la uva y similares que se citan, respectivamente, en los anexos I al IX.
Cuando no existan métodos oficiales para determinados análisis y hasta que sean estudiados por el grupo de trabajo correspondiente, podrán ser utilizados los adoptados por Organismos nacionales e internacionales de reconocida solvencia.
Quedan derogadas las disposiciones de igual o inferior rango que se opongan a la presente Orden.
La presente disposición entrará en vigor a los treinta días de su publicación en el «Boletín Oficial del Estado».
Lo digo a VV. EE. para su conocimiento y efectos.
Dios guarde a VV. EE. muchos años.
Madrid, 1 de diciembre de 1981.
RODRIGUEZ INCIARTE
Excmos. Sres. Ministros de Defensa, de Hacienda, de Administración Territorial, de Trabajo, Sanidad y Seguridad Social, de Industria y Energía, de Economía y Comercio, de Educación y Ciencia, de Obras Públicas y Urbanismo y de Agricultura y Pesca.
Anexo I. Aceites y grasas
50 (a). Anilina (por cromatografía de gases).
50 (b). Anilina (por cromatografía de líquidos de alta eficacia).
50 (c). Anilina como acetanilida (por cromatografía de gases).
50 (d). Anilina como acetanilida (por cromatografía de líquidos de alta eficacia).
51. Anilidas grasas (por cromatografía de gases).
52. Anilidas grasas (por cromatografía de líquidos de alta eficacia).
53. Colorantes artificiales.
Anexo II. Aguas
15 (b). Boro.
Toma de muestras de aguas.
Anexo III. Carne y productos cárnicos
1. Preparación de la muestra para el análisis.
3. Almidón,
Anexo IV. Fertilizantes
3. Agua total.
9. Nitrógeno nítrico (método de Robertson).
Anexo V. Productos fitosanitarios
19 (a). Tiram.
Anexo VI. Leche y productos lácteos
20. Residuo insoluble de sangre.
Anexo VII. Productos orgánicos fertilizantes
3 (a). Materia orgánica total (por calcinación).
6. pH.
Anexo VIII. Suelos
12. Necesidades de cal de los suelos ácidos.
Anexo IX. Productos derivados de la uva y similares
Alcoholes.
4. Compuestos sulfurados.
50 (a). Anilina (por cromatografía de gases)
Dilución con hexano, extracción con ácido clorhídrico y cloroformo y posterior determinación por cromatografía de gases.
50 (a).2.1 Cromatógrafo de gases con detector de llama.
50 (a)2.2. Columna de vidrio de carbowax 20M 4 por 100 + KOH 0,8 por 100 sobre carbopack, tres metros de longitud.
Condiciones cromatográficas indicativas:
50 (a).2.3. Temperatura del horno: 180° C.
50 (a).2.4. Temperatura del inyector: 250° C.
50 (a).2.5. Temperatura del detector: 300° C.
50 (a).2.6. Flujo gas portador aproximadamente 25 ml. Na/minuto.
50 (a).2.7. Tiempo de retención aproximadamente cinco minutos.
50 (a).3.1. Hexano.
50 (a).3.2. Acido clorhídrico 2N.
50 (a).3.3. Hidróxido sódico 2N.
50 (a).3.4. Cloroformo.
50 (a).3.5. Sulfato sódico anhidro.
50 (a).3.6. Anilina.
Tomar 25 ml de aceite y añadir 50 ml de hexano, extraer dos veces con 10 ml. cada vez de ácido clorhídrico 2N. Alcalinizar la solución clorhídrica a pH aproximadamente 8 con hidróxido sódico 2N y extraer dos veces con 10 ml. de cloroformo cada vez. Enrasar a 25 ml y desecar con sulfato sódico anhidro.
Inyectar en el cromatógrafo.
Los resultados se expresan en mg/l por comparación con los obtenidos en un aceite al que se ha añadido cantidades conocidas de anilina La concentración de la muestra y parámetros cromatográficos y atenuaciones se combinarán para obtener un límite de detección de 0,5 mg/l.
50 (a).6 1. Para concentraciones < 0,5 mg/l se repetirá el método partiendo de mayores cantidades de muestra.
50 (b). Anilina (por cromatografía de líquidos de alta eficacia)
Dilución con hexano, extracción con ácido clorhídrico y metanol y posterior determinación por cromatografía de líquidos de alta eficacia.
50 (b).2.1. Cromatógrafo de líquidos de alta eficacia.
50 (b).2.2. Columna RP-18.
50 (b).2.3. Solvente acetonitrilo/metanol (70/30).
50 (b).2.4. Longitud de onda: 254 nm.
50 (b).2.5. Flujo 1 ml/min.
50 (b).3.1. Hexano.
50 (b).3.2. Acido clorhídrico 2N.
50 (b).3.3. Hidróxido sódico 2N.
50 (b).3.4. Metanol.
50 (j).3.5. Acetonitrilo.
50 (b).3.6. Sulfato sódico anhidro.
50 (b).3.7. Anilina.
50 (b).3.8. Cloruro de metileno.
Tomar 25 ml de aceite y añadir 25 ml de hexano, extraer dos veces con 10 ml cada vez de ácido clorhídrico 2N. Alcalinizar la solución clorhídrica con hidróxido sódico 2N a pH = 8 y extraer dos veces con 10 ml de cloruro de metileno cada vez en embudo de decantación, haciendo pasar las dos extraociones por un embudo con sulfato sódico anhidro. Se evapora a sequedad y se recoge con 25 ml de metanol. Inyectar seguidamente en el cromatógrafo.
Los resultados se expresan en mg/l por comparación con los obtenidos en un aceite al que se ha añadido cantidades conocidas de anilina. La concentración de la muestra y parámetros cromatográficos y atenuaciones se combinarán para obtener un límite de detección de 0,5 mg/l.
50 (b).6.1. Para concentraciones < 0,5 mg/l se repartirá el método partiendo de mayores cantidades de muestra.
50 (c). Anilina como acetanilida por cromatografía de gases
Dilución con hexano, extracción con ácido clorhídrico, neutralización con hidróxido sódico, extracción de la anilina libre con cloruro de metileno, derivación a acetanilida, concentración y posterior determinación por cromatografía de gases.
50 (c).2.1. Cromatógrafo de gases con detector específico de nitrógeno.
50 (c).2.2. Condiciones cromatográficas:
Flujo de N₂: 60 ml.
Temperatura de la columna: 230° C.
Temperatura de inyección: 300° C.
Temperatura del detector: 300° C.
Columna carbowax 20 M 6 por 100 sobre G.C.Q. 60/80 3 m × ¼''.
50 (c).3.1. Hexano.
50 (c).3.2. Acido clorhídrico 2,5M.
50 (c).3.3. Hidróxido sódico 2,5M.
50 (c).3.4. Cloruro de metileno.
50 (c).3.5. Sulfato sódico anhidro.
50 (c).3.6. Anhídrido acético.
50 (c).3.7. Acetona.
50 (c).3.8. Anilina.
50 (c).3.9. Acetanilida.
Diluir 25 ml de aceite con 25 ml de hexano. Proceder a extraer dos veces con 20 ml. de la disolución de ácido clorhídrico en embudo de decantación. Recogidas las fracciones ácidas se neutralizan con la disolución de hidróxido sódico, añadiendo en exceso de ésta para que quede alcalina. Seguidamente extraer la anilina libre con cloruro de metileno (30 × 20 × 20 ml) en embudo de decantación, haciendo pasar las tres extracciones a través de un embudo con sulfato sódico anhidro. A continuación derivar la anilina a acetanilida con anhídrido acético (0,2 ml), procediendo a evaporar a sequedad en rotavapor (50° C). Recoger la acetanilida con acetona, lavando adecuadamente con más acetona hasta completar 20 ml., pasándolos, seguidamente a una probeta, y concentrar a un volumen de 5 ml la muestra así preparada se inyecta en el cromatógrafo de gases con detector específico de nitrógeno.
Los resultados se expresan en mg/l por comparación con los obtenidos en un aceite al que se ha añadido cantidades conocidas de anilina. La concentración de la muestra y parámetros cromatográficos y atenuaciones se combinarán para obtener un límite de detección de 0,5 mg/l.
50 (c).6.1. Para concentraciones < 0,5 mg/l se repetirá el método partiendo de mayores cantidades de muestra.
50 (c).6.2. En las muestras que una vez inyectadas se obtengan picos con tiempo de retención que no coincidan exactamente con el de la acetanilida, hay que repetir el procedimiento, pero sin derivación, y si siguen apareciendo los mismos picos no se debe considerar que originalmente exista anilina en la muestra.
50 (c).6.3. El «disolvente blanco» (acetona 5 ml. + 0,2 ml. de anhídrido acético) debe inyectarse después de cualquier muestra que dé positiva la acetanilida (anilina derivada) y comprobar que no vuelve a salir el pico correspondiente a ésta.
50 (d) Anilina como acetanilida (por cromatografía de líquidos de alta eficacia)
Dilución con hexano, extracción con ácido clorhídrico, neutralización con hidróxido sódico, extracción de la anilina libre con cloruro de metileno, derivación a acetanilida, exaporación y posterior determinación por cromatografía de líquidos de alta eficacia.
50 (d).2.1. Cromatógrafo de líquidos.
50 (d).2.2. Columna RP-8.
50 (d).2.3. Solvente (metanol/agua ácido acético (60/39/1).
50 (d).2.4. Flujo un ml/minuto.
50 (d).3.1. Hexano.
50 (d).3.2. Acidó clorhídrico 2,5M.
50 (d).3.3. Hidróxido sódico al 2,5M.
50 (d).3.4. Cloruro de metileno.
50 (d).3.5. Sulfato sódico anhidro.
50 (d).3.6. Anhídrido acético.
50 (d).3.7. Metanol.
50 (d).3.8. Anilina.
50 (d).3.9. Acetanilida.
Diluir 25 ml. de aceite con 25 ml. de hexano. Extraer dos veces con 20 ml. de ácido clorhídrico 2,5M en embudo de decantación. Recogidas las fracciones acidas se neutralizan con hidróxido sódico 2,5M, añadiendo en exceso para que quede alcalina. Seguidamente extraer la anilina libre con cloruro de metileno (30 × 20 × 20 ml.) en embudo de decantación, haciendo pasar las tres extracciones a través de un embudo con sulfato sódico anhidro. A continuación derivar la anilina a acetanilida con anhídrido, acético (0,2 ml.), procediendo a evaporar a sequedad en rotavapor (50° C). Recoger el residuo con 10 ml. de metanol e inyectar 10 µl. en el cromatógrafo.
Los resultados se expresan en mg/l. por comparación con los obtenidos en un aceite al que se ha añadido cantidades conocidas de anilina. La concentración de la muestra y parámetros cromatográficos y atenuaciones se combinarán para obtener un límite de detección de 0,5 mg/l.
50 (d).6.1. Para concentraciones < 0,5 mg/l. se repetirá el método partiendo de mayores cantidades de muestra.
51. Anilidas grasas (por cromatografía de gases)
Extracción con metanol, transesterificación con hidróxido potásico en metanol y hexano y posterior determinación cromatográfica por cromatografía de gases.
51.2.1. Cromatógrafo de gases con detector de ionización de llama.
51.2.2. Columna cromatográfica 2 m. × ⅛'' en acero inoxidable de SE-30 al 5 por 100 sobre Chromosorb AW-DMCS 80-100 mesh.
51.2.3. Condiciones cromatográficas indicativas:
Temperatura del homo 280° C.
Flujo gas portador (nitrógeno), 20 ml. por minuto.
51.3.1. Metanol.
51.3.2. Hidróxido potásico 2M en metanol.
51.3.3. Hexano.
Extraer 750 ml. de aceite con dos porciones de 75 ml. de metanol. Se reúnen ambos extractos, concentrándose a sequedad. La totalidad del residuo se transesterifica con 0,5 ml. de hidróxido potásico 2M en metanol y 5 ml. de hexano, agitando a temperatura ambiente durante treinta segundos. Esperar veinte minutos antes de inyectar 2 µl. en el cromatógrafo.
Tiempo de retención aproximado de la oleilanilida... ocho minutos.
Los, resultados se expresarán en mg/l. por comparación con los obtenidos con un aceite al que se ha añadido cantidades conocidas de oleilanilida. La concentración de la muestra, parámetros cromatográficos o atenuaciones se combinarán para obtener un límite de detección de 10 mg/l.
51.6.1. Preparación del patrón de oleilanilida.
Calentar a ebullición durante tres horas en matraz con refrigerante unos 50 g. de anilina e igual cantidad de ácido oleico. Enfriar y disolver en unos 100 ml. de éter etílico. Lavar tres veces con 50 ml. de hidróxido sódico 2M para eliminación del ácido oleico libre y, a continuación, con otras tres porciones de 80 ml. de ácido clorhídrico 2M para quitar la anilina libre. El exceso de ácido clorhídrico se elimina lavando con agua hasta pH neutro. Secar la fase etérea con SO₄Na₂ anhidro y evaporar a sequedad.
52. Anilidas grasas (por cromatografía de líquidos de alta eficacia)
Extracción con metanol, concentración del extracto, disolución del residuo y posterior determinación por cromatografía de líquidos de alta eficacia.
52.2.1. Cromatógrafo de líquidos.
52.2.2. Columna RP-8.
52.2.3. Condiciones cromatográficas:
Columna: RP-8.
Longitud de onda: 254 nm.
Solvente: Metanol.
Flujo: 1 ml/minuto.
52.3.1. Metanol.
52.3.2. Agua destilada.
52.3.3. Acido acético.
Extraer 100 ml. de aceite en dos porciones de 50 ml. de metanol. Reunir ambos extractos concentrándose a sequedad. Disolver la totalidad del residuo en 10 ml de metanol e inyectar 10 µl. en el cromatógrafo.
Como en 51.5.
53. Colorantes artificiales
Dilución con ciclohexano, extracción por columna de alúmina, lavado con alcohol etílico, concentración y posterior determinación por cromatografía en capa fina.
53.2.1. Espectrodensitómetro.
53.2.2. Columnas de vidrio para cromatografía de 20 cm. de longitud y 2 cm. de diámetro interno.
53.2.2. Placas de gel de sílice comerciales de 0,25 mm. de espesor.
53.2.4. Cubetas de desarrollo para cromatografía.
53.3.1. Ciclohexano.
53.3.2. Eter de petróleo.
53.3.3. Alcohol etílico.
53.3.4. Benceno
53.3.5. Tetracloruro de carbono.
53.3.6. Alúmina neutra. Actividad 1.
Tomar 20 ml. de aceite y diluir con 20 ml. de ciclohexano. Hacer pasar la mezcla lentamente por columna cromatográfica de alúmina previamente embebida con ciclohexano. Una vez que ha pasado todo el aceite por la columna, lavar varias veces con éter de petróleo hasta que éste salga incoloro. A continuación pasar por la columna 25 ml. de alcohol etílico que se recogen en un matraz. Concentrar hasta casi sequedad y colocar en placa de silicagel manchas de 3 μl. frente a patrones. Introducir la placa en una cubeta de cromatografía previamente saturada con eluyente (benceno-tetracloruro de carbono) 50/50.
Los colorantes desarrollados en la placa se identifican inicialmente por su Rf., confirmándose por espectrodensitometría (en caso de no disponer de este aparato, será necesario raspar la placa y realizar un barrido en espectro U.V.).
15 (b). BORO
(Azometina)
Determinación de la cantidad de boro, mediante espectro fotometría, previa reacción con azometina.
15 (b).2.1. Matraces aforados de 100 ml. de capacidad.
15 (b).2.2. Espectrofotómetro capaz de leer a 410 nm.
15 (b).3.1. Acido clorhídrico concentrado.
15 (b).3.2. Solución de 1.000 mg/l. de boro: Pesar 5,7160 g. de BO₃H₃ y llevar a 1.000 ml. con agua.
15 (b).3.3. Solución de 10 mg/l. de boro: Preparada por apropiada dilución de la anterior y conservada en frasco de polietileno.
15 (b).3.4. Acido ascórbico.
15 (b).3.5. Solución tampón-enmascarante: Disolver 250 g. de acetato amónico en 500 ml. de agua desionizada, añadir 125 ml. de ácido acético de 99 por 100. Disolver en esta disolución 6,7 g. de sal disódica del ácido etilen-diamino-tetracético y 6 ml. de ácido tioglicólico al 80 por 100.
15 (b).3.6. Solución de azometina: Introducir 0,9 g. de azometina-H en un matraz aforado de 100 ml. Agregar unos 75 ml. de agua, agitar y si es necesario introducir el matraz durante unos instantes en un baño de agua templada hasta su completa disolución, A continuación agregar 2 g. de ácido ascórbico y completar a volumen con agua desionizada. Esta solución puede guardarse en refrigerador durante catorce días.
15 (b).4.1. Preparación de la curva de calibrado.
Introducir 0, 4, 8, 12 y 16 ml. de la solución de 10 mg/l. de boro en cinco matraces aforados de 100 ml. y enrasar con agua desionizada. Las concentraciones serán, por tanto, de 0, 0,4, 0,8, 1,2 y 1,6 mg/l. de boro, respectivamente. Introducir en tubos de ensayos 5 ml. de cada una de estas soluciones, añadir 4 ml. de solución tampón-enmascarante y 2 ml. dé solución de azometina. Tapar los tubos, invertirlos 3-4 veces para mezclar y homogeneizar perfectamente. Medir la absorbencia a 410 nm., en cubeta, de 1 cm., entre las una y dos horas después de agregado el reactivo, frente al patrón que no contiene solución de boro.
Con los datos obtenidos construir la curva patrón.
15 (b).4.2. Determinación: Tomar 5 ml. de la muestra y operar exactamente igual que en 15 (b).4.1.
Calcular el contenido en boro expresado en mg/l. mediante comparación con la curva de calibrado, teniendo en cuenta, si es necesario, la dilución de la muestra.
15 (b).6.1. El método sigue la ley de Lambert-Beer hasta 3 mg. de boro por litro. La cantidad mínima detectable de boro es de 0,2 mg/litro.
15 (b).6.2. Para sintetizar la azometina-H proceder como se indica a continuación: En un vaso de 1.000 nm. introducir 10 g. de la sal monosódica del ácido 4 amino-5hidroxi-2,7-naftalendisulfónico, agregar 500 ml. de agua y agitar con varilla. Introducir en la suspensión obtenida los eléctrodos de un pH-metro y agregar NaOH al 10 por 100, poco a poco y agitando hasta obtener un pH = 7; seguidamente, sin dejar de agitar, añadir ácido clorhídrico concentrado hasta pH = 1,5. Agregar 10 ml. de salicilaldehído e introducir el vaso en un baño a 45° C, agitando enérgicamente con ayuda de un agitador mecánico dé varilla.
Al cabo de una hora dar por finalizada la operación, dejando en reposo durante un mínimo de doce horas. Se obtiene un precipitado amarillo, el cual se filtra y se lava con alcohol etílico. El residuo se seca a 100-105° C durante tres horas. El polvo obtenido de color amarillo-naranja debe conservarse en desecador.
1. «Estudio sobre la determinación de boro en plantas con azometina-H». M. Lachica. Estación Experimental del Zaidín (Granada).
TOMA DE MUESTRAS DE AGUAS
Obtener de un agua una muestra representativa de la misma para poder determinar a partir de ella sus características físicas y químicas.
Este método de toma de nuestras se aplicará a todos los tipos de muestreo de aguas, cualquiera que sea su procedencia, ya sean de manantiales, pozos, ríos, lagos, redes de distribución de aguas, depósitos, etc.
Muestras simples: Son aquellas tomadas en un tiempo y lugar determinado para su análisis individual.
Muestras compuestas: Son las obtenidas por mezcla y homogeneización de muestra simples recogidas en el mismo punto y en diferentes tiempos.
Muestras integradas: Son las obtenidas por mezclas de muestras simples recogidas en puntos diferentes y simultáneamente.
Ejemplar de la muestra para el laboratorio: Cada una de las partes obtenidas por reducción de la muestra.
Exceptuando el material específico que pueda utilizarse para determinaciones especiales, los recipientes en que se recojan las muestras deberán ser de vidrio neutro o material plástico y tendrán que cumplir los siguientes requisitos:
a) No desprender materia orgánica, elementos alcalinos, boro, sílice u otros que puedan contaminar la muestra recogida.
b) Que la adsorción ejercida por sus paredes sea mínima sobre cualquiera de los componentes presentes en la muestra de agua.
c) Que el material constituyente del recipiente no reaccione con los componentes de la muestra.
d) Deberán poderse cerrar y sellar herméticamente.
Los envases de plástico se utilizarán para tomar las muestras en las que se deban determinar elementos alcalinos y/o radiactividad.
Los envases de vidrio borosilicatado se utilizarán cuando se analicen gases y deberán ser de color topacio cuando se investiguen elementos alterables por la luz.
Todo el material que se use para la toma de muestras deberá estar escrupulosamente limpio, debiendo enjuagarse con agua destilada o desmineralizada.
Los equipos o aparatos que se utilicen serán función de las condiciones físicas del lugar de muestreo, así como de los parámetros a determinar, y se hallarán comprendidos en los siguientes:
4.1. Directamente mediante la botella o recipiente que se va a enviar a laboratorio o que se utilice para las determinaciones «in situ».
4.2. Equipo tomamuestras representado en la figura número 1 o similar.
4.3. Equipo tomamuestras representado en la figura número 2 o similar.
4.4. Equipos automáticos. Para casos especiales no pueden establecerse normas fijas debido a la amplia gama de posibilidades de trabajo: Toma de porciones en función del tiempo, del caudal circulante, de las características de la muestra, con almacenamiento en un único o múltiples recipientes, con refrigeración, con adición de reactivos, etc.
El objetivo fundamental es conseguir que la porción de agua tomada sea representativa y dado que la toma de muestras, de hecho, presenta una enorme variedad de situaciones diferentes, en todos aquellos casos en que sea posible se fijarán para cada uno de ellos las condiciones más apropiadas, en entrevista mantenida entre el personal de laboratorio y el responsable de tomar la muestra.
En fuentes, redes de distribución, pozos dotados de bomba de extracción y casos similares será necesario dejar fluir el agua, durante el periodo de tiempo que se estime conveniente para conseguir que la muestra sea verdaderamente representativa.
En ríos, embalses, etc., será preciso considerar diversos factores, tales como profundidad, flujo de corriente, distancia a la orilla, etc., recomendándose en estos casos la obtención de muestras integradas y de no ser posible se tomará una muestra simple en el centro de la corriente o varias muestras simples en los lugares más apropiados de la masa de agua.
Siempre que sea posible, el recipiente se enjuagará con el agua objeto del muestreo.
El equipo especificado en 4.1 se utilizará para toma de muestras en grifos de redes de distribución, canales de riego, fuentes, arroyos de poca profundidad, pozos dotados de bomba de extracción y casos similares.
En ríos, embalses, pozos sin bomba, grandes depósitos de almacenamiento y casos similares se utilizará el equipo especificado en 4.2, siempre que la profundidad no exceda de 30 metros.
Si la profundidad es mayor de 30 metros se utilizará el equipo descrito en 4.3, mediante un torno.
En caso necesario la muestra se dividirá en el número de partes precisas, constituyendo cada una de ellas un ejemplar de la muestra para el laboratorio.
El volumen de la muestra estará en función del número de ensayos y determinaciones que se pretendan realizar.
No es posible alcanzar una completa y perfecta conservación, pues nunca se consigue una total estabilización de cada constituyente; como máximo las técnicas de conservación retrasan los procesos químicos o biológicos, los cuales después de tomada la muestra continúan.
En cuanto al tiempo entre la recogida y su análisis puede decirse, como norma general, que cuanto menor sea este intervalo mejores serán los resultados del análisis.
Siempre que sea posible, se utilizará la tabla siguiente, en la que se especifica: Determinación a realizar, tipo de envase a utilizar (plástico o vidrio); volumen mínimo de muestra; procedimiento de conservación y tiempo máximo que debe transcurrir desde la toma de muestra hasta el momento de comenzar el análisis.
| Determinación | Tipo de envase |
Volumen mínimo — ml. |
Conservación | Tiempo máximo |
|---|---|---|---|---|
| Acidez. | P o V | 100 | Refriger. 4° C. | 24 horas. |
| Alcalinidad. | P o V | 100 | Refriger. 4° C. | 24 horas. |
| Amonio. | P o V | 500 | Refriger. 4° C o SO₄H₂; pH 2. | 24 horas. |
| Anhídrido carbónico. | V | 100 | — | Inmediato. |
| Boro. | P | 50 | — | — |
| Carbono orgánico total. | P o V | 50 | Refriger. 4° C o SO₄H₂; pH 2. | 24 horas. |
| Cianuros. | P o V | 500 | Refriger. 4° C o NaOH; pH 12. | 24 horas. |
| Cloro en sus diferentes. | P o V | 500 | — | Inmediato. |
| Clorofilas. | P o V | 500 | Congelación y oscuridad. | 30 días. |
| Cloruros. | P o V | 100 | — | 7 días. |
| Color. | V | 100 | Refriger. 4° C. | 24 horas. |
| Conductividad. | P o V | 100 | Refriger. 4° C. | 24 horas. |
| Demanda bioquímica de oxígeno (DBO). | P o V | 1.000 | Refriger. 4° C. | 6 horas. |
| Demanda química de oxígeno. | P o V | 100 | SO₄H₂; pH < 2. | Lo antes posible. |
| Detergentes. | V | 2.000 | 20 mg/l. de Cl₂Hg. | 1 día. |
| Fenoles. | V | 500 | PO₄H₃; PH < 4 1 gr/l SO₄Cu. | 24 horas. |
| Fluoruros. | P | 200 | — | 7 días. |
| Fosfatos: | ||||
| Ortofosfatos disueltos. | V | 100 | Filtrar «in situ» y refriger. 4° C. | 24 horas. |
| Fósforo hidrolizable. | V | 100 | SO₄H₂; pH < 2. | 24 horas. |
| Fósforo total. | V | 100 | Refriger. 4° C o SO₄H₂; pH < 2. | 7 días. |
| Grasas y aceites. | V | 2.000 | SO₄H₂; pH < 2 y refriger. 4° C. | 24 horas. |
| Ioduros. | P o V | 100 | Refriger. 4° C. | 24 horas. |
| Metales disueltos. | P o V | 200 | Filtrar «in situ» NO₃H; pH < 2. | 6 meses. |
| Metales totales. | P o V | 200 | NO3H; pH < 2. | 6 meses. |
| Nitratos. | P o V | 100 | Refriger. 4° C o SO₄H₂; pH < 2. | 24 horas. |
| Nitritos. | P o V | 100 | Refriger. 4° C o SO₄H₂; pH < 2. | 24 horas. |
| Olor. | V | 500 | Refriger. 4° C. | Lo antes posible. |
| Oxígeno disuelto. | V | 250 | — | Inmediato. |
| Ozono. | V | 250 | — | Inmediato. |
| pH. | P o V (B) | 50 | Refriger. 4° C. | 6 horas. |
| Residuos. | P o V | 500 | Refriger. 4° C. | 7 días. |
| Sabor. | V | 500 | — | Inmediato. |
| Sílice. | P | 100 | Refriger. 4° C. | 7 días. |
| Sulfatos. | P o V | 500 | Refriger. 4° C. | 7 días. |
| Sulfitos. | P o V | 500 | — | Inmediato. |
| Sulfuros. | P o V | 500 | 2 ml. acetato de cinc, 2N. | 24 horas. |
| Temperatura. | — | — | — | Inmediato. |
| Turbiedad. | P o V | 100 | Refriger. 4° C. | Lo antes posible. |
|
P = Plástico; V= Vidrio; V (B) = Vidrio borosilicatado. |
||||
Obtenidas las muestras se cerrarán convenientemente y se precintarán, en su caso, de forma que quede garantizada su inviolabilidad, etiquetándolas para su perfecta identificación.
1. «Standar Methods for the examination of water and wastewater», 14.ª Ed. APHA, AWWA WPCF.
2. «Methods for Chemical analysis of water and wastes», EPA, 1972.
3. «Analyse des eaux. Methodes et instructions», AFNOR.
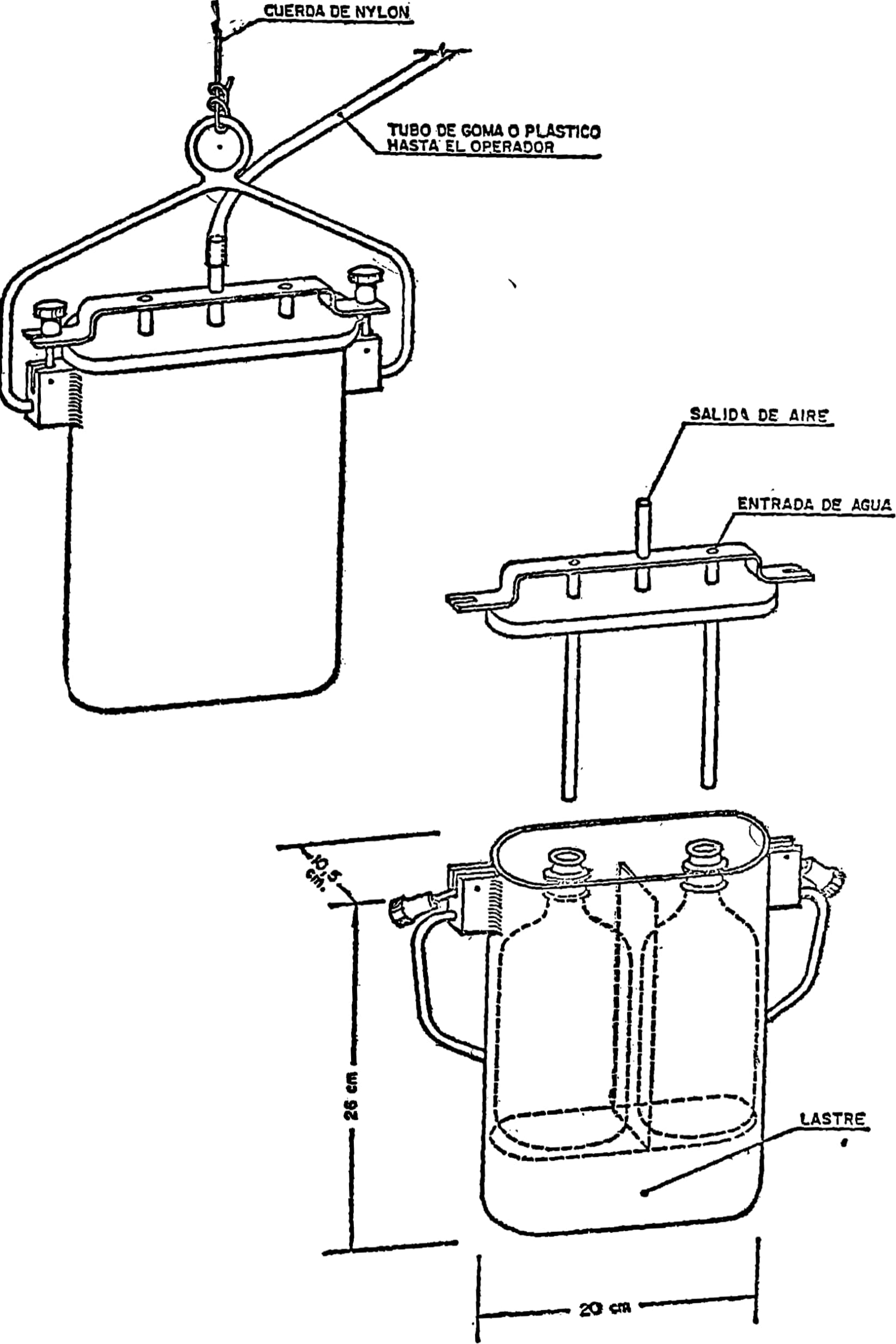
Figura número 1
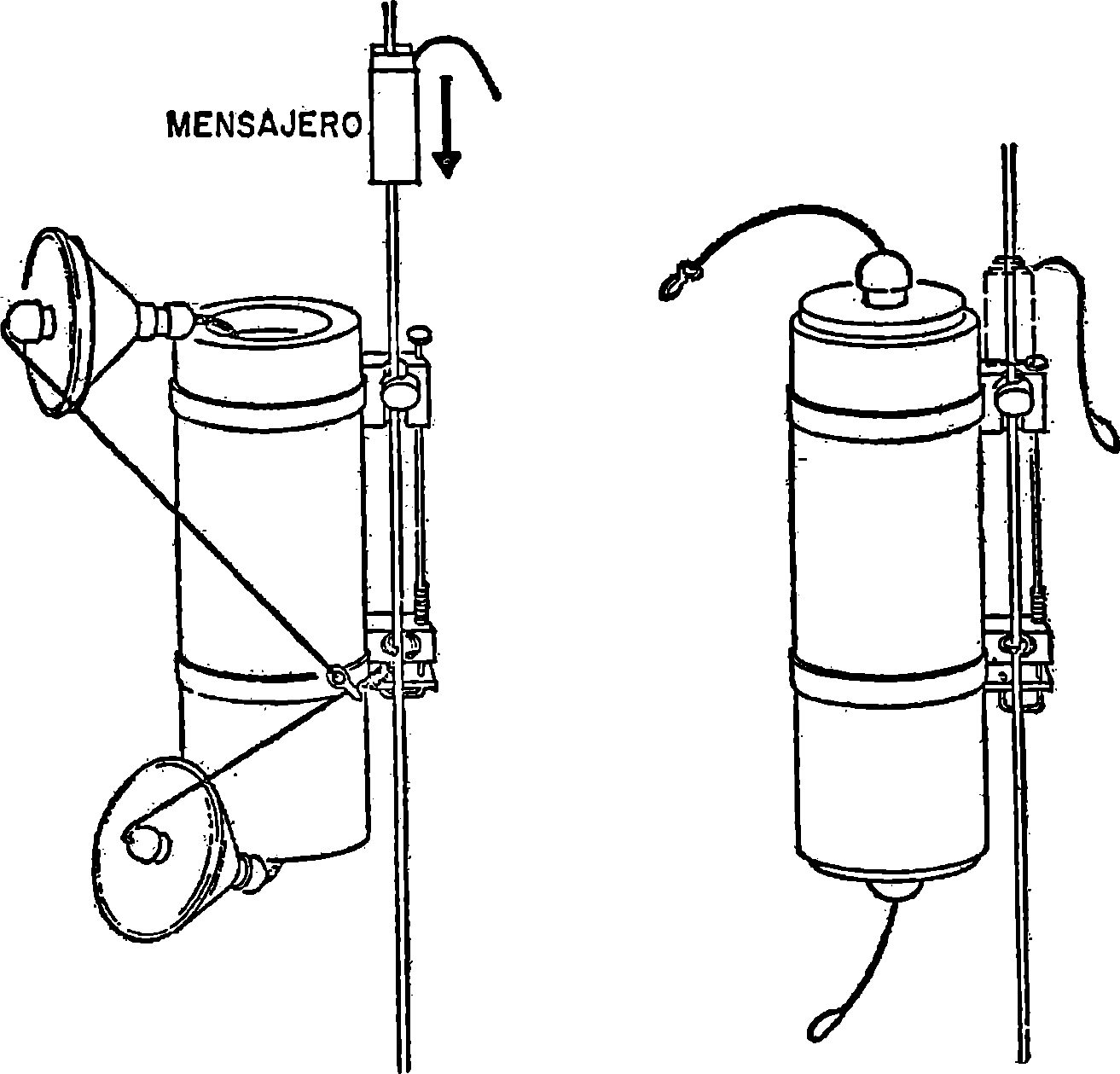
Figura número 2
1. Preparación de la muestra para el análisis
Las operaciones descritas a continuación tienen por finalidad conseguir una muestra para el análisis lo más homogénea posible: Por ello, toda simplificación o tratamiento insuficiente en esta operación puede conducir a unos resultados que no sean representativos.
1.2.1. Cuchillo.
1.2.2. Trituradoras eléctricas de distinto grado de finura en el picado.
1.2.3. Frascos de vidrio de 250 ml. de capacidad, de color topacio, boca ancha y tapón esmerilado.
1.2.4. Cápsulas de porcelana de 20 cm. de diámetro.
1.3.1. Quitar las diferentes capas protectoras del producto si las tuviere (piel, gelatina, grasa, etc.).
1.3.2. Tomar una muestra representativa de 200 gramos.
1.3.3. Partirla con cuchillo en rodajas o trozos de 0,5-1 centímetros.
1.3.4. Cortar los trozos en pequeños cubos.
1.3.5. Pasarlos varias veces por trituradora hasta conseguir una mezcla homogénea.
1.3.6. La muestra, bien homogeneizada debe guardarse inmediatamente en los frascos, limpios y secos, de forma que queden llenos, para prevenir pérdidas de humedad.
1.3.7. Conservarlos en refrigeración de forma que evite su deterioro y cualquier cambio en su composición.
1.3.8. Tomar las muestras para las diferentes determinaciones, a ser posible dentro de las veinticuatro horas siguientes.
3. Almidón (método cuantitativo)
Extracción de azúcares simples con etanol caliente 80 por 100, permaneciendo el almidón. El residuo de almidón se solubiliza con ácido perclórico diluido, y medida a 630 nm. del color desarrollado al calentarlo con el reactivo antronasulfúrico.
3.2.1. Balanza analítica.
3.2.2. Matraces aforados de 100 mi, y 200 ml.
3.2.3. Tubos de centrífuga, cónicos, de 100 ml.
3.2.4. Pipetas graduadas de 10 ml., 25 ml. 5 ml. y 2 ml.
3.2.5. Centrífuga de 2.000 r.p.m.
3.2.6. Baño de agua.
3.2.7. Baño de agua termostatable hasta 25° C.
3.2.8. Probeta graduada de 25 ml.
3.2.9. Papel de filtro Albet número 238 o equivalente.
3.2.10. Espectrofotómetro capaz de lecturas de 630 nm.
3.3.1. Disolución de ácido sulfúrico-antrona. Disolver 0,2 g. de antrona en 100 ml. de H₂SO₄. El reactivo sirve para 3-4 días conservándolo a 0° C.
3.3.2. Glucosa patrón. Disolver 0,1 g. de glucosa anhídrida en 100 de agua.
3.3.3. Acido perclórico al 52 por 100.
3.3.4. Etanol al 80 por 100.
3.4.1. Extracción de azúcar y de grasa. Pasar 2,0 g. de carne triturada, preparada como en el método 1, en un tubo centrífugo cónico de 100 ml. Añadir 25,0 ml. de disolución etanol-éte de petróleo (1-3), tapar con tapón, agitar vigorosamente y centrifugar a 2.500 r.p.m, durante cinco minutos. Decantar y dejar a un lado la disolución alcohol-éter. Añadir 10 ml. de etanol caliente al 80 por 100, agitar y centrifugar a 2.500 r.p.m, durante cinco minutos. Dejar a un lado la disolución alcohólica y repetir la extracción alcohólica con etanol caliente.
3.4.2. Extracción de almidón. Añadir 5,0 ml. de agua al residuo y remover. Añadir 6,5 ml. de disolución de ácido perclórico diluido (52 por 100); remover o agitar durante cinco minutos. Dejar reposar durante quince minutos. Añadir 20,0 ml. de agua y centrifugar durante cinco minutos. Verter la disolución, de almidón en un frasco volumétrico de 100 ml. Añadir 6,5 ml. de disolución de ácido perclórico (52 por 100) y remover. Dejar reposar durante treinta minutos. Remover y lavar el contenido entero del tubo en el frasco volumétrico que contiene el primer extracto. Llevar a volumen y filtrar por papel filtro (3.2.9).
3.4.3. Determinación de almidón. Diluir 5 ml. de disolución de almidón filtrada en 200 ml. con agua destilada. Pipetear 5 ml. de dicha disolución en un tubo, enfriar en baño de agua y añadir 10 ml. de antrona reactivo (3.3.1). Mezclar completamente y calentar durante cinco minutos a 100° C. Quitar el tubo del baño, enfriar con rapidez a 25° C y determinar la absorbancia a 630 nm. El color permanece fijo durante treinta minutos.
3.5.1. Curva patrón de glucosa. Diluir 1, 2, 5 y 10 ml. de glucosa patrón (3.3.2) hasta 100 ml. con agua destilada. A partir de las lecturas obtenidas dibujar la curva patrón.
3.5.2. Contenido en almidón de la muestra:
Glucosa (porcentaje) = 0,04 P.
Almidón (porcentaje) = 1,06 M.
siendo:
P = μg. de, glucosa leídos en la curva patrón.
M = porcentaje total de glucosa obtenido,
3.6.1. Teniendo en cuenta que el carregenato se determina como almidón, se deberá deducir del valor obtenido de almidón el correspondiente de carragenato.
3.6.2. En los productos que declaren contener carragenato y hasta que no exista técnica oficial para su determinación cuantitativa se permitirá una tolerancia del 1,4 por 100 en el valor obtenido de almidón.
3.6.3. Cuando la naturaleza de la muestra lo requiera se podrán repetir las extracciones, hasta que el producto quede completamente libre de grasas y azúcares.
1. W. Glover, H. Kirschenbaum y A. Caldwell (Departamento de Consumo y Comercialización, Ministerio de Agricultura de los Estados Unidos. Laboratorio de Inspección de Carne, Nueva York, N. Y. 10011).
3. Agua total
El agua que contiene la muestra se determina por desecación en estufa.
El método no es aplicable a muestras que producen sustancias volátiles diferentes del agua a la temperatura de desecación.
3.2.1. Pesasustancias capaz para 2 gramos.
3.2.2. Estufa con regulación de temperatura a 100-130° C.
3.2.3. Desecador.
Calentar 2 g. de muestra debidamente preparada para análisis durante cinco horas en una estufa a 100 ± 1° C. En el caso de NO₃Na, SO₄(NH₄)₂ y sales de K, calentar hasta peso constante a 130° ± 1° C. Expresar el porcentaje de pérdida en peso a la temperatura utilizada como contenido de agua.
1. «Association of Official Analytical Chemist. Official Methods of Analysis». Ed. 1980, 2.012
9. Nitrógeno nítrico (método de Robertson)
Determinar el N total y el N insoluble en agua. La diferencia entre ambos es el N soluble.
En la disolución de N soluble, eliminar el N nítrico al estado de óxido nítrico por medio de sulfato ferroso. Una vez eliminado, determinar el N total en el residuo y la diferencia entre el N soluble y este último es el N nítrico.
Aplicable en presencia de cianamida cálcica y urea.
Como en 6 (a).2.
9.3.1. SO₄Fe 7 H₂O.
9.3.2. Como en 6 (a).3.
9.4.1. Modalidad A: Caso general en que es necesario determinar el N insoluble en agua.
Determinar el N total por los métodos 6 (b) o 6 (c).
9.4.2. Separar y determinar el N insoluble en agua por el método correspondiente.
9.4.3. En la disolución obtenida en 9.4.2 eliminar el N nítrico y determinar el N restante.
Para ello, colocar el filtrado procedente del apartado anterior en un matraz Kjeldahl de 500 ml. y añadir 2 g. de SO₄Fe. 7H₂O y 20 ml. de SO₄H₂ (si el N total es mayor del 5 por 100, poner 5 g. de SO₄Fe 7 H₂O).
9.4.4. Ponerlo a la llama hasta evaporar el agua y aparezcan humos blancos. Continuar la digestión por lo menos diez minutos más para expulsar todo el N nítrico. Si se produce una fuerte vaporización, añadir 10 ó 15 perlas de vidrio.
9.4.5. Agregár 0,65 g. de Hg o 0,7 de HgO y continuar la digestión hasta que toda la materia orgánica se haya oxidado.
Enfriar, diluir y continuar como en 6 (a).4.2 y siguientes.
9.4.6. Modalidad B: Modificación de Jones para el caso en que no hace falta determinar el N insoluble en agua por ser todo él soluble.
Determinar el N total por los métodos 6 (b) o 6 (c).
9.4.7. Eliminar el N nítrico y determinar el.N restante.
Para ello, pesar 0,5 g. del problema, colocarlos en un matraz Kjeldahl de 500 mililitros, añadir 50 ml. de agua y agitar suavemente.
Agregar 2 g. de SO₄Fe 7H₂O y 20 ml. de SO₄H₂, continuando como en 9.4.4 y 9.4.5.
Modalidad A:
N total − N insoluble = N soluble.
N soluble − N obtenido en 9.4.5 = N nítrico.
Modalidad B:
N total antes de eliminar el N nítrico menos N total después de su eliminación = N nítrico.
1 Association of Official Analytical Chemist, «Official Methods of Analysis». Ed. 1980, 2049-2051.
19 (a). Tiram
Extracción con cloroformo y posterior cuantíficación espectrofotométrica a 280 nm.
19 (a).2.1. Espectrofotómetro capaz de efectuar lecturas a 280 nm.
19 (a).3.1. Cloroformo.
19 (a).4.1. Preparación de la gráfica patrón.
Pesar, con precisión de 0,1 mg. 100 mg. de Tiram patrón y disolver en 100 ml. de cloroformo. Diluir a 1/10 con cloroformo y pipetear, en seis matraces, a 100 ml., 4, 6, 8. 10, 12 y 14 ml., completando el volumen con cloroformo. Los matraces contienen respectivamente, 4, 6, 8, 10, 12 y 14 μg/ml. de Tiram. Leer las absorbancias a 280 nm. y construir la gráfica patrón.
19 (a).4.2. Determinación.
Pesar, con aproximación de 0,1 mg., la cantidad de muestra necesaria para tener aproximadamente 0,1 g. de Tiram. Agitar durante cinco minutos con 60 ml. de cloroformo [19 (a).6.1], dejar en reposo hasta adquirir temperatura ambiente y enrasar hasta 100 con cloroformo. Pipetear una alícuota de 10 ml. a otro matraz aforado de 100 ml., enrasando con cloroformo y de éste pipetear una nueva alícuota de 10 ml. en matraz de 100 ml. enrasándolo con más cloroformo. Leer las absorbancias utilizando como referencia un blanco de cloroformo.

siendo:
A = contenido de Tiram en µ/ml. de la dilución final, leído en la gráfica (corregido según la pesada y riqueza del patrón).
P = peso en g., de la muestra.
19 (a).6.1. En caso de producto coloreado, añadir 0,3-0,4 g. de carbón activo y filtrar con papel de filtro, lavando éste con el cloroformo necesario para enrasar a 100 ml.
1. «Laboratorios y Análisis». Ministerio de Agricultura.
20. Residuo insoluble de sangre
Mediante dilución de la sangre en suero fisiológico, posterior filtración y desecación se obtiene el residuo insoluble, cuyo porcentaje se determina por pesada.
Este método es aplicable a leches en polvo, que pueden contener harina de sangre de las denominadas comercialmente solubles.
20.2.1. Embudo cilíndrico esmerilado con placa filtrante, soldada al misimo de 65 mm, de diámetro y porosidad 1. Su peso debe ser inferior al límite máximo de pesada de las balanzas analíticas, lo que normalmente se consigue con una altura del, cilindro igual o inferior a 4 centímetros.
20.2.2. Agitador electromagnético.
20.2.3. Estufa de desecación.
20.3.1. Suero salino fisiológico. Se obtiene disolviendo 9 g. de cloruro de sodio en 1.000 ml. de agua destilada.
20.3.2. Acetona.
20.3.3. Agua oxigenada de 100 volúmenes.
20.3.4. Acido clorhídrico concentrado (d = 1,19).
20.4.1. Tomar aproximadamente un gramo de muestra, medida con la precisión del miligramo, y diluir en un vaso de precipitados de 250 ml. con 50 ml. de suero salino fisiológico, agitando con un agitador electromagnético durante cinco minutos.
A continuación verter y filtrar el líquido poco a poco en el embudo cilíndrico con placa filtrante, previamente desecado y pesado con la precisión del miligramo y filtrar con ayuda de vacío.
Lavar agitador y vaso con pequeñas cantidades de suero fisiológico, que se vierten luego en el embudo hasta completar la filtración con una cantidad aproximada de 200 ml. de líquido de lavado. Lavar a continuación embudo y placa dos veces con agua destilada y añadir después 10 ml. de acetona, de forma que empapen bien la placa. Someter a vacío durante unos minutos.
A continuación pasar el embudo a una estufa y desecar a 100-105° C hasta peso constante (precisión del miligramo).
20.4.2. Limpieza del embudo cilíndrico con placa filtrante. Acoplar el embudo cilíndrico en un erlenmeyer y verter sobre la placa filtrante unos 50 ml. de agua oxigenada de 100 volúmenes y de 1 a 5 ml. de ácido clorhídrico concentrado. Dejar reposar durante unas veinticuatro horas y añadir, si es necesario, más agua oxigenada y ácido clorhídrico para ultimar la limpieza, A continuación lavar repetidamente cilindro y placa con agua desionizada.

siendo:
R = tanto por ciento de residuo insoluble.
P₁ = peso, en gramos, del residuo obtenido por diferencia de pesadas del embudo cilíndrico.
P₂ = peso, en gramos, de la muestra.
3 (a). Materia orgánica total (por calcinación)
En el presente método se determina la materia orgánica por calcinación tras extraer mediante lavados sucesivos sustancias que no son materia orgánica (sales amónicas, carbonatos, etc.).
3 (a).2.1. Matraz erlenmeyer de 200 ml. de capacidad.
3 (a).2.2. Agitador magnético.
3 (a).2.3. Estufa de precisión de ± 5° C.
3 (a).2.4. Desecador.
3 (a).2.5. Cápsula o crisol.
3 (a).2.6. Embudo de 10 cm. de diámetro.
3 (a).2.7. Balanza con precisión de 0,1 mg.
3 (a).2.8. Horno o mufla.
3 (a).2.9. Papel de filtro de cenizas conocidas.
3 (a).3.1. Acido clorhídrico concentrado (d = 1,19).
3 (a).3.2. Agua destilada desionizada.
3 (a).3.3. Solución de ácido clorhídrico al 5 por 100 (v/v).
Preparar la muestra según el método oficial número 1, desecando a 100-105° C.
Pesar, con precisión de 0,1 mg. 3-4 g. de la muestra e introducirla en un matraz de 200 ml. de capacidad. Añadir a continuación 50 ml. de reactivo 3 (a) 3.3. manteniéndolo en contacto durante quince minutos, agitando cada cinco minutos.
Transcurridos los quince minutos filtrar sobre filtro de cenizas conocidas, desecado y tarado, lavando el residuo que queda en el filtro con 100 ml. del reactivo 3 (a) 3.3 con alícuotas de 10-15 ml. cada vez hasta completar aproximadamente los 105 ml. de la solución de agua clorhídrico. Desecar el filtro a 100-105° C. Enfriar en desecador y pesar. Incinerar a la temperatura de 540° C. hasta obtener cenizas blancas, utilizando si es necesario 1 ml. de ácido nítrico para facilitar la calcinación total.

siendo:
a = peso, en g., de la muestra seca.
b = peso, en g., de la muestra lavada y seca.
c = peso, en g., dé la muestra lavada, seca y calcinada.
1. AFNOR. 1976. «Produits organiques». Supports et Milieux de culture. Norma U44 —160.
6. pH (1/25)
Medida del potencial eléctrico que se crea en la membrana de vidrio de un electrodo, función de las, actividades de iones hidrógeno a ambos lados de la membrana utilizando como referencia un electrodo de calomelanos con puente salino.
6.2.1. Potenciómetro (pH-metro) y juego de electrodos de vidrio y calomelanos.
6.2.2. Vasos de precipitados de 250 ml.
6.2.3. Varillas agitadoras o agitador magnético.
6.3.1. Solución ClKO.1 M. Disolver 7,456 g. de ClK en 100 ml. de agua destilada y diluir hasta 1 litro.
6.3.2. Solución tampón de biftalato potásico 0,05 M. Secar la sal durante dos horas a 110° C. Disolver 1P,21 g. de sal en agua destilada y diluir hasta 1 litro. Como, conservador añadir a la solución tampón 1 ml. de cloroformo o un cristal de timol de unos 10 mm. de diámetro. Esta solución tiene un pH de 4,00 en el intervalo de temperatura de 15 a 30° C.
6.3.3. Solución tampón de PO₄H₂K, 0,025 M y PO₄H Na₂ 0,025 M. Secar las dos sales durante dos horas a 110° C. Disolver 3,44 g. de PO₄H₂K y 3,55 g. de PO₄H Na₂ en agua destilada y diluir hasta 1 litro Como conservador añadir 1 ml. de cloroformo o un cristal de timol de unos 10 mm. de diámetro a la solución tampón. Esta solución tiene un pH de 6,90 a 15° C., de 6,88 a 20° C., de 6,86 a 25° C. y de 6,85 a 30° C.
6.4.1. Medida del pH en agua.
Pesar 4 g. de substrato (según el método número 1, procedimiento de preparación muestra) y añadir 100 ml. de agua destilada. Agitar vigorosamente con varilla o agitador magnético durante treinta minutos, dejándola reposar durante treinta minutos aproximadamente. Ajustar los electrodos de acuerdo que el electrodo de vidrio quede bien sumergido en la parte sedimentada de la suspensión y el electrodo de calomelanos quede en la solución-suspensión sobrenadante para que se establezca buen contacto eléctrico a través del capilar salino. La suspensión debe ser agitada inmediatamente antes de introducir los electrodos, pero no durante la medida.
La lectura del pH se mide de acuerdo con las instrucciones específicas del potenciómetro utilizando para la calibración del aparato soluciones tampón próximas a las lecturas del pH del substrato.
6.4.2. Medida del pH en ClK.
El procedimiento es igual que para el 6.4.1, utilizando el reactivo ClK (6.3.1) en lugar del agua destilada.
6.5. Expresión de los resultados.
6.5.1. Los resultados se expresan de acuerdo con la medida que nos da el potenciómetro (pH-metro), refiriendo su valor a la suspensión (substrato-agua o ClK) medida 1:25 en agua y 1:25 en ClK 0,1N, respectivamente.
1. «Métodos oficiales de análisis de suelos y aguas (1974)». Ministerio de Agricultura.
12. Necesidades de cal de los suelos ácidos
La necesidad de cal de un suelo ácido se define como la cantidad de cal necesaria para elevar el pH del suelo a un nivel deseado. El método consiste en una titulación directa con hidróxido cálcico.
12.2.1. Potenciómetro (pH-metro).
12.2.2. Vasos de precipitados de 100 ml.
12.3.1. Solución de hidróxido de calcio. Añádase 1 g. de óxido de calcio ó 1,5 g. de hidróxido cálcico por cada litro de agua desionizada libre de dióxido de carbono. Mézclese y déjese en reposo, protegiéndolo del aire, hasta que el exceso se sedimente. Aspírese la solución y manténgase en un recipiente protegida del dióxido de carbono del aire.
Introducir 10 g. de suelo en cada uno de siete vasos de precipitados de 100 ml. y agregar 0, 5, 15, 20, 30, 40 y 50 ml. de la solución de hidróxido cálcico (12.3.1) a los vasos de precipitados. Añadir agua suficiente para hacer que cada vaso tenga una razón de suelo al agua de 1:5. Dejar en reposo durante tres días y determinar el valor de pH de la suspensión suelo-agua. Hacer una gráfica del pH en función de los miliequivalentes de calcio añadido por 100 g. de suelo y determinar los millequivalentes de calcio necesarios para que el pH llegue al nivel deseado.
Necesidad de Cal en Tm. de Ca Co₃/Ha = 0,05 A.h.D. siendo:
A = meq de Ca/100 g. de suelo.
h = profundidad en cm. de suelo que se desea tratar.
D = densidad aparente del suelo.
En los cálculos de la necesidad de cal de los suelos es frecuente considerar, 15 cm. de profundidad de suelo.a tratar y una densidad aparente de 1,35.
1. «Methods of analysis for soils, plants and waters». University of California,
Alcoholes
4. Compuestos sulfurados
Formación de precipitado negro de sulfuro de mercurio.
4.2.1. Tubo de ensayo.
4.3.1. Alcohol.
4.3.2. Mercurio.
Introducir en el tubo 4.3.1 alrededor de 1 ml. de mercurio y 20 ml. de alcohol. Agitar durante uno a dos minutos. Observar la superficie del mercurio.
Si la superficie de mercurio permanece brillante sin aparición de un velo negruzco, el alcohol está exento de compuestos sulfurados.
1. «Codex Oenologique International. Alcool rectifie alimentaire».
Estatuko Aldizkari Ofiziala Estatu Agentzia
Manoteras Etorb., 54 - 28050 Madril




